

Abstract
Live attenuated vaccine strains, such as type I nonreplicating uracil auxotroph mutants, are highly effective in eliciting lifelong immunity to virulent acute infection by Toxoplasma gondii. However, it is currently unknown whether vaccine-elicited immunity can provide protection against acute infection and also prevent chronic infection. To address this problem, we developed nonreverting, nonreplicating, live attenuated uracil auxotroph vaccine strains in the type II Δku80 genetic background by targeting the deletion of the orotidine 5′-monophosphate decarboxylase (OMPDC) and uridine phosphorylase (UP) genes. Deletion of OMPDC induced a severe uracil auxotrophy with loss of replication, loss of virulence in mice, and loss of the ability to develop cysts and chronic infection. Vaccination of mice using type II Δku80 Δompdc mutants stimulated a fully protective CD8+ T cell-dependent immunity that prevented acute infection by type I and type II strains of T. gondii, and this vaccination also severely reduced or prevented cyst formation after type II challenge infection. Nonreverting, nonreplicating, and non-cyst-forming Δompdc mutants provide new tools to examine protective immune responses elicited by vaccination with a live attenuated type II vaccine.
INTRODUCTION
Toxoplasma gondii is an obligate intracellular parasite with an extremely broad host range inclusive of most birds and virtually all mammals (1). T. gondii chronically infects approximately one-third of the human population (2). Three major strain types are found in North America and Europe, and these type I, II, or III strains differ with respect to their virulence traits in mice (3). Type I strains are considered to be highly virulent, type II strains have low virulence, and type III strains are considered to be avirulent. Following acute infection by type II strains of T. gondii, a chronic infection is established. Chronic infection is characterized by slow-growing bradyzoite forms within tissue cysts in brain, muscle, and eye (4), and infection is considered to be lifelong (5, 6). Reactivated infection during AIDS or immune suppression may cause a severe and difficult to treat toxoplasmic encephalitis (7) or recurrent ocular toxoplasmosis (8). Vaccination against Toxoplasma is difficult due to the existence of multiple antigenically distinct strain types and multiple antigenically distinct developmental stages. Toxoplasma also can effectively escape immunity by the development of a chronic encysted stage that is not easily cleared by immune responses.
Antigen-based vaccine formulations provide only a partially protective immunity and do not prevent the establishment of tissue cysts and chronic infection following exposure to T. gondii (9). Immunization with T. gondii extracts or killed noninvasive parasites also fails to elicit significant immunity to reinfection with T. gondii (10, 11). Suboptimal vaccine priming using antigen-based vaccine formulations is not surprising in view of the requirement for populations of CD8+ and CD4+ T cells that produce the interferon gamma (IFN-γ) that is necessary to establish immunity and control of natural infection (12). In contrast to the suboptimal immunity elicited by antigen-based vaccines, immunization with whole transgenic organisms elicits potent Th1-biased immune responses associated with control of infection, and this vaccination strategy has enabled key experimental insights into host responses that prime immunity (13).
Disruption of the de novo pyrimidine synthesis pathway has led to remarkably effective vaccination strategies based on live attenuated uracil auxotroph vaccine strains (14). Disruption of carbamoyl phosphate synthetase II in the virulent type I RH strain (the CPS mutant) induced a severe uracil auxotrophy (15, 16). CPS is a live attenuated mutant that invades host cells normally but fails to proliferate within its vacuole due to nutrient limitation, and this loss of replication ability causes a complete absence of virulence during infection (15). Disruption of the orotidine 5′-monophosphate decarboxylase gene (OMPDC) in the virulent type I RH Δku80 strain (17) also produced a nonreplicating uracil auxotroph mutant that elicited protective immunity to virulent acute infection (18). The extreme attenuation and avirulence of type I T. gondii uracil auxotrophs are exemplified by survival of severely immune-deficient IFN-γ−/− knockout mice (15, 18, 19) and interleukin-2 receptor γ knockout (NOD/SCID/IL-2Rγ−/−) mice (20) that were infected with high-dose challenges of CPS.
Vaccination with CPS elicits a lifelong CD8+ T cell-dependent immunity against infection with type I strains (15, 18, 19, 21, 22). This immunity is also effectively elicited in tyk2−/− (23) or in MyD88−/− signaling-deficient mice (24). Immunity elicited by vaccination with CPS requires production of interleukin-12 (IL-12) (24, 25), IFN-γ (15, 19), and CD8+ T cell populations (25, 26), which possess cytolytic activity in vitro and in vivo (22). Previous studies have implicated a key role for active T. gondii invasion of host cells in mechanisms that promote T cell immunity to Toxoplasma (27–29). Recent studies using the CPS vaccination model have found a role for active parasite invasion in the activation of myeloid cells and the priming of CD4+ and CD8+ T cell immunity (30–32). Furthermore, phagocytosis of CPS by myeloid cells elicited surprisingly insignificant CD4+ and CD8+ T cell responses and ineffective immunity to Toxoplasma (21). In contrast, active invasion of myeloid cells by CPS triggered potent CD4+ and CD8+ T cell responses and highly effective protective immunity to Toxoplasma (21).
Previously, vaccination with CPS was shown to protect mice against a virulent acute infection after challenge with a type I or type II strain of T. gondii. However, this CPS vaccination did not completely prevent the development of cysts and chronic infection in mice that were challenged with a type II strain (33). Here, we developed nonreverting uracil auxotroph vaccine strains in the type II Δku80 genetic background by deleting the orotidine 5′-monophosphate decarboxylase gene (OMPDC). The resulting type II Pru Δku80 Δompdc mutants exhibit a severe uracil auxotrophy with loss of replication and virulence and undetectable cyst formation. Vaccination of mice with the type II Pru Δku80 Δompdc mutant stimulates a potent protective immunity against infection by type I and type II strains of Toxoplasma gondii.
MATERIALS AND METHODS
Primers.
All oligonucleotide primers used in this study for plasmid construction and PCR validation of genotypes of mutant strains are listed in Tables S1 and S2 in the supplemental material.
Plasmid constructs.
Yeast recombination was used to fuse 3 distinct genetic elements (a 5′ target flank, a hypoxanthine-xanthine-guanine phosphoribosyltransferase gene [HXGPRT] selectable marker, and a 3′ target flank) in their correct order in the yeast shuttle plasmid vector pRS416. This recombination was performed using 29- to 36-bp crossovers common to pRS416 and the HXGPRT selectable marker or to the gene targeting flanks (see Table S1 in the supplemental material) (18, 34). Gene knockout targeting plasmids were engineered to delete a short region of the gene's predicted 5′ untranslated region (UTR) and the entire predicted coding region of the targeted genomic locus (www.ToxoDB.org, version 12.0). Targeting plasmids were first verified by restriction enzyme digest and were sequenced to verify 100% gene homology to the available type II ME49 genome sequence (17).
Plasmid pOMPP4 was constructed to delete nucleotides 2709536 to 2712859 in the OMPDC locus defined as TGME49_259690 (55.m04842) on chrVIIb of the ToxoDB database. The HXGPRT minigene cassette (35, 36) was fused between a 1,055-bp 5′ genomic targeting flank and a 994-bp 3′ genomic targeting flank amplified from DNA isolated from the Pru Δku80::HXGPRT strain (37).
Plasmid pOMPPC4 was constructed to remove HXGPRT from the disrupted chromosomal OMPDC locus of the Pru Δku80 Δompdc::HXGPRT strain. Plasmid pOMPP4 was digested with PmeI to remove the HXGPRT cassette fragment followed by self-religation.
Plasmid pNUPP3 was designed to delete nucleotides 1497792 to 1500995 of the uridine phosphorylase (UP) locus on chrXI annotated as TGME49_310640. The HXGPRT minigene cassette was fused between a 1,024-bp 5′ targeting fragment and a 934-bp 3′ targeting fragment.
Plasmid pgUPROMP was designed to complement uracil auxotrophy while disrupting the uracil phosphoribosyltransferase locus (UPRT). A chromosomal segment of 3,258 bp corresponding to nucleotides 2712430 to 2709172 (−) on chrVIIb (the OMPDC gene) was flanked with a 1,151-bp 5′ UPRT targeting DNA flank and a 1,119-bp 3′ UPRT targeting DNA flank. Plasmid pGUPROMT was designed to replace nucleotides corresponding to 2718655 to 2722697 of the annotated UPRT chromosomal locus TGME49_312480 chrXI.
Strains, culture conditions, and PFU assays.
The parental strains of T. gondii used in this study are Pru Δku80::hxgprt and Pru Δku80 Δhxgprt (17). The RH strain (38), designated the RH(EP) strain (39), was used as a virulent type I strain in challenge infections. All strains that were used or developed in this study are listed in Table 1. Parasites were maintained by serial passage in diploid human foreskin fibroblasts (HFF) at 35°C (40). PFU assays were performed over 8 days (41) unless otherwise stated. Uracil, uridine, cytidine, deoxyuridine, deoxycytidine, xanthine, mycophenolic acid (MPA), and 5-fluorodeoxyuridine (FUDR) were obtained from Sigma, Inc. 6-Thioxanthine (6TX) was obtained from Acros Organics (Thermo Fisher Scientific).
TABLE 1.
Strains used in this study
| Strain characteristic(s) | Parent strain characteristic(s) | Source or reference |
|---|---|---|
| RH | RH(ERP) | 38, 41 |
| RH Δku80 Δompdc::HXGPRT | RH Δku80 Δhxgprt | 18 |
| Pru Δku80::HXGPRT | Pru Δhxgprt | 37 |
| Pru Δku80 Δhxgprt | Pru Δku80::HXGPRT | 37 |
| Pru Δku80 Δompdc::HXGPRT | Pru Δku80 Δhxgprt | This study |
| Pru Δku80 Δup::HXGPRT | Pru Δku80 Δhxgprt | This study |
| Pru Δku80 Δompdc Δhxgprt | Pru Δku80 Δompdc::HXGPRT | This study |
| Pru Δku80 Δompdc Δup::HXGPRT | Pru Δku80 Δompdc Δhxgprt | This study |
| Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC | Pru Δku80 Δompdc::HXGPRT | This study |
Pyrimidine starvation viability assays.
Pyrimidine auxotrophs were serially diluted and inoculated into multiple 25-cm2 HFF flasks. Parasites were allowed to invade for 2 h in uracil medium at 35°C, and cell monolayers were then rinsed 3× with cold phosphate-buffered saline (PBS) to remove uracil and extracellular parasites. Infected cultures were incubated for various periods of time without uracil to initiate uracil starvation conditions, and then uracil was added back to the culture medium at various times and PFU were scored 8 days later (18). The percentage of initial viability remaining after different times of uracil starvation was determined.
Genomic DNA isolation and PCR.
Genomic DNA was purified using the DNA blood minikit (Qiagen). PCR products were amplified using a 4:1 mixture of Roche High Fidelity Expand Long and Expand Long Taq polymerases.
Transformation of Toxoplasma gondii and knockout verification strategy.
Electroporations were performed on the model BTX600 electroporator with 1.33 × 107 freshly isolated tachyzoites in the presence of ∼15 μg of linearized targeting plasmid DNA as described previously (17, 34, 37). Following selection of parasite clones, the genotype of the selected clones was validated in PCR assays to measure the following: (i) PCR 1, targeted deletion of the coding region of the targeted gene (DF and DR primers); (ii) PCR 2, correct targeted 5′ integration (CXF and 5′DHFRCXR primers); and (iii) PCR 3, correct targeted 3′ integration (3′DHFRCXF and CXR primers) using a previously described strategy (34).
Single and double knockouts at the OMPDC (Δompdc) and UP (Δup) loci.
The Pru Δku80 Δhxgprt strain was transfected with SpeI-linearized pOMPP4, or with SpeI-linearized pNUPP3, and knockouts were continuously selected in MPA (25 μg/ml), xanthine (250 μM), and uracil (250 μM) to select and isolate the cloned Pru Δku80 Δompdc::HXGPRT and Pru Δku80 Δup::HXGPRT strains, respectively. To remove the HXGPRT minigene cassette from the Pru Δku80 Δompdc::HXGPRT strain, parasites were transfected with SpeI-linearized pOMPPC4 and selected in 6TX (250 μg/ml) and uracil (250 μM). The Pru Δku80 Δompdc Δhxgprt strain was validated using PCR 4 with a forward primer (CLOMF) designed into the 3′ side of the 5′ targeting flank and the CXR primer. The Pru Δku80 Δompdc Δhxgprt strain was transfected with SpeI-linearized pNUPP3 to isolate the Pru Δku80 Δompdc Δup::HXGPRT double mutant strain following selection in MPA and xanthine.
Functional complementation of the RH Δku80 Δompdc::HXGPRT strain.
The Pru Δku80 Δompdc::HXGPRT strain was transfected with PmeI-linearized plasmid pgUPROMP containing the 3,258-bp genomic OMPDC gene. Following transfection, the culture was maintained in the presence of uracil (250 μM) for 24 h, and then uracil medium was removed, and the selection was continued in the absence of uracil. Parasites emerging from this selection were subcloned, and individual isolates were evaluated for their genotype to verify targeted deletion of UPRT and the simultaneous insertion of a functional allele of OMPDC at the UPRT locus. The genotype of the expected gene replacement at the UPRT locus was verified using PCR 5 to measure the deletion of the UPRT locus and by using PCR 6 and 7 to assay for correct 5′ and 3′ targeted integration of the genomic allele of OMPDC (using primers UPRTCXF with OMXEXR and OMXEXF with UPRTCXR). Individual clonal isolates were also examined for functional deletion of UPRT by measuring resistance to 50 μM FUDR to demonstrate loss of UPRT activity.
Mice, immunizations, T cell depletion, and challenge infections.
Adult 6- to 8-week-old C57BL/6 and IFN-γ−/− knockout mice (002287) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were maintained at the Dartmouth-Hitchcock Medical Center (Lebanon, NH) mouse facility, and mice were cared for and handled according to Animal Care and Use Program of Dartmouth College using National Institutes of Health-approved Institutional Animal Care and Use Committee guidelines. Mice were immunized twice 14 days apart with 3 × 106 tachyzoites by intraperitoneal injection. Purified anti-CD8 (clone 2.43) and isotype control (rat IgG2a) antibodies were purchased from BioXCell. Antibody (500 μg) was administered 1 day prior to challenge infections, and 250 μg of antibody was administered 0 and 3 days after challenge infection. CD8+ T cells were depleted by greater than 99%. Challenge infections were performed 30 days after vaccination.
Cyst burden measurements.
Brain cyst burdens were determined as previously described (33, 37). The Pru Δku80 background expresses green fluorescent protein (GFP) under the control of a bradyzoite-specific LDH2 promoter. Cysts were screened at ×150 magnification to detect GFP+ bradyzoites encased within a cyst wall structure.
Statistical analysis.
Unless noted otherwise, all experiments were repeated at least two times and results were similar between repeats. Statistical analysis was performed using Graphpad Prism 6 software. Data for PFU and cyst analysis was calculated using the 2-tailed unpaired Student t test. Student t tests were conducted under the assumption of equal variance. Survival experiments were analyzed using the log-rank Mantel-Cox test. A P value of <0.05 was considered to be statistically significant. P values of ≤0.05, 0.01, 0.001, or 0.0001 are indicated by *, **, ***, and ****, respectively, in Fig. 5 to 7.
FIG 5.
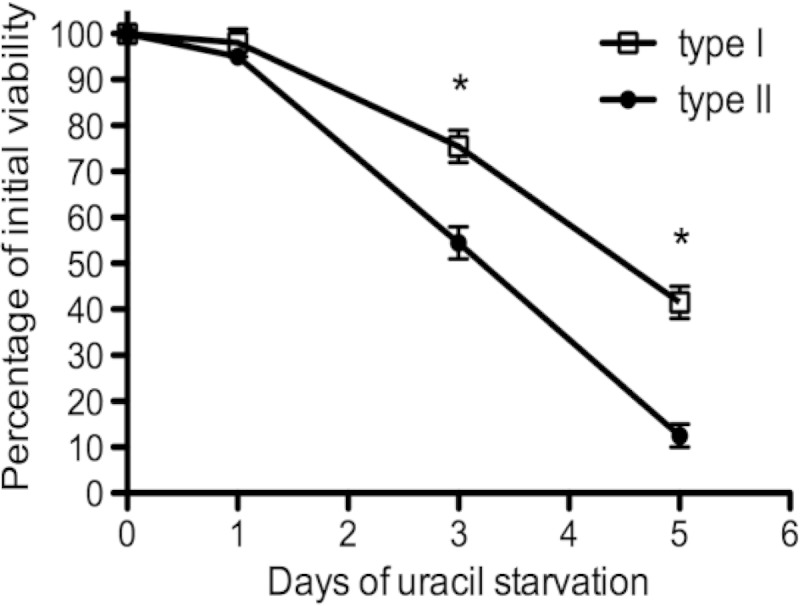
Pyrimidine starvation causes a rapid loss of parasite viability in type I and type II uracil auxotrophs. PFU assays were used to measure how rapidly pyrimidine auxotrophs lost viability intracellularly under conditions of pyrimidine starvation in a 5-day starvation assay. Viability profiles for the type II Pru Δku80 Δompdc::HXGPRT mutant (solid circles) and the type I RH Δku80 Δompdc::HXGPRT mutant (open squares) are shown. *, P < 0.05.
FIG 7.

Type II uracil auxotrophs elicit protective immunity to type I and type II T. gondii infection. Groups of C57BL/6 mice (n = 8) were vaccinated with the type II Pru Δku80 Δompdc::HXGPRT mutant, or groups of mice were left unvaccinated (naive). Mice were challenged with type I (RH) or type II (Pru Δku80::HXGPRT) infections 30 days after vaccination, and survival was monitored for 30 days. (A) Pru Δku80 Δompdc::HXGPRT mutant-vaccinated mice (○) or unvaccinated naive mice (▼) were challenged i.p. with 1000 tachyzoites of the virulent type I RH strain. (B) Pru Δku80 Δompdc::HXGPRT mutant-vaccinated mice (○) or unvaccinated naive mice were challenged intraperitoneally with 200 (▲) or 2 × 107 (▼) tachyzoites of the type II Pru Δku80::HXGPRT strain. (C) Naive or vaccinated mice (n = 4), as indicated, were treated with anti-CD8 antibody or isotype control antibody at the time of challenge infection with 4 × 107 type II Pru Δku80::HXGPRT tachyzoites. (D) Cyst burdens present in the brains of Pru Δku80 Δompdc::HXGPRT mutant-vaccinated mice that were rechallenged with 2 × 107 tachyzoites of the Pru Δku80::HXGPRT strain (n = 8) or present in naive mice that were challenged with 200 tachyzoites of the Pru Δku80::HXGPRT strain (n = 7). Cyst measurements were performed 31 days postinfection. **, P < 0.01; ****, P < 0.0001.
RESULTS
Type II pyrimidine pathway mutants.
To functionally define the predicted roles of the de novo pyrimidine synthesis and the pyrimidine salvage pathways (Fig. 1) in type II strains of T. gondii, we targeted the last enzymatic activity of the de novo pathway encoded by the orotidine monophosphate decarboxylase (OMPDC) gene that produces uridine-5′-monophosphate (UMP) from orotidine-5′-monophosphate to establish the pyrimidine nucleotide pool (14, 18). The Pru Δku80 Δhxgprt strain was targeted to delete the OMPDC gene to develop the Pru Δku80 Δompdc::HXGPRT mutant (Fig. 2A). The uridine phosphorylase (UP) gene encodes the salvage enzyme that converts uridine or deoxyuridine to uracil (42, 43) prior to its being salvaged into the nucleotide pool by conversion of uracil to UMP via the uracil phosphoribosyltransferase (UPRT) enzyme activity (Fig. 1) (39). The UP gene was deleted using a similar targeting strategy (Fig. 2A) to develop the Pru Δku80 Δup::HXGPRT mutant. The Pru Δku80 Δompdc::HXGPRT mutant was then retargeted at the deleted OMPDC locus to excise the HXGPRT selectable marker to develop the Pru Δku80 Δompdc Δhxgprt mutant (Fig. 2B), which was retargeted to delete the UP locus to develop the Pru Δku80 Δompdc Δup::HXGPRT double mutant (Table 1). The Δompdc Δup double mutant was developed to assess the ability of type II strains to salvage pyrimidine nucleosides from the host.
FIG 1.

Pyrimidine synthesis and salvage pathways in Toxoplasma gondii. The six-step de novo pyrimidine synthesis pathway (red) and the pyrimidine salvage enzyme pathway (blue) show both pathways produce uridine-5′-monophosphate (UMP) (boxed). Intermediate metabolites are shown (phosphoribosyl-1-pyrophosphate [PRPP]). (Top) The biosynthetic enzymes mediating each step of the de novo pathway are carbamoyl phosphate synthetase II (CPSII), aspartate carbamoyltransferase (ACT), dihydroorotase (DHO), dihydroorotate dehydrogenase (DHODH), orotate phosphoribosyltransferase (OPRT), and orotidine-5′-monophosphate (OMP) decarboxylase (OMPDC). (Bottom) The pyrimidine salvage enzymes mediating each step of the salvage pathway are cytidine deaminase (CTD), uridine phosphorylase (UP), and uracil phosphoribosyltransferase (UPRT).
FIG 2.

Targeted gene deletion at the orotidine 5′-monophosphate decarboxylase locus (OMPDC). (A) Strategy for deletion and validation of the endogenous OMPDC locus via targeted integration of the HXGPRT marker at the OMPDC locus of the Pru Δku80 Δhxgprt strain. Approximate locations of PCR products used to validate genotypes are depicted. The Pru Δku80 Δhxgprt parental strain was positive for the PCR 1 product, while the targeted OMPDC knockout was positive for PCR 2 and PCR 3 (see Materials and Methods). (B) Strategy for cleanup of the deleted OMPDC locus. The HXGPRT marker was removed from the disrupted OMPDC locus using the depicted strategy that utilized the ability to negatively select against the HXGPRT marker using 6TX selection. Successfully cleaned up strains with HXGPRT deleted were validated using PCR 4 (see Materials and Methods).
Functional analysis of pyrimidine synthesis and salvage in type II T. gondii.
The type II Δompdc, Δup, and Δompdc Δup strains were examined in PFU assays under various growth conditions to assess parasite growth and nutrient rescue profiles. The Δup mutant and the parental Pru Δku80 strains replicate normally in the absence or in the presence of uracil, uridine, or cytidine (data not shown). In contrast, the type II Δompdc (Fig. 3A) and Δompdc Δup (Fig. 3B) mutants exhibited a severe uracil auxotrophy and replication deficiency in the absence of uracil supplementation. In contrast to the complete rescue of growth observed with uracil supplementation (Fig. 3A and B), the replication of the Pru Δompdc mutant was only weakly rescued with high concentrations of uridine, as shown by the formation of very small plaques (Fig. 3C). In addition, the replication of the Pru Δompdc mutant was not significantly rescued in high concentrations of cytidine (Fig. 3C). Similar rescue profiles were observed using deoxyuridine and deoxycytidine, respectively (data not shown). The Pru Δompdc Δup double mutant was not detectably rescued by addition of either uridine or cytidine (Fig. 3D), indicating that pyrimidine pathways in type II strains mirror the pathways known to exist in type I strains (18). However, the rescue of growth in type II strains using uridine or cytidine supplementation was reduced in comparison to rescue of the corresponding type I mutants, suggesting a reduced pyrimidine salvage capacity for pyrimidine nucleosides is present in type II strains.
FIG 3.
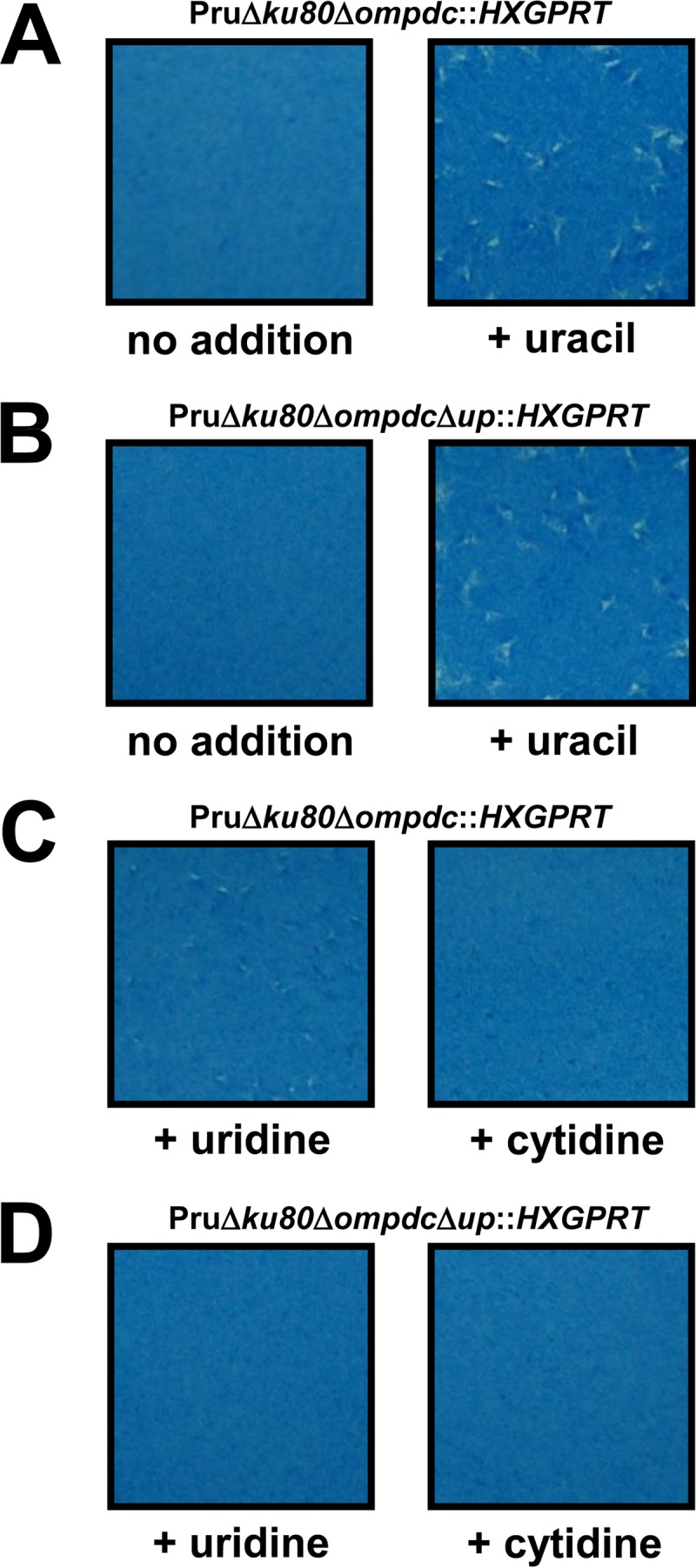
Functional rescue of type II Toxoplasma gondii uracil auxotrophs. Approximately 100 tachyzoites of the Pru Δku80 Δompdc::HXGPRT (A and C) or Pru Δku80 Δompdc Δup::HXGPRT (B and D) strain were inoculated into a PFU assay in the absence of pyrimidine supplementation (A and B), in the presence of uracil supplementation (200 μM) (A and B), in the presence of uridine supplementation (200 μM) (C and D), or in the presence of cytidine supplementation (200 μM) (C and D) of the growth medium. The culture monolayer was stained 8 days later to reveal PFU and HFF monolayers.
Complementation of type II pyrimidine auxotrophy.
The Pru Δompdc mutant was complemented by insertion of a functional wild-type allele of the OMPDC gene at the UPRT locus (Fig. 4A). The Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC complemented strain was resistant to FUDR (data not shown), and the growth rate of the complemented strain was normal whether uracil was absent or present in the culture medium (Fig. 4B).
FIG 4.

Complementation of the Pru Δompdc mutant. (A) Strategy for complementation of the Pru Δompdc mutant by simultaneous knockout of UPRT and targeted insertion of the genomic allele of OMPDC at the UPRT locus. The Pru Δku80 Δompdc::HXGPRT strain was transfected with plasmid pGUPROMP and parasites that were complemented for de novo pyrimidine biosynthesis were selected by growth in media without uracil supplementation. Approximate locations of PCR validation product PCR 5 (targeted deletion of UPRT), and PCR 6 and PCR 7 (targeted integration of the genomic allele of OMPDC) are shown (see Materials and Methods). Correctly targeted replacement clones were positive for the PCR 6 and PCR 7 products and were negative for PCR 5. (B) Functional rescue of complemented strain Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC. Approximately 100 tachyzoites of the Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC strain were assayed in an 8-day PFU assay in the absence (no addition) or presence of uracil.
Pyrimidine starvation.
PFU assays were used to measure how rapidly type II pyrimidine auxotrophs lost viability intracellularly under conditions of uracil starvation. When subjected to uracil starvation, the Pru Δompdc Δup mutant lost viability at a markedly higher rate than the Pru Δompdc mutant (data not shown). In addition, the type II Pru Δompdc mutant lost viability at a significantly higher rate than the corresponding type I RH Δompdc mutant (Fig. 5).
Pyrimidine biosynthesis is required for type II virulence and cyst development.
While the Pru Δku80::HXGPRT strain exhibits an ∼75% lethal dose (LD75) after a 2 × 105-tachyzoite challenge dose (Fig. 6A) and is uniformly lethal at higher doses (Fig. 6), C57BL/6 mice infected with the type II Pru Δku80 Δompdc::HXGPRT mutant did not exhibit acute virulence or signs of infection after receiving challenge doses of 2 × 105 (Fig. 6A), 2 × 106 (Fig. 6B), or 2 × 107 (Fig. 6C) tachyzoites, and IFN-γ−/− knockout mice survived a 2 × 105 challenge dose (Fig. 6D). In contrast, the complemented Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC strain exhibited the same acute virulence phenotype as the Pru Δku80::HXGPRT strain (Fig. 6).
FIG 6.

Pyrimidine biosynthesis is required for type II virulence and cyst development. Groups (n = 8) of C57BL/6 mice were inoculated i.p. with (A) 2 × 105 tachyzoites, (B) 2 × 106 tachyzoites, or (C) 2 × 107 tachyzoites of the Pru Δku80::HXGPRT strain (solid squares), the Pru Δku80 Δompdc::HXGPRT mutant (open circles), or the complemented Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC strain (solid circles), and survival was monitored for 30 days. (D) IFN-γ−/− knockout mice (n = 4) were infected with 2 × 105 tachyzoites of the parental Pru Δku80::HXGPRT strain (solid squares) or the Pru Δku80 Δompdc::HXGPRT mutant (open circles), and survival was monitored for 30 days (single experiment). (E) Cyst burdens in the brains of infected mice. Groups (n = 8) of mice were inoculated with 200 tachyzoites of the Pru Δku80::HXGPRT parental strain, with 200 tachyzoites of the complemented Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC strain, or with 200 or 2 × 106 tachyzoites of the Pru Δku80 Δompdc::HXGPRT mutant, and cyst burdens were determined 3 weeks postinfection. Cyst burdens were also determine in mice infected with 2 × 107 tachyzoites of the Pru Δku80 Δompdc::HXGPRT mutant 31 days postinfection. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Mice infected with 200 or 2 × 106 tachyzoites of the Pru Δku80 Δompdc::HXGPRT mutant failed to establish detectable brain cyst burdens by 3 weeks postinfection (Fig. 6E). (Three weeks postinfection is the peak time point of cyst formation in the Pru Δku80 background [37].) Cysts were also not detected in mice infected with this mutant or the Pru Δku80 Δompdc Δup::HXGPRT double mutant at any time point evaluated up to 7 weeks postinfection (data not shown). In contrast, mice infected with 200 tachyzoites of the complemented Pru Δku80 Δompdc::HXGPRT Δuprt::gOMPDC strain exhibited typical cyst burdens compared to the Pru Δku80::HXGPRT strain (Fig. 6E). In addition, cysts were not detected in mice that survived the 2 × 107 challenge dose (Fig. 6C) of the Pru Δku80 Δompdc::HXGPRT mutant (Fig. 6E). Collectively, these results demonstrate that type II uracil auxotroph mutants exhibit an avirulent phenotype and a severe defect in their ability to establish cysts and chronic infection.
Type II pyrimidine auxotrophs elicit protective immunity against type I and type II T. gondii infection.
We examined type II pyrimidine auxotrophs as live attenuated vaccine strains. C57BL/6 mice were vaccinated with the Pru Δku80 Δompdc::HXGPRT strain, and 30 days later, mice were challenged with 1,000 tachyzoites of virulent type I strain RH (LD1000) (Fig. 7A). All Pru Δku80 Δompdc::HXGPRT-vaccinated mice survived type I virulent challenge infection, whereas no naive unvaccinated mice survived the challenge infection. We also evaluated protection from type II infection. Pru Δku80 Δompdc::HXGPRT-vaccinated mice survived a type II virulent challenge of 2 × 107 tachyzoites, whereas no naive unvaccinated mice receiving this challenge dose survived (Fig. 7B). We performed CD8+ T cell depletion experiments to determine whether this protective immunity was dependent on CD8+ T cell populations. Vaccinated mice depleted of CD8+ T cells rapidly succumbed to type II challenge infection, whereas vaccinated mice treated with control isotype antibody survived challenge infection (Fig. 7C). In addition, 31 days after type II challenge infection, we examined the brains of mice that survived type II challenge infection to assess whether brain cysts (chronic infection) were present. While naive mice that received a 200-tachyzoite challenge of the Pru Δku80::HXGPRT strain produced high numbers of cysts, cyst burdens were not detected in Pru Δku80 Δompdc::HXGPRT-vaccinated mice that were rechallenged with 2 × 107 tachyzoites of the PruΔ ku80::HXGPRT strain (Fig. 7D).
DISCUSSION
Type I uracil auxotroph vaccines were previously described to stimulate potent T cell-mediated immunity to virulent type I challenge infection (15, 18, 19, 21–24). While the type I CPS uracil auxotroph vaccine also protected vaccinated mice from virulent type II challenge infection and reduced chronic infection (cyst burdens) by ∼98%, vaccinated mice were incompletely protected from chronic infection (33). Our results using a nonreverting, nonreplicating, type II vaccine strain suggest that vaccination with a type II strain may provide a more secure immunity to type II infection by preventing cyst development and chronic infection. Type II uracil auxotroph vaccines protected mice from infection by both type I and type II strains.
Our investigation of pyrimidine auxotrophy in the type II background of T. gondii indicates that the pyrimidine synthesis and salvage pathways mirror the pathways previously reported in type I strains (14–16, 18). The uridine phosphorylase activity of type II is used to some extent for physiological salvage of uridine and deoxyuridine based on the observation of the formation of very small plaques when using either of these nucleosides to rescue the growth of the Pru Δku80 Δompdc::HXGPRT vaccine strain. While visible plaques were observed in type I uracil auxotrophs using biochemical rescues with cytidine or deoxycytidine (18), visible plaques were not observed after cytidine or deoxycytidine rescue of the type II uracil auxotrophs in plaque assays. However, inspection of these cultures prior to staining did reveal tiny zones of infection involving just several infected cells without detectable lysis of the monolayer. These results suggest that cytidine and deoxycytidine can be converted by type II strains into uridine or deoxyuridine, respectively, by the action of a cytidine deaminase activity. Nonetheless, the ability to rescue type II uracil auxotrophs with pyrimidine nucleosides was notably reduced in comparison to rescue profiles previously reported for type I uracil auxotrophs (18). In addition, we demonstrated that pyrimidine starvation also occurs at a higher rate in type II uracil auxotrophs than in corresponding type I mutants. Together, these results indicate that type II strains rely more heavily on the de novo pyrimidine synthesis pathway due to a reduced capacity to salvage pyrimidine nucleosides by comparison to type I uracil auxotroph mutants (18). While these findings may be explained by a reduced metabolic capacity at the cytidine deaminase, uridine phosphorylase, or uracil phosphoribosyltransferase steps of salvage (18), it is possible that this metabolic difference may also be affected by the inherent capacity of type I and type II strains to transport nutrients from the host cell into the vacuole or into the parasite, where nucleobases and nucleosides become available for incorporation into the UMP pool.
Deletion of OMPDC in the type II background of T. gondii induced a severe uracil auxotrophy, with loss of replication and loss of virulence in mice. Furthermore, cyst burdens and chronic infection were not established following high-dose infection or vaccination of mice with type II Δompdc uracil auxotroph mutants. Vaccination of mice with the Pru Δku80 Δompdc mutant stimulated a fully protective CD8+ T cell-dependent immunity that prevented virulent acute infection by type I and type II strains of T. gondii, and this vaccination also severely reduced or potentially prevented cyst formation after type II challenge infection in C57BL/6 mice. In our cyst burden analysis after challenge infections, we scored ∼50% of the brain homogenates for cysts using dark-field microscopy to detect GFP+ cysts. Extremely low-level cyst burdens may be present, although we estimate cyst burdens in rechallenged vaccinated mice to be below 1 cyst per brain.
Acute virulence in type I T. gondii uracil auxotrophs is attenuated by at least 8 logs (18), and type II uracil auxotroph virulence appears to be similarly attenuated. Type I uracil auxotrophs were previously demonstrated to elicit remarkably potent CD4+ and CD8+ T cell responses following vaccination of mice (15, 18-25, 27, 30-33). Consequently, nonreverting, nonreplicating, and non-cyst-forming Δompdc type II uracil auxotroph mutants provide new and improved vaccination tools to elicit protective immunity to T. gondii and to examine the host immune responses that are elicited by vaccination with a live attenuated type II vaccine strain.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants AI041930, AI097018, and AI105563. ToxoDB, PlasmoDB, and EuPathDB are part of the NIH/NIAID-funded Bioinformatics Resource Center.
The valuable work of the developers of the Toxoplasma gondii Genome Resource at www.ToxoDB.org is gratefully acknowledged.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02756-14.
REFERENCES
- 1.Dubey JP. 2007. The history and life cycle of Toxoplasma gondii, p 1-18. In Weiss LM, Kim K (ed), Toxoplasma gondii: the model apicomplexan parasite: perspectives and methods. Elsevier, London, United Kingdom. [Google Scholar]
- 2.Jones JL, Kruszon-Moran D, Wilson M, McQuillan G, Navin T, McAuley JB. 2001. Toxoplasma gondii infection in the United States: seroprevalence and risk factors. Am J Epidemiol 154:357–365. doi: 10.1093/aje/154.4.357. [DOI] [PubMed] [Google Scholar]
- 3.Howe DK, Sibley LD. 1995. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis 172:1561–1566. doi: 10.1093/infdis/172.6.1561. [DOI] [PubMed] [Google Scholar]
- 4.Weiss LM, Kim K. 2000. The development and biology of bradyzoites of Toxoplasma gondii. Front Biosci 5:D391–D405. doi: 10.2741/Weiss. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denkers EY, Bzik DJ, Fox BA, Butcher BA. 2012. An inside job: hacking into JAK/STAT signaling cascades by the intracellular protozoan Toxoplasma gondii. Infect Immun 80:476–482. doi: 10.1128/IAI.05974-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dubey JP, Lindsay DS, Speer CA. 1998. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev 11:267–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luft BJ, Remington JS. 1992. Toxoplasmic encephalitis in AIDS. Clin Infect Dis 15:211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- 8.Jones JL, Holland GN. 2010. Annual burden of ocular toxoplasmosis in the US. Am J Trop Med Hyg 82:464–465. doi: 10.4269/ajtmh.2010.09-0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang NZ, Chen J, Wang M, Petersen E, Zhu XQ. 2013. Vaccines against Toxoplasma gondii: new developments and perspectives. Expert Rev Vaccines 12:1287–1299. doi: 10.1586/14760584.2013.844652. [DOI] [PubMed] [Google Scholar]
- 10.Butcher BA, Denkers EY. 2002. Mechanism of entry determines the ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect Immun 70:5216–5224. doi: 10.1128/IAI.70.9.5216-5224.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subauste CS, Wessendarp M. 2000. Human dendritic cells discriminate between viable and killed Toxoplasma gondii tachyzoites: dendritic cell activation after infection with viable parasites results in CD28 and CD40 ligand signaling that controls IL-12-dependent and -independent T cell production of IFN-gamma. J Immunol 165:1498–1505. doi: 10.4049/jimmunol.165.3.1498. [DOI] [PubMed] [Google Scholar]
- 12.Dupont CD, Christian DA, Hunter CA. 2012. Immune response and immunopathology during toxoplasmosis. Semin Immunopathol 34:793–813. doi: 10.1007/s00281-012-0339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dzierszinski FS, Hunter CA. 2008. Advances in the use of genetically engineered parasites to study immunity to Toxoplasma gondii. Parasite Immunol 30:235–244. doi: 10.1111/j.1365-3024.2007.01016.x. [DOI] [PubMed] [Google Scholar]
- 14.Fox BA, Chaudhary K, Bzik DJ. 2007. Toxoplasma: molecular and cellular biology. Horizon Bioscience, Norwich, United Kingdom. [Google Scholar]
- 15.Fox BA, Bzik DJ. 2002. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature 415:926–929. doi: 10.1038/415926a. [DOI] [PubMed] [Google Scholar]
- 16.Fox BA, Ristuccia JG, Bzik DJ. 2009. Genetic identification of essential indels and domains in carbamoyl phosphate synthetase II of Toxoplasma gondii. Int J Parasitol 39:533–539. doi: 10.1016/j.ijpara.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. 2009. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot Cell 8:520–529. doi: 10.1128/EC.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox BA, Bzik DJ. 2010. Avirulent uracil auxotrophs based on disruption of orotidine-5′-monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii. Infect Immun 78:3744–3752. doi: 10.1128/IAI.00287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gigley JP, Fox BA, Bzik DJ. 2009. Cell-mediated immunity to Toxoplasma gondii develops primarily by local Th1 host immune responses in the absence of parasite replication. J Immunol 182:1069–1078. doi: 10.4049/jimmunol.182.2.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baird JR, Byrne KT, Lizotte PH, Toraya-Brown S, Scarlett UK, Alexander MP, Sheen MR, Fox BA, Bzik DJ, Bosenberg M, Mullins DW, Turk MJ, Fiering S. 2013. Immune-mediated regression of established B16F10 melanoma by intratumoral injection of attenuated Toxoplasma gondii protects against rechallenge. J Immunol 190:469–478. doi: 10.4049/jimmunol.1201209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupont CD, Christian DA, Selleck EM, Pepper M, Leney-Greene M, Harms Pritchard G, Koshy AA, Wagage S, Reuter MA, Sibley LD, Betts MR, Hunter CA. 2014. Parasite fate and involvement of infected cells in the induction of CD4+ and CD8+ T cell responses to Toxoplasma gondii. PLoS Pathog 10:e1004047. doi: 10.1371/journal.ppat.1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordan KA, Wilson EH, Tait ED, Fox BA, Roos DS, Bzik DJ, Dzierszinski F, Hunter CA. 2009. Kinetics and phenotype of vaccine-induced CD8+ T-cell responses to Toxoplasma gondii. Infect Immun 77:3894–3901. doi: 10.1128/IAI.00024-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw MH, Freeman GJ, Scott MF, Fox BA, Bzik DJ, Belkaid Y, Yap GS. 2006. Tyk2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting IFN-gamma-dependent IL-10 reactivation. J Immunol 176:7263–7271. doi: 10.4049/jimmunol.176.12.7263. [DOI] [PubMed] [Google Scholar]
- 24.Sukhumavasi W, Egan CE, Warren AL, Taylor GA, Fox BA, Bzik DJ, Denkers EY. 2008. TLR adaptor MyD88 is essential for pathogen control during oral Toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol 181:3464–3473. doi: 10.4049/jimmunol.181.5.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson DC, Matthews S, Yap GS. 2008. IL-12 signaling drives CD8+ T cell IFN-gamma production and differentiation of KLRG1+ effector subpopulations during Toxoplasma gondii infection. J Immunol 180:5935–5945. doi: 10.4049/jimmunol.180.9.5935. [DOI] [PubMed] [Google Scholar]
- 26.Wilson DC, Grotenbreg GM, Liu K, Zhao Y, Frickel EM, Gubbels MJ, Ploegh HL, Yap GS. 2010. Differential regulation of effector- and central-memory responses to Toxoplasma gondii infection by IL-12 revealed by tracking of Tgd057-specific CD8+ T cells. PLoS Pathog 6:e1000815. doi: 10.1371/journal.ppat.1000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dzierszinski F, Pepper M, Stumhofer JS, LaRosa DF, Wilson EH, Turka LA, Halonen SK, Hunter CA, Roos DS. 2007. Presentation of Toxoplasma gondii antigens via the endogenous major histocompatibility complex class I pathway in nonprofessional and professional antigen-presenting cells. Infect Immun 75:5200–5209. doi: 10.1128/IAI.00954-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldszmid RS, Coppens I, Lev A, Caspar P, Mellman I, Sher A. 2009. Host ER-parasitophorous vacuole interaction provides a route of entry for antigen cross-presentation in Toxoplasma gondii-infected dendritic cells. J Exp Med 206:399–410. doi: 10.1084/jem.20082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gubbels MJ, Striepen B, Shastri N, Turkoz M, Robey EA. 2005. Class I major histocompatibility complex presentation of antigens that escape from the parasitophorous vacuole of Toxoplasma gondii. Infect Immun 73:703–711. doi: 10.1128/IAI.73.2.703-711.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baird JR, Fox BA, Sanders KL, Lizotte PH, Cubillos-Ruiz JR, Scarlett UK, Rutkowski MR, Conejo-Garcia JR, Fiering S, Bzik DJ. 2013. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res 73:3842–3851. doi: 10.1158/0008-5472.CAN-12-1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox BA, Sanders KL, Bzik DJ. 2013. Non-replicating Toxoplasma reverses tumor-associated immunosuppression. Oncoimmunology 2:e26296. doi: 10.4161/onci.26296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fox BA, Sanders KL, Chen S, Bzik DJ. 2013. Targeting tumors with nonreplicating Toxoplasma gondii uracil auxotroph vaccines. Trends Parasitol 29:431–437. doi: 10.1016/j.pt.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gigley JP, Fox BA, Bzik DJ. 2009. Long-term immunity to lethal acute or chronic type II Toxoplasma gondii infection is effectively induced in genetically susceptible C57BL/6 mice by immunization with an attenuated type I vaccine strain. Infect Immun 77:5380–5388. doi: 10.1128/IAI.00649-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rommereim LM, Hortua Triana MA, Falla A, Sanders KL, Guevara RB, Bzik DJ, Fox BA. 2013. Genetic manipulation in Deltaku80 strains for functional genomic analysis of Toxoplasma gondii. J Vis Exp e50598. doi: 10.3791/50598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donald RG, Carter D, Ullman B, Roos DS. 1996. Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J Biol Chem 271:14010–14019. [DOI] [PubMed] [Google Scholar]
- 36.Donald RG, Roos DS. 1998. Gene knock-outs and allelic replacements in Toxoplasma gondii: HXGPRT as a selectable marker for hit-and-run mutagenesis. Mol Biochem Parasitol 91:295–305. doi: 10.1016/S0166-6851(97)00210-7. [DOI] [PubMed] [Google Scholar]
- 37.Fox BA, Falla A, Rommereim LM, Tomita T, Gigley JP, Mercier C, Cesbron-Delauw MF, Weiss LM, Bzik DJ. 2011. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot Cell 10:1193–1206. doi: 10.1128/EC.00297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabin AB. 1941. Toxoplasmic encephalitis in children. JAMA 116:801–807. doi: 10.1001/jama.1941.02820090001001. [DOI] [Google Scholar]
- 39.Pfefferkorn ER. 1978. Toxoplasma gondii: the enzymic defect of a mutant resistant to 5-fluorodeoxyuridine. Exp Parasitol 44:26–35. doi: 10.1016/0014-4894(78)90077-2. [DOI] [PubMed] [Google Scholar]
- 40.Fox BA, Gigley JP, Bzik DJ. 2004. Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int J Parasitol 34:323–331. doi: 10.1016/j.ijpara.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Pfefferkorn ER, Pfefferkorn LC. 1976. Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp Parasitol 39:365–376. doi: 10.1016/0014-4894(76)90040-0. [DOI] [PubMed] [Google Scholar]
- 42.Iltzsch MH. 1993. Pyrimidine salvage pathways in Toxoplasma gondii. J Eukaryot Microbiol 40:24–28. doi: 10.1111/j.1550-7408.1993.tb04877.x. [DOI] [PubMed] [Google Scholar]
- 43.Iltzsch MH, Klenk EE. 1993. Structure-activity relationship of nucleobase ligands of uridine phosphorylase from Toxoplasma gondii. Biochem Pharmacol 46:1849–1858. doi: 10.1016/0006-2952(93)90592-K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.