

Abstract
Adherent-invasive Escherichia coli (AIEC) pathogroup isolates are a group of isolates from the intestinal mucosa of Crohn's disease patients that can invade intestinal epithelial cells (IECs) or macrophages and survive and/or replicate within. We have identified the ibeA gene in the genome of AIEC strain NRG857c and report the contribution of IbeA to the interaction of AIEC with IECs and macrophages and colonization of the mouse intestine. An ibeA deletion mutant strain (NRG857cΔibeA) was constructed, and the in vitro effect on AIEC adhesion and invasion of nonpolarized and polarized Caco-2 cells, the adhesion and transcytosis of M-like cells, the intracellular survival in THP-1 macrophages, and the contribution to intestinal colonization of the CD-1 murine model of infection were evaluated. A significant reduction in invasion was observed with the ibeA mutant in Caco-2 and M-like cells, whereas adhesion was not affected. Complementation of the mutant reestablished Caco-2 invasive phenotype to wild-type levels. Reduction in invasion did not significantly affect transcytosis through M-like cells at early time points. The absence of ibeA significantly affected AIEC intramacrophage survival up to 24 h postinfection. No significant changes associated with IbeA were found in AIEC colonization across the murine gastrointestinal tract, but a slight reduction of gamma interferon was observed in the ceca of mice infected with the ibeA mutant. In addition, a decrease in the pathology scores was observed in the ilea and ceca of mice infected with the ibeA mutant. Our data support the function of IbeA in the AIEC invasion process, macrophage survival, and inflammatory response in the murine intestine.
INTRODUCTION
The adherent-invasive Escherichia coli (AIEC) pathogroup was initially characterized in isolates from the ileal mucosa of Crohn's disease (CD) patients. Members of this group are able to efficiently invade a wide range of human epithelial cell lines in vitro (1, 2) and, additionally, they can survive and replicate within human and murine macrophages without causing cell death (3, 4). These traits were established as the key feature of AIEC, and it has been proposed that these properties would enable the bacteria to colonize/invade the intestinal mucosa, while at the same time causing sustained inflammation (5). The data in vivo, using different murine models of infection, suggest that AIEC might participate in the development of colitis in either a normal or a susceptible host. In germfree mice with an innate immune deficiency, such as those lacking the Toll-like receptor 5, colonization with AIEC, which was detectable up to 10 days postinfection, resulted in chronic colitis and persistence of inflammation for months, as an effect of an altered microbiota composition (6). This observation was not exclusive for mice with an altered immune response since the AIEC was able to persist in transgenic mice expressing the human receptor CEACAM (7) or in a wide array of streptomycin-treated mice (e.g., CD-1, DBA/2, 129e, and C3H), receiving both AIEC prototype strains LF82 and NRG857c, causing a chronic proinflammatory response in the ileum, cecum and colon (8).
Although there are some compelling data suggesting that AIEC, upon transient colonization, can instigate chronic inflammation, a key feature of the pathogenic process, the invasive properties are still not fully elucidated. Genetic analysis indicates that the prototype strains LF82 and NRG857c lack any of the traditional determinants associated with invasion found in other pathogenic E. coli strains and enteric pathogens, such as the invasion plasmid antigens (Ipa proteins) in the plasmid of Shigella spp. (9) or the proteins encoded in the tia locus in enterotoxigenic E. coli (1). Comparative genomic analysis, of the prototype strains LF82 (10) and NRG857c (11), identified some potential virulence determinants which might play a role in invasion. Both AIEC prototype isolates, as well as a third AIEC strain, UM146 (12), carried the gimA locus. The presence of the gimA locus was evaluated in a collection of extraintestinal pathogenic E. coli strains, and only 16.1% of them were found to carry it (13). Further, the gimA locus occurs almost exclusively in the B2 phylogenetic group. Within this locus, the presence of the ibeA (invasion of the brain endothelium protein A) gene has been described and initially identified in a newborn meningitis-causing E. coli (NMEC) isolate (14). IbeA has been reported to contribute to the pathology of NMEC (15), and the ibeA sequence has been linked to the reductive evolution exerted in this pathogen, suggesting a high degree of conservation (13).
In other pathogenic E. coli strains, a role for IbeA in pathogenicity has been reported. For example, in the case of avian pathogenic E. coli (APEC), 26% of highly pathogenic strains have been reported to be ibeA positive (16), and the absence of ibeA was associated with reduced mortality (from 27 to 4%) in chickens (17). Further, IbeA has been associated with early pathogenic events of APEC infections in chickens (18) and with the traversal of the human blood-brain barrier and subsequent access to the bloodstream in NMEC infections (14). The role of IbeA in resistance to oxidative stress has been reported in APEC strain BEN2908; however, its contribution to the pathogenic process is not clear (19). Scarce information exists regarding the molecular mechanisms mediating IbeA interactions with host cells. However, IbeA is described as a 50-kDa outer membrane protein (20), with seven predicted transmembrane domains with extended β-sheets that traverse through the membrane into the extracellular space (20). Two reports suggest that IbeA can bind host proteins as potential receptors, including vimentin and PTB-associated splicing factor (PSF) (21, 22). The ibeA gene is located in the same operon with two other genes, ibeR and ibeT. The ibeR gene reportedly encodes an RpoS-like regulator with a narrow functional spectrum that is believed to contribute to bacterial virulence adaptation in certain NMEC strains (23). Whether ibeR regulates ibeA is not known; however, invasion of brain endothelial cells is affected in its absence (23). The ibeT gene, located immediately downstream of ibeA, has been reported to affect adhesion and invasion of brain endothelial cells, even though it shows sequence homology to Na+/H+ antiporters (24).
We found intriguing that IbeA, the described invasin of the ibeRAT operon, is present in two prototypic AIEC strains, LF82 and NRG857c (10, 11), as well as in AIEC strains UM146 (12) and KD-1 (25). This might point toward an important role of IbeA within the AIEC pathogroup. Because the role of IbeA in the interaction of AIEC strains with the human gut has not been elucidated, the goal of the present study was to determine whether IbeA contributes to AIEC invasion of the intestinal epithelium and mediates interaction with the host cells. To address this question, (i) an ibeA deletion mutant in strain NRG857c (NRG857cΔibeA) was generated, (ii) the effect on the interaction of NRG857c and its ibeA mutant with intestinal epithelial cells and M-like cells was measured, (iii) the IbeA participation in the transcytosis process of NRG857c across M-like cells was evaluated, (iv) the function of IbeA on the interaction of NRG857c with human macrophages was examined and, lastly, (v) the effect on ICR (CD-1) mouse gut colonization was also assessed.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Strains and plasmids are listed in Table 1. Strains were routinely grown on Luria-Bertani (LB) broth or LB agar at 37°C. When required, growth medium was supplemented with antibiotics at the following concentrations: chloramphenicol, 30 μg ml−1; carbenicillin, 100 μg ml−1; streptomycin, 100 μg ml−1; and gentamicin, 100 and 20 μg ml−1. The AIEC strain NRG857c was used in the present study (2, 11). An ibeA mutant derivative strain was constructed by disruption of the ibeA gene via lambda red-mediated gene replacement (26) with the chloramphenicol acetyltransferase gene (cat) amplified from the plasmid pKD3 (27). The primers were IbeA2-F and IbeA2-R. The cat PCR product flanked by 40 bases upstream and downstream homologous to ibeA was subsequently electroporated into Red+Gam-producing AIEC NRG857c (with plasmid pKM201) (26, 28). The presence of the cat gene was further confirmed by PCR (see Fig. S1 in the supplemental material). For complementation, the ibeA gene and putative promoter region were amplified from NRG857c with the primers RCC20-F and RCC20-R. The primer pair amplified the ibeA region from NRG857c while containing restriction sites for XmaI and BamHI. The digested product was cloned into the low-copy-number plasmid pACYC177, and the resulting plasmid, pRCC20, was transformed into NRG857cΔibeA. The plasmid pRCC20 carries a 4.4-kb fragment containing the first two genes of the operon, ibeR and ibeA, plus the putative promoter region (positions 4662970 to 4666955 of the NRG857c genome, GenBank accession number CP001855 [11]). The mutant strain complemented with the plasmid pRCC20 was renamed RCC23-1. Disruption of the wild-type (wt) ibeA gene was confirmed at the genetic and at the transcriptional level and then compared to the control E. coli K-12 strain MG1655 and the human commensal isolate E. coli HS (29) (see Fig. S1A and B in the supplemental material). The absence of ibeA and its complementation did not affect growth of the different strains (see Fig. S1C in the supplemental material). We also confirmed that disruption of the ibeA gene did not affect the other two members of the operon (ibeR and ibeT) (see Fig. S2 in the supplemental material).
TABLE 1.
Bacterial strains, plasmids, and primers used in this study
| E. coli strain, plasmid, or primer | Relevant characteristic(s) and/or sequence (5′–3′)a | Source or reference(s) |
|---|---|---|
| E. coli strains | ||
| NRG857c | AIEC strain isolated from Crohn's disease patient; Tcr Apr Smr Cmr; O83:H1 serotype | Lab stocks (2, 11) |
| NRG857c (Cu) | AIEC strain isolated from Crohn's disease patient; antibiotic sensitive; O83:H1 serotype | 28 |
| NRG857cΔibeA | ibeA disrupted by the cat gene amplified from pKD3; Cmr | This study |
| RCC23-1 | NRG857cΔibeA strain transformed with the plasmid pRCC20 (Cmr Apr) | This study |
| MG1666 | Prototype E. coli K-12 strain; nonpathogenic | 60 |
| HS | Commensal E. coli from a human subject | 61 |
| DH5α | E. coli DH5α laboratory strain | Lab stocks |
| Plasmids | ||
| pKM201 | Temperature-sensitive Red-Gam-expressing plasmid; Apr | 26 |
| pKD3 | Template plasmid containing the cat gene template; Cmr | 27 |
| pACYC177 | 3.9-kb cloning vector | 62 |
| pRCC20 | ibeA amplified from NRG857c genomic DNA and cloned into pACYC177 (XmaI and BamHI sites) | This study |
| Primers | ||
| RCC04-F | ATGGGCCGAAGATGGCATTG; forward primer to amplify ibeR; product length, 1,084 bp | This study |
| RCC04-R | CCCTTGTTGCACGTACTCAC; reverse primer to amplify ibeR | This study |
| RCC05-F | ATTGCCGCAGCAATGAGTG; forward primer to amplify ibeA; product length, 1,075 bp | This study |
| RCC05-R | GCGGAATCATTACGCCATAAG; reverse primer to amplify ibeA | This study |
| RCC06-F | GCGTTTTCCTGCATTATTGC; forward primer to amplify ibeT; product length, 1,084 bp | This study |
| RCC06-R | TTCGGGCTAAGACTAACGG; reverse primer to amplify ibeT | This study |
| IbeA2-F | TAA ATA TGG AGA CTG GGG GGC GGA TGA AGA AAA TAA AAA CGT GTA GGC TGG AGC TGC TTC; for lambda red replacement | This study |
| IbeA2-R | ATT CAA ATA ATT CGC ATC ACC ATA CTC GGT GAC CGT ACT CAT GGG AAT TAG CCA TGG TCC; for lambda red replacement | This study |
| RCC20-F | TGTACACCCGGGTATCGACGGCCTGGAAATC; amplify ibeA with XmaI site; for cloning into pACYC177 | This study |
| RCC20-R | GTGGTCGGATCCACCGATGCCAATAACCAAC; amplify ibeA with BamHI site; for cloning into pACYC177 | This study |
Cmr, chloramphenicol resistance; Tcr, tetracycline resistance; Apr, ampicillin resistance; Smr, streptomycin resistance.
PCR and RT-PCR of ibeA.
The presence of ibeA in the strain NRG857c was confirmed via PCR by using the primers RCC05-F and RCC05-R. For confirmation of ibeA expression, reverse transcription-PCR (RT-PCR) was used. RNA was extracted from all strains with an RNeasy minikit (Qiagen) according to the manufacturer's instructions. Subsequently, 1 μg of RNA was reverse transcribed to cDNA with the QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's instructions. The ibeA gene was amplified from cDNA using the primers RCC05-F and RCC05-R. As a negative control, strain MG1655 was used. 16S rRNA was amplified by using 16S universal primers and used as an expression control.
Establishment of Caco-2 and M-like cell cultures.
Caco-2 (ATCC HTB-37) cells were seeded at a concentration of 5 × 105 on the upper side of polystyrene Transwell inserts (3-μm pore size, 12-mm filters; Corning) in 500 μl of complete growth medium and cultured until fully differentiated. Caco-2 complete growth medium contains minimum essential medium (MEM; Gibco) supplemented with 2 mM glutamine, 1 mM sodium pyruvate, 1× nonessential amino acids, penicillin-streptomycin (100 U/ml and 100 μg/ml), and 10% fetal bovine serum. The basolateral side of the insert is filled with 1.5 ml of complete growth medium. At 14 days after differentiation, 5 × 105 Raji B lymphocytes were added to the basolateral chamber in 1 ml of complete growth medium and maintained for 6 days (30, 31). The corresponding monocultures were used as a model for normal enterocytes. The integrity of the cell monolayer was measured by transepithelial resistance (32) before and after the experiments with an STX2 electrode/EVOM2 epithelial voltmeter (World Precision Instruments). Successful establishment of M-like cells was confirmed by transmission electron microscopy. M-like cell cultures showed sections with reduced or absent microvilli compared to Caco-2 monoculture (see Fig. S3 in the supplemental material), a finding consistent with previous reports (33). The percentage of Caco-2 cells that acquire M-like cell morphology can range from 15 to 30% of the monolayer (33, 34).
Bacterial adhesion and invasion to Caco-2 and M-like cells. (i) Cell preparation.
For adhesion, Caco-2 and M-like cells were cultivated in 12-well plates. The cells were washed twice with MEM without any supplement prior to infection. For bacterial suspension preparation, the strains were grown on LB medium to logarithmic phase (i.e., an optical density at 600 nm [OD600] of 1.0) and diluted in MEM to a concentration of 108 CFU/ml. The medium from the cell monolayers was then aspirated, and 500 μl of bacterial suspension was added (∼5 × 107 CFU; multiplicity of infection [MOI] of 100) (35, 36) to the apical transwell compartment. MOIs of 1 and 10 were used only in the indicated experiments. The bacterial suspension was serially diluted and plated to confirm the bacterial input.
(ii) Measurement of adhesion.
After 3 h, the monolayers were washed four times with phosphate-buffered saline (PBS) and then lysed with 200 μl of 0.1% Triton X-100 and plated on LB agar plates with the corresponding antibiotic. Time points of 10, 60, and 120 min postinfection were evaluated as indicated.
(iii) Measurement of invasion.
A gentamicin protection assay was performed after 3 h of infection, in which the monolayers were washed twice with PBS, and MEM containing gentamicin (100 μg ml−1) was added to the apical and basolateral chambers of the transwell system. Monolayers were incubated for 1 h and then were washed twice with PBS. The monolayer was then lysed with 200 μl of 0.1% Triton X-100, and released intracellular bacteria were plated for quantification.
(iv) Readout.
The adherent and invasive bacteria were calculated as percentage of initial inoculum (input) and then adjusted to be expressed as percent change compared to the wild type or as total recovered CFU.
Bacterial transcytosis across M-like cells.
For the M-like cell transcytosis experiments, the monolayers and bacteria were prepared as described above. An MOI of 100 (5 × 107 CFU) was added to the apical transwell compartment, and after infection, the basolateral medium was collected at 0, 1, 2, and 3 h postinfection, and the bacteria were enumerated on LB agar plates. The monolayer integrity was confirmed by measuring the transepithelial electrical resistance (TEER). Values above 300 Ω suggest integrity of the monolayer (34). As a negative control for M-cell-specific transcytosis, polarized Caco-2 monolayers were used, and transcytosis was also measured at 0, 1, 2, and 3 h postinfection. Caco-2 monolayers have a reduced transcytotic capability compared to M cells. All results are expressed as a percentage of the CFU number used for the infection.
Bacterial uptake, survival and replication in macrophages. (i) Cell preparation.
The human macrophage-like monocyte cell line THP-1 (ATCC TIB-202) was maintained in RPMI 1640 supplemented with 2 mM glutamine (Gibco catalog no. 11875-93), 1 mM sodium pyruvate, 10 mM HEPES, 1× nonessential amino acids, penicillin-streptomycin (100 U/ml and 100 μg/ml), and 10% fetal bovine serum. THP-1 cells were activated to macrophages (37) by using a concentration of 200 nM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich). THP-1 cells (2 × 105) were seeded on a 24-well plate and differentiated for 72 h in the presence of 200 nM PMA in 1 ml of RPMI 1640. After activation, the medium was removed, and the cells were left to rest for an additional 72 h (37) prior to infection.
(ii) Bacterial suspension preparation.
The bacterial strains were grown on LB media to logarithmic phase (OD600 of 1.0) and diluted in RPMI 1640 to a concentration of 2 × 107 CFU/ml. The cell monolayers were washed twice with PBS prior to receiving 1 ml of the bacterial suspension (2 × 107 CFU; MOI of 10) (35, 36). The bacterial concentration was confirmed and used as the bacterial input. After 1 h of infection (bacterial uptake), monolayers were washed twice with PBS, followed by incubation with RPMI 1640 plus gentamicin (100 μg ml−1) for 1 h. For later incubation times, the medium was replaced with RPMI 1640 plus 20 μg of gentamicin ml−1, followed by incubation for 4, 16, 24, and 48 h.
(iii) Readout.
The monolayers were lysed with 200 μl of 0.1% Triton X-100, and the released intracellular bacteria were serially diluted and plated on L agar. Survival was expressed as the mean percentage of the number of bacteria recovered at 4, 16, 24, and 48 h compared to that at 1 h after gentamicin treatment, which was defined as 100%. The uptake values were defined at 1 h after gentamicin treatment and expressed as the percentage of the infection inoculum.
In vivo bacterial infections in mice. (i) Mice and treatment.
Eight- to ten-week-old female CD-1 (ICR) mice were purchased from Charles Rivers Laboratories. Animals were housed in a specific-pathogen-free barrier under biosafety level 2 conditions. At 24 h before the infection, mice were given 25 mg of streptomycin via oral gavage.
(ii) Infection dose.
AIEC strains NRG857c and NRG857cΔibeA and the K-12 strain MG1655 were grown on LB medium overnight at 37°C. A bacterial suspension of 109 CFU was centrifuged and resuspended in 400 μl of PBS. Each animal received 109 CFU in 400 μl of PBS via oral gavage, and control groups received sterile PBS. Colonization was determined at 4 days (six mice per group), and persistence was determined at 14 days (seven mice per group) postinfection.
(iii) Readout.
After infection, the number of bacteria was monitored in the fecal pellets daily for the first 4 days and subsequently every other until 14 days postinfection. Feces were resuspended in PBS by vortexing, and bacteria were plated for enumeration. For quantification of bacteria in tissues, sections of the ileum (terminal), cecum, and colon were collected at 4 and 14 days postinfection in 15-ml tubes containing PBS and then homogenized using Covidien (Minneapolis, MN) precision disposable tissue grinder systems. The resuspended feces and tissue homogenates were then serially diluted and plated on MacConkey agar containing streptomycin (100 μg ml−1). After overnight incubation at 37°C, colonies were counted and expressed as either CFU per gram of feces or CFU per organ.
Cytokine quantification.
The colon and cecum of each mouse were removed, and the fecal contents were collected. To measure the local IFN response, tissue sections (three small punches) were then incubated in 1 ml of complete RPMI (10% fetal bovine serum, 1% l-glutamine, and 50 μg of gentamicin ml−1) for 18 h. The gamma interferon (IFN-γ) levels were determined by using a mouse IFN-γ ELISA Ready-Set-Go kit (eBioscience) according to the manufacturer's instructions.
Histopathological evaluation of mouse tissues.
Sections of mouse small intestine (terminal), cecum, and colon were excised at 14 days postinfection and washed with PBS. The sections were fixed in buffered 10% formalin, paraffin embedded, sectioned into 5-μm slices, and then stained with hematoxylin and eosin at the Histopathology Core at the University of Texas Medical Branch (UTMB). Three sections from the same tissue (small intestine, cecum, and colon) from each animal were stained and scored according to the histopathological scoring criteria used previously by Small et al. (8). According to these criteria, the lumen, the surface of the epithelium, the mucosa, and the submucosa are scored separately. The histopathological scoring was performed blindly by Elena Sbrana (UTMB).
Statistical analysis.
One-way analysis of variance (ANOVA), followed by Tukey's post-test analysis, was performed when comparing more than two groups, and multiplicity-adjusted P values are reported. Kruskal-Wallis one-way ANOVA, followed by Dunn's multiple-comparison test, was used for comparisons in the animal experiments to the variance of the populations. A 95% confidence interval was used for most of the analyses. For the macrophage survival assay and transcytosis experiments, a two-way ANOVA or two-way ANOVA repeated measures were used with a Bonferroni post-test analysis. All analysis was performed using GraphPad Prism 6.0 (GraphPad Software, Inc.). A P value of ≤0.05 was considered significant.
Electron microscopy.
Transwell inserts containing Caco-2 cells or M-like cells were washed gently with PBS and fixed in a mixture of 2.5% formaldehyde, 0.1% glutaraldehyde, 0.03% trinitrophenol, and 0.03% CaCl2 in 0.05 M cacodylate buffer (pH 7.2). Samples were processed further by postfixing in 1% OsO4, stained en bloc in 1% uranyl acetate (in 0.1 M maleate buffer [pH 5.2]), and embedded in Poly/Bed 812 (Polysciences, Warrington, PA). Specimens were examined by using a Philips 201 electron microscope.
RESULTS
Contribution of IbeA to invasion and adhesion of the intestinal epithelium.
The role of IbeA during adherence and invasion assays was evaluated comparing the wild-type (wt) strain NRG857c, NRG857cΔibeA, and the complemented strain RCC23-1 using Caco-2 and M-like cells. Reduction or increases in adhesion or invasion were expressed as a comparison to the levels observed for the strain NRG857c, which we defined as 100%. We initially studied the interaction of AIEC strains with nonpolarized Caco-2 cells, where different MOIs were tested. We found that in the strain NRG857cΔibeA adhesion seemed to be slightly increased compared to the wild type at an MOI of 1 (an increase of 24%, P = 0.052); this difference was not detected at an MOI of 100 (Fig. 1A). Regarding invasion, NRG857cΔibeA showed a decrease compared to the wild type at all MOIs tested (Fig. 1B). When studying the invasion of NRG857cΔibeA at different time points (10, 60, 120, and 180 min), we found consistently that the invasion was reduced compared to the wild-type strain NRG857c. We found that larger differences were observed at earlier time points. At 3 h postinfection, a reduction of 70% compared to the wild type (P < 0.0001) was still observed (Fig. 1C). We also found that after 2 h postinfection, the numbers of intracellular bacteria did not increase substantially (Fig. 1D). Furthermore, the deletion of ibeA in NRG857c caused a transient phenotype with significantly increased adherence at early time points but diminished thereafter (180 min) (Fig. 1E). The amount of adhered bacteria as a percentage of the inoculated bacteria was similar for the wild type and strain NRG857cΔibeA (Fig. 1F). It is possible that in the absence of IbeA certain adhesins are transiently overexpressed. It is important to mention that even though at early time points (10 and 60 min), we observed increased adherence of NRG857cΔibeA compared to the wild type, which was not reflected in increased invasion, impaired invasion was constant throughout the time course evaluated.
FIG 1.

Effect of time and multiplicity of infection (MOI) on adhesion and invasion of AIEC to intestinal epithelial cells. AIEC adhesion (A) and invasion (B) were measured in nonpolarized Caco-2 cells at MOIs of 1, 10, and 100, and bacteria were recovered and quantified at 3 h postinfection. The kinetics of invasion (C) and adhesion (E) were measured for up to 3 h at an MOI of 100. Adhesion and invasion were also represented as a percentage of initial inoculum used (D and F). The wt AIEC strain NRG857c is displayed in black bars, and NRG857cΔibeA (AIEC ΔibeA) is indicated in white bars, while the complemented RCC23-1 strain (ΔibeA/ibeA+) is indicated in gray bars. For the invasion assays, gentamicin was added and incubation proceed for an extra hour. The data are expressed as means ± the standard errors from two independent experiments (n = 10). ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (for comparisons between groups; one-way ANOVA, followed by Tukey's multiple-comparison tests, was used).
The invasion levels in the strain NRG857cΔibeA were reduced to 64.5% ± 3.1% compared to the levels observed in the wt strain (P < 0.0001) at an MOI of 100. Complementation with the ibeA-expressing plasmid partially restored the invasion levels, to 87% of the wt (P = 0.029). When measuring the adhesion levels, we found a slight reduction to 81.8% ± 5.8% (P = 0.11) (Fig. 2A and D). These results suggested that IbeA played a major role in the invasion of intestinal epithelial cells by AIEC, even though adhesion was not significantly affected at 3 h postinfection. When the same strains were tested in polarized Caco-2 cells, we found the differences to be even more prominent. The invasion levels of strain NRG857cΔibeA were reduced to 33.4% ± 7.1% of the wt levels (P = 0.0002), and the complemented strain restored the invasion to 113.7% ± 26% of the wt level (P = 0.63). The adhesion levels were consistent with those observed in nonpolarized Caco-2 cells, and they were not significantly different at 120.3% ± 7.1% of the wt levels (P = 0.17) (Fig. 2B and E). Overall, these results confirmed the role of IbeA in the AIEC invasion process. Polarization of Caco-2 cells has been reported to effectively affect the bacterium-epithelial cell interaction, making the monolayer surface less accessible and the barrier more restrictive (38). When measuring the invasion levels of the wild-type NRG857c strain, it was observed that the CFU recovered in polarized Caco-2 cells were 2.55 × 103 CFU, while in nonpolarized Caco-2 cells the values were 1 log higher, i.e., 1.70 × 104 CFU. Interestingly, this difference in CFU levels was not observed when adhesion was measured, 1.39 × 106 CFU in nonpolarized versus 2.99 × 106 CFU in polarized Caco-2 cells, supporting the idea that polarization had an impact on restricting the invasion event (Table 2). Our data showing that IbeA participates in AIEC invasion (Fig. 2E) also demonstrated that differences were not associated with damage to the cell monolayer, since the transepithelial resistance was not significantly affected between the different groups tested.
FIG 2.

Effect of IbeA on adhesion and invasion of AIEC on the intestinal epithelium. AIEC adhesion (A, B, and C) and invasion (D, E, and F) were measured in nonpolarized (A and D) and 14-day-polarized (B and E) Caco-2 cells. The role of IbeA in the interaction of AIEC with M-like cells was also evaluated (C and F). The wt AIEC strain NRG857c is represented in gray bars, and NRG857cΔibeA (AIEC ΔibeA) is represented in white bars, while the complemented RCC23-1 strain (ΔibeA/ibeA+) is represented in checkered bars. For all of the experiments, an MOI of 100 was used, and bacteria were recovered and quantified at 3 h postinfection. For the invasion assays, monolayers were incubated with gentamicin for an extra hour. The data are expressed as the means ± the standard errors from three independent experiments (n = 9). ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (for comparisons between groups; one-way ANOVA, followed by Tukey's multiple-comparison tests, was used).
TABLE 2.
Bacterial levels on Caco-2 and M-like cells during adhesion and invasion experiments
| Cell type | Mean ± SEa |
|||||
|---|---|---|---|---|---|---|
| NRG857c (wild type) |
NRG857cΔibeA |
RCC23-1 (ΔibeA/ibeA+) |
||||
| CFU recovered | % wild type | CFU recovered | % of wild type | CFU recovered | % of wild type | |
| Adhesion | ||||||
| Caco-2 (nonpolarized) | 1.39E+06 ± 1.48E+05 | 100 ± 8.1 | 1.19E+06 ± 9.34E+04ns | 81.8 ± 5.8ns | 9.67E+05 ± 8.19E+04* | 70.3 ± 7.3* |
| Caco-2 (polarized) | 2.99E+ 06 ± 3.35E+05 | 100 ± 11.24 | 3.45E+06 ± 2.05E+05ns | 120.3 ± 7.1ns | 3.75E+06 ± 3.40E+05ns | 121.9 ± 11.6ns |
| M-like cells | 8.46E+06 ± 1.20E+06 | 100 ± 14.23 | 8.76E+06 ± 1.35E+04ns | 107.7 ± 16.5ns | 6.51E+06 ± 4.35E+05ns | 78.5 ± 5.22ns |
| Invasion | ||||||
| Caco-2 (nonpolarized) | 1.70E+04 ± 1.44E+03 | 100 ± 3.9 | 8.95E+03 ± 8.16E+02*** | 64.5 ± 3.1*** | 1.54E+04 ± 9.21E+02* | 87.1 ± 3.8* |
| Caco-2 (polarized) | 2.55E+03 ± 3.99E+02 | 100 ± 12.19 | 9.78E+02 ± 2.57E+02*** | 33.42 ± 7.1*** | 2.31E+03 ± 5.12E+02ns | 113.7 ± 26ns |
| M-like cells | 2.71E+03 ± 3.82E+02 | 100 ± 23.1 | 4.32E+02 ± 3.18E+01* | 19.67 ± 1.05* | 1.13E+03 ± 7.31E+01ns | 44.4 ± 6.8ns |
Values represent the means at each time point. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns, not significant (statistical significance compared to NRG857c). The CFU recovered for the wild-type strain NRG857c was set as 100% for each experiment. A multiplicity of infection of 100 (5 × 107 CFU) was used.
Bacterial adhesion and invasion were also measured in M cells, which represent ca. 10% of the follicle-associated epithelium (39). Caco-2 cells were differentiated into M-like cells by performing coculture with Raji B lymphocytes. We found a slight reduction in the adhesion levels of NRG857cΔibeA to 78.5% ± 5.2% of the wild-type levels (P = 0.73). In addition, the levels of intracellular bacteria recovered in the ibeA mutant accounted for 19.6% ± 1.05% of the wild-type levels (P = 0.013), while the complemented strain restored the phenotype to 44.4% ± 6.8% (P = 0.060) (Fig. 2C and F). Interestingly, the reduction of AIEC invasion levels due to disruption of ibeA in M cells (19.6% of the wt) was more prominent than that observed with polarized Caco-2 cells (33.42% of the wt). Finally, nonquantitative visual representation of the changes in AIEC invasion associated with IbeA can be observed in Fig. 3. In Caco-2 monolayers treated with gentamicin, larger accumulates of intracellular bacteria can be observed within the cytosol of the cell infected with the wt and complemented strains than in the cell infected with the ibeA mutant (Fig. 3).
FIG 3.
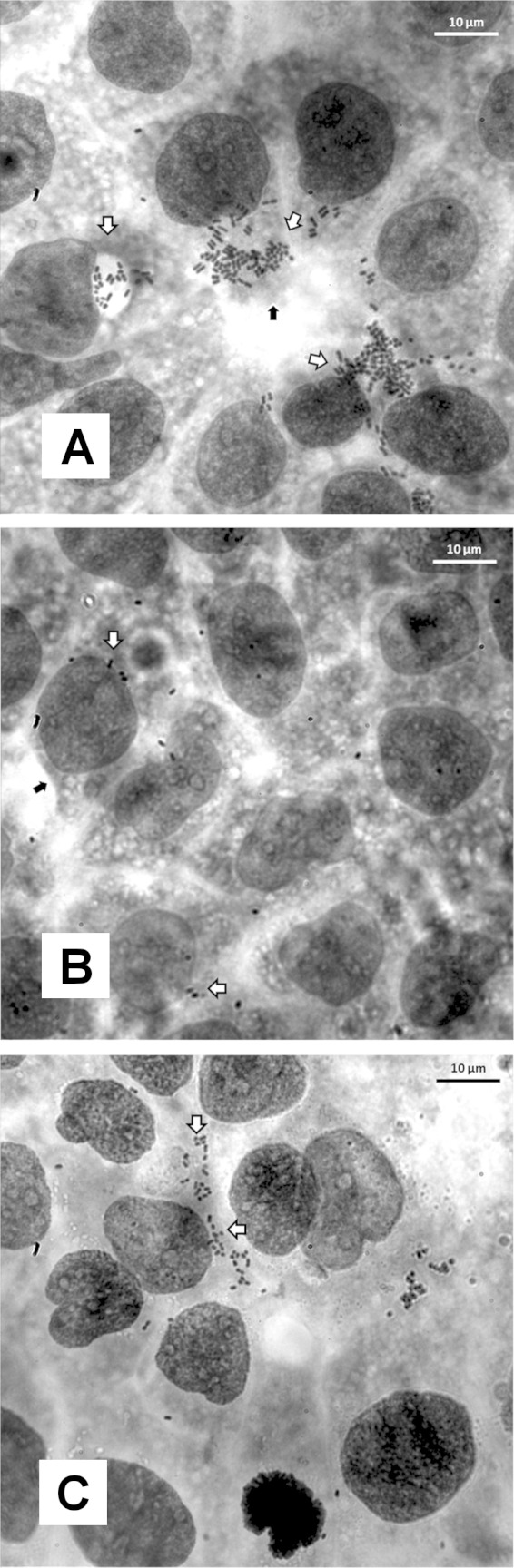
Visual examination of invasion of AIEC on intestinal epithelium. After infection for 3 h, monolayers were treated with gentamicin, and the monolayers were stained with Giemsa. Monolayers were visualized at ×100 magnification. Monolayers were infected with the wild-type strain NRG857c (A), NRG857cΔibeA (B), or the complemented RCC23-1 strain (ΔibeA/ibeA+) (C). White arrows mark sections of the monolayer where bacterial aggregates were present. Black arrows mark the cell membrane.
Contribution of IbeA to transcytosis across M cells.
Although M cells are important for sampling luminal antigens in the gastrointestinal tract, property exploited by a wide range of gut pathogens (39), transcytosis has been shown to be strain specific. For example, E. coli O157 (TUV 93-0) had translocation levels 4-fold higher than enteropathogenic E. coli O127 (E2348/69) and O26 (ZAP1139) (40). Scarce data are available regarding AIEC but one study showed that the transcytosis of the AIEC strain LF82 was 5-fold higher than that of a nonpathogenic E. coli K-12 strain (34). Therefore, the ability of AIEC to go across M cells was analyzed next to determine whether invasion of M cells by NRG857cΔibeA correlated with impaired transcytotic capability. As a control, the transcytosis was also measured across monolayers of polarized Caco-2 cells (Fig. 4B). At 0 h postinfection, no transcytosed bacteria were found in the basolateral compartment of polarized Caco-2 monolayers or M-like cell monolayers, suggesting that the integrity of the monolayer is intact (data not shown). When the transcytosed bacteria were measured at 1 and 2 h postinfection, no significant differences were found across the groups (Fig. 4A). At 3 h postinfection, the differences increased and the percentages of transcytosed bacteria were 0.65% ± 0.18% for NRG857c and 0.33% ± 0.07% for NRG857cΔibeA (P < 0.05) (Fig. 4A). Lower levels of transcytosed bacteria in NRG857cΔibeA were expected, since the numbers of intracellular bacteria were reduced; however, the reduction in transcytosis was not as marked as the reduction observed in invasion of M cells by AIEC. It is possible then that AIEC might possess other mechanisms of translocation through the monolayer besides transcytosis, such as paracellular transport. Surprisingly, at 3 h postinfection, lower levels of transcytosis were observed in the strain RCC23-1. Complementation partially restored the AIEC wild-type phenotype in M cells (Fig. 2F), but this trend was not observed in the translocation experiment, which suggests that increased IbeA production in this strain might interfere with the process of transcytosis.
FIG 4.
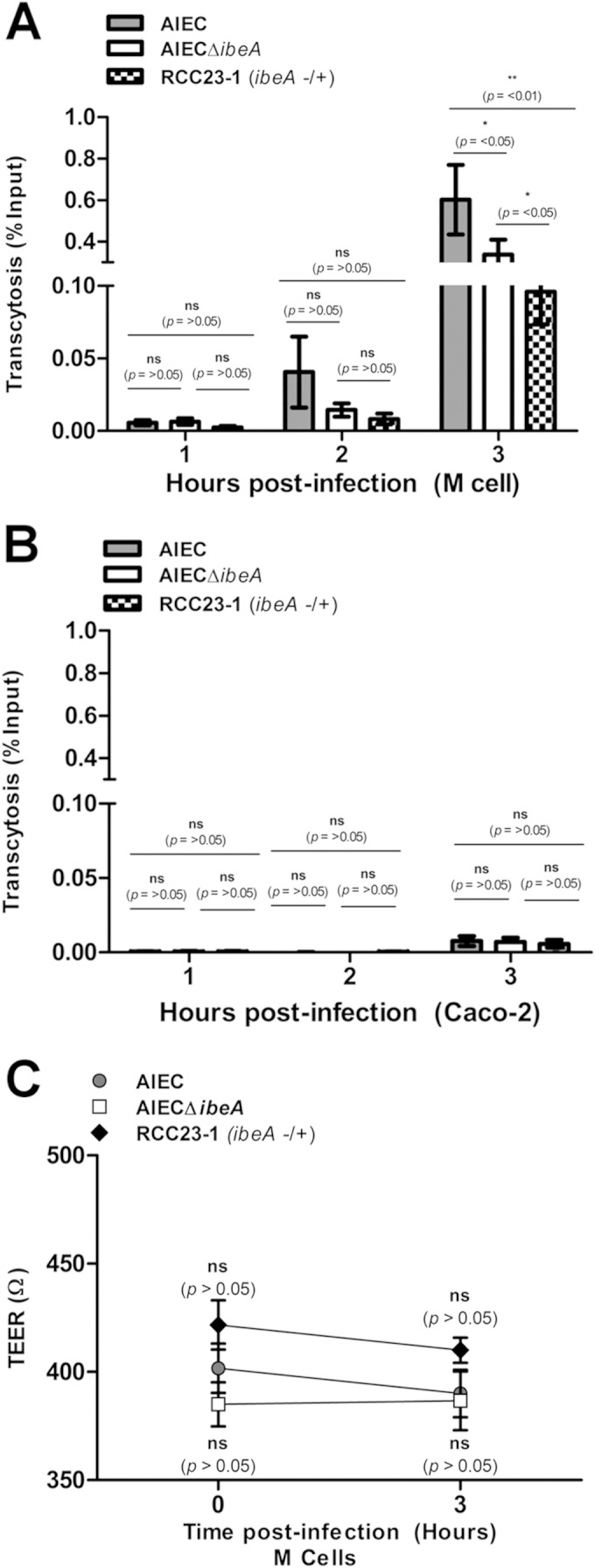
Transcytosis of the AIEC strains across Caco-2 and M-like cells. Transcytosis of NRG857c (AIEC), NRG857cΔibeA (AIEC ΔibeA), and the complemented RCC23-1 strain (ΔibeA/ibeA+) was performed. Monolayers of 5 × 105 Caco-2 or M-like cells were infected at an MOI of 100. (A) Assays were performed in a transwell chamber as described in Materials and Methods, and bacteria were collected from the basolateral media at 1, 2, and 3 h postinfection. (B) Caco-2 monolayers were used as controls where minimal transcytosis was observed. (C) TEER was measured before infection and at the final time point (3 h) to monitor monolayer integrity. Comparisons were made at each time between groups. Numbers are expressed as a percentage of the infection inoculum (5 × 107 CFU). The data are expressed as the means ± the standard errors from three independent experiments (n = 8). ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (for comparisons between groups; two-way repeated-measures ANOVA, followed by the Bonferroni post-test, was used).
Although coculture increased the permeability of the monolayer, we did not find significant changes in TEER values between infected Caco-2 or M cell monolayers (data not shown). Additionally, no significant TEER differences were observed between groups, suggesting that differences in translocation did not correlate with TEER values, as observed when comparing NRG857c samples postinfection. The TEER mean for NRG857c was 390 ± 10 Ω and that for NRG857cΔibeA was 386 ± 13 Ω, while that for the complemented strain RCC23-1 was 410 ± 5 Ω (Fig. 4C). Only a slight reduction of ∼10 Ω was observed when we compared the TEER preinfection (0 h) to postinfection (3 h) for all groups. As previously reported, all cultures where the TEER values fell below 300 Ω were excluded from experimentation (34).
Effect of ibeA on AIEC intramacrophage survival.
It has been shown that AIEC is taken up by macrophages, where it can survive and possibly contribute to sustain inflammation (5). To gain a more comprehensive understanding of the IbeA role in pathogenesis, survival within macrophages was determined as one of the hallmarks of AIEC infection (3, 4). Differentiated macrophage-like THP-1 cells were used as a model (37), and bacterial uptake (Fig. 5A) and bacterial survival and replication (Fig. 5B) were measured as previously described (41). The bacterial uptake by macrophages was determined and found to be ca. 20% of the initial inoculum (2 × 106 CFU) for all of the groups: NRG857c (20.69% of input), NRG857cΔibeA (17.39%) and the control E. coli DH5α (22.07%) (Fig. 5A). Survival or replication was measured at 4, 16, 24 and 48 h after gentamicin treatment. At 4 h postinfection, the percentages of intramacrophage survival were significantly different between the wild-type NRG857c (54%) and NRG857cΔibeA (27.31%) (P < 0.001). These differences were consistent throughout the time course of the experiment up to 24 h (32.42% for NRG857c versus 4.34% for NRG857cΔibeA) (P < 0.001) and 48 h (16.525% for NRG857c versus 1.85% for NRG857cΔibeA) (P < 0.001) (Fig. 5B). At later time points, the survival of AIECΔibeA was similar to that of a nonpathogenic E. coli DH5α strain. These results are consistent with previous reports where AIEC can survive within human macrophages (3, 4). Significant differences were found between NRG857c and NRG857cΔibeA at an early time point (4 h), as well as later time points (48 h), suggesting that IbeA might not only play a role in the early interaction between bacteria and macrophages but also facilitate bacterial survival. No intracellular replication was observed in this experiment compared to previous reports (4). It is worth noting that further insight into the mechanism by which IbeA contributes to AIEC intramacrophage survival needs to be studied, since a report of an APEC strain, BEN2908, suggests that ibeA deletion makes the strain more sensitive to killing by 25 mM H2O2 and implicates IbeA in the resistance to the reactive oxygen species (ROS) response (19).
FIG 5.

AIEC intramacrophage survival. THP-1 macrophages were infected with NRG857c (AIEC), NRG857cΔibeA (AIEC ΔibeA), and nonpathogenic E. coli DH5α at an MOI of 10 for 1 h. (A) After infection, cells were incubated with gentamicin and, after 1 h of treatment, the bacterial uptake was quantified. (B) The level for uptaken bacteria (calculated as CFU) determined after 1 h of treatment with gentamicin was defined as 100%. Intramacrophage survival of bacteria was calculated at 4, 16, 24, and 48 h. The data are expressed as the means ± the standard errors from two independent experiments (n = 7). Two-way analysis of variance with a Bonferroni post-test analysis was performed. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (all of the groups were compared to wt AIEC at the different time points).
Effect of ibeA deletion on AIEC intestinal colonization of mice.
A model of AIEC colonization in vivo has recently been reported, where our AIEC strain NRG857c persisted and caused inflammation in the intestines of conventional mouse strains (8). In order to determine the contribution of IbeA to AIEC-mediated colonization and inflammation in vivo, the aforementioned model was used. Two different time points were established to evaluate the role of IbeA in the early stage of colonization (4 days postinfection) (Fig. 6) and initial persistence (14 days postinfection) (Fig. 7). In all of the experiments, mouse groups treated with PBS or infected with E. coli K-12 MG1655 (11) were used as negative controls.
FIG 6.

NRG857c (AIEC) and NRG857cΔibeA (AIEC ΔibeA) bacterial counts in feces and intestine during early stage of colonization. Groups of six female streptomycin-pretreated CD-1 (ICR) mice were infected with 109 CFU of NRG857c (AIEC), NRG857cΔibeA (AIEC ΔibeA), or MG1655 (nonpathogenic K-12 E. coli) or treated with PBS (negative control) via oral gavage as described in Materials and Methods. (A) Feces were collected daily, and sections of the ileum, cecum, and colon were homogenized; CFU were determined at day 4 postinfection. Solid lines indicate the arithmetic means for each of the infected groups. (B) The CFU levels per organ and in the feces are also reported. The data are expressed as means ± the standard errors from six mice per group; findings representative of one experiment are shown. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01 (compared to the wt strain NRG857c; Kruskal-Wallis one-way ANOVA, followed by Dunn's multiple-comparison tests, was used).
FIG 7.

Bacterial levels of AIEC and AIEC ΔibeA in feces and intestine during persistence. Groups of seven female streptomycin-pretreated CD-1 (ICR) mice were infected with 109 CFU of NRG857c (AIEC), NRG857cΔibeA (AIEC ΔibeA), or MG1655 (nonpathogenic E. coli K-12) or treated with PBS (negative control) via oral gavage as described in Materials and Methods. (A) Feces were collected daily and subsequently every other day up to 14 days posttreatment. (B) Sections of the ileum, cecum, and colon were homogenized, and the CFU were determined at day 14 postinfection. The percentage of mice that were infected, based on our threshold of bacterial detection, was also determined. The results are expressed as the percentages of the infected mice. (C) The resulting curves were compared via log-rank test. The data are expressed as the means ± the standard errors from seven mice per group; representative results from one experiment are shown. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (compared against the wt strain NRG857c; Kruskal-Wallis one-way analysis of variance, followed by Dunn's multiple-comparison tests, was used).
At 4 days postinfection, no significant differences in the bacterial levels across the intestine between the wild-type strain NRG857c and NRG857cΔibeA were found. When comparing the NRG857c strain with E. coli MG1655, statistically significant differences were found in the ileum (P ≤ 0.01, ANOVA P value = 0.0028), the cecum (P ≤ 0.05, ANOVA P value = 0.0059), and the colon (P ≤ 0.05, ANOVA P value = 0.0160) (Fig. 6A). As expected, MG1655 was less efficient at colonizing and persisting than NRG857c. The differences observed between the bacterial levels were not attributed to changes in the percentage of infected mice. It was only in day 4 that the group infected with MG1655 had 50% of the mice with nondetectable bacteria (Fig. 6B).
Next, the IbeA contribution to persistence was measured. Because the bacterial numbers did not have a normal distribution, the mean number of CFU per gram of feces was compared against the wild-type strain NRG857c at each time point by a nonparametric one-way ANOVA. There were no statistically significant differences between mice infected with NRG857c and mice infected with NRG857cΔibeA during the course of infection. In contrast, the differences between NRG857c and MG1655 were statistically significant for the first 4 days of the study (Fig. 7A), and statistical analysis could not be applied at later time points because mice cleared MG1655 from the intestine. For example, at day 8 postinfection the bacterial levels of NRG857c were 1.56 × 103 CFU/g of feces, versus no detectable levels for MG1655 (see Table S1 in the supplemental material). In all cases, the bacterial levels in the feces were consistent with those found across the intestine (Fig. 7B). In each of the intestinal sections, NRG857c and NRG857cΔibeA had similar levels of colonization, while strain MG1655 was not detected in the intestine at 14 days postinfection (Fig. 7B).
At 14 days postinfection, the group infected with NRG857c showed bacteria detectable in the feces of 28% of mice, while the group infected with NRG857cΔibeA showed bacteria detectable in the feces of 37% of mice. Although no significant differences were found between NRG857c and NRG857cΔibeA (P = 0.83 [log-rank test]), differences in the curves were statistically significant compared to strain MG1655 (P = 0.002 and P = 0.001, respectively [log-rank test]) (Fig. 7C). These results suggest that the absence of IbeA did not seem to affect the ability of NRG857c to persist within the mouse intestine.
AIEC infection-associated inflammatory markers.
Inflammatory local immune response was measured in the ceca and colons of infected animals at 14 days postinfection. The inflammatory mediator IFN-γ was selected because it is known to be elevated in the lamina propria of Crohn's disease lesions (42). Further, in a model of inflammation mediated by the AIEC strain NRG857c, IFN-γ was elevated in the colon and the cecum of infected CD-1 (ICR) mice (8). Mice infected with NRG857c showed a significant increase in the levels of IFN-γ in the cecum (Fig. 8A) but not in the colon. The levels of IFN-γ in the ceca of animals infected with NRG857c were 123.4 ± 36.5 pg/ml, while the PBS control group had IFN-γ levels of 10.7 ± 0.79 pg/ml (P = 0.021), which corresponds to an increase of >10-fold. The colon presented a very different scenario (Fig. 8B), with levels of IFN-γ detected in the group infected with AIEC of 46.6 ± 13.8 pg/ml and no significant differences from the other groups.
FIG 8.

IFN-γ local inflammatory response in the colon and cecum after infection with AIEC. Groups of seven female CD-1 (ICR) mice were infected with NRG857c (AIEC), NRG857cΔibeA (AIEC ΔibeA), and MG1655 (nonpathogenic E. coli K-12) or treated with PBS (negative control), and tissue sections of the colon and cecum were obtained at 14 days postinfection. IFN-γ levels were measured by enzyme-linked immunosorbent assay in the cecum (A) and colon (B). The data are expressed as the means ± the standard errors from seven mice per group; representative results from one experiment are shown. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01 (compared against the wt strain NRG857c; an unpaired Student t test with Welch's correction, due to differences in the variance between sample populations, was used).
When comparing the IFN-γ levels in the groups infected with AIEC NRG857c and NRG857cΔibeA, a slight reduction was found in the cecum (from 123.4 ± 36.5 pg/ml to 44.5 ± 13 pg/ml), but the reduction did not reach statistical significance (P = 0.08). However, five of the seven mice infected with NRG857c had IFN-γ values higher than 100 pg/ml in the cecum, which was not observed in any of the mice infected with NRG857cΔibeA. Furthermore, IFN-γ values observed in mice infected with NRG857cΔibeA (44.5 pg/ml) resembled more closely the values observed in the group infected with the nonpathogenic MG1655 (43.9 ± 15 pg/ml; P = 0.97) (Fig. 8A). The results suggest that in the absence of IbeA, the IFN-γ response in the cecum was diminished, with NRG857cΔibeA resembling more closely the inflammatory pattern observed with the nonpathogenic MG1655. In contrast, the IFN-γ responses observed in the colon between the AIEC wild-type and NRG857cΔibeA strains did not differ drastically in their means (46.46 ± 13 pg/ml versus 46.84 ± 22 pg/ml, P = 0.39) (Fig. 8B), suggesting that the IFN-γ inflammatory response against AIEC in the colon was not as pronounced as in the cecum. It should be noted that IFN-γ levels were not detectable in mice sacrificed at 4 days postinfection (data not shown).
AIEC pathology in the murine intestine.
Histological analysis of hematoxylin-eosin-stained small intestines, ceca, and colons was performed in all seven animals from each AIEC-infected group, even if bacteria were not detected after 14 days (Fig. 7A and B). In the ilea of animals infected with AIEC NRG857c, a high pathology score (4.5) was obtained as a result of the damage in the epithelium. Desquamation of the surface epithelium (Fig. 9A) with focal ulceration of the mucosa and complete loss of villi and crypts was observed. The damage in the lumen of the ileum was not as marked as that observed in surface epithelium (black bars versus dark gray bars in Fig. 9A). In the group infected with NRG857cΔibeA (a 2.33 ileum pathology score) a disruption of the surface epithelium with desquamation and no major damage in the lumen was observed. The PBS-treated group consisted mostly of unremarkable mucosa with a slightly hypercellular lamina propria (a 0.5 ileum pathology score). Similar results were observed in the cecum, with the difference that the lumen showed moderate to dense presence of necrotic epithelial cells only when infected with NRG857c (dark gray bars in Fig. 9B and E). The pathology observed in the colon, consistent with our IFN-γ results, was unremarkable, and the differences in the pathology score means were not drastically different between the AIEC-infected groups (1.50 and 1.167) or the PBS-treated groups (1.50 versus 0.75) (Fig. 9C). This result contrasted with those observed in the small intestine (4.5 versus 0.5) and colon (4.16 versus 1.75). When we compared the pathology scores between NRG857c-infected and NRG857cΔibeA-infected animals, lower numbers were obtained for the latter group in the ileum (4.5 versus 2.33) and the cecum (4.16 versus 1.83) but not in the colon (1.50 versus 1.167). Overall, the results showed that in the mice colonized with NRG857cΔibeA, there is a reduction in the damage to the intestinal tissue, particularly in the ileum and cecum. This result is consistent with what has been previously shown with AIEC infections leading to tissue pathology (11) and supports the role of AIEC bacteria as a promoter of inflammatory disorders in the gastrointestinal tract (43).
FIG 9.

Ileal, cecal, and colonic pathologies after NRG857c (AIEC) or NRG857cΔibeA (AIEC ΔibeA) infection. The pathology scores were calculated for the small intestine (A), cecum (B), and colon (C) at 14 days postinfection after staining with hematoxylin and eosin. ICR (CD-1) mice were infected or treated as follows: AIEC (n = 6), AIEC ΔibeA (n = 6), or PBS (n = 3). Scores represent an average of three views per section, and data are expressed as means with standard deviations for each group. Panels D, E, and F are representative for small intestinal sections that were stained with hematoxylin and eosin and used to calculate pathology scores. Ilea of ICR (CD-1) mice infected with AIEC (D) or AIEC ΔibeA (E) or treated with PBS as a control (F) are displayed. The extensive damage to the intestinal epithelium can be observed (black arrows). The data are representative of the results from one experiment. The pathology scores for each intestinal segment are presented in parentheses. Pathology score means were compared using a one-way ANOVA, followed by Tukey's multiple-comparison tests. ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (for comparisons between groups).
DISCUSSION
It has become increasingly relevant to study AIEC, since evidence supporting the association of an imbalanced microbiota in the development of Crohn's disease (CD) has been accumulating in recent years (44). Among the microorganisms that have been reported to be enriched in patients with CD and possibly involved in the onset or aggravation of symptoms, Gram-negative bacteria such as Campylobacter species (45) or AIEC (1) have been reported. Several mechanisms have been proposed describing the interaction of AIEC isolates (2, 46, 47) and the host gut. Special attention has been devoted to identify virulence-associated factors in AIEC (10, 11), and we have paid special attention to invasion, as one of the characteristic properties of AIEC (1). As such, it is important to define which factors correlate with the pathogenic potential of AIEC. This is the first report associating the IbeA protein in the interaction of AIEC with intestinal epithelial cells and macrophages, and our results indicate that although there is a contribution of IbeA to AIEC invasion of intestinal epithelial cells and survival within macrophages, no major effect was associated with AIEC colonization in a mouse model, which suggests that AIEC is equipped with additional adhesins and/or invasins that are mediating these interactions.
Among the virulence factors of AIEC that might contribute to the interaction with the host are the type 1 pili (46), flagella (47), and long polar fimbriae (48, 49), which are reported to act in concert to promote the interaction of AIEC with the intestinal epithelium. However, flagella are ubiquitous in pathogenic and nonpathogenic E. coli strains (50) and not a unique factor for AIEC strains. Although type 1 pili in AIEC strains (7, 8) might contribute to increased interaction with abnormally expressed CEACAM in the host, AIEC prototype strains LF82 and NRG857c were still able to colonize the gut of conventional mouse strains such as ICR (CD-1), DBA/2, and C3H that lack CEACAM receptors (8). In addition, long polar fimbriae have been reported to primarily mediate the interaction of AIEC M cells (48). Based on this evidence, we decided to search for other virulence determinants associated with invasion in AIEC. We found the presence of the ibeA gene in the fully sequenced AIEC genomes (10–12) and based on the fact that reports have shown that ibeA is not ubiquitous among E. coli and is absent in nonpathogenic strains (13, 17), we characterized its function. Interestingly, a recent study screened E. coli strains associated with inflammatory bowel disease (IBD) for classic virulence genes associated with diarrheagenic and extraintestinal E. coli and did not detect the ibeA gene (51). However, not all of the strains of this collection were considered AIEC, even if associated with IBD; therefore, further studies are necessary to define the distribution of ibeA in AIEC strains.
The described role of IbeA as an invasin in other pathogenic E. coli isolates such as NMEC (14) and APEC (16) is consistent with our findings with AIEC strain NRG857c, where it plays an important role in the invasion of intestinal epithelial cells. The absence of ibeA accounted for a reduction in invasion of ca. 67% compared to wt; however, intracellular bacteria were still detectable, suggesting that IbeA is not the sole invasion determinant of AIEC. In addition, the effect of IbeA on AIEC invasion seems to be more prominent upon full Caco-2 polarization, a more restrictive epithelial barrier than the nonpolarized model (52). It is possible that IbeA invasion depends on the natural configuration of the monolayer to mediate the invasive process. Whether this is dependent on the spatial rearrangement of a potential receptor remains to be elucidated. The absence of IbeA also caused a significant reduction in intracellular AIEC in M-like cells; however, the wt phenotype was partially restored upon complementation. As mentioned above, since intracellular levels of bacteria were still found upon disruption of ibeA, it is important to define other virulence determinants associated with AIEC invasion. Genes for a complete type VI secretion system (T6SS), which has been reported to contribute to invasion in other enteric pathogens (53), have been found in NRG857c (11), but it is unknown whether they play any functional role in this process.
The fact that IbeA might contribute to AIEC invasion of intestinal epithelial cells, as well as M cells, where the bacteria could persist (1), is interesting because it has been reported that some pathogens can use M cells to translocate through the host intestinal tract (39). Our results did not find significant differences in transcytosis through M cells associated with IbeA at early time points, even though the percentage of transcytosed bacteria was lower in the ibeA mutant than in the wt strain. Therefore, it is plausible to propose that other factors, such as the long polar fimbriae, are participating in the transcytosis through M cells (48). Our results further support the idea that AIEC might have more than one mechanism to interact with M cells and access the underlying macrophages present in the lymphoid tissue. The survival within murine macrophages by AIEC isolates has been well described (3), where the survival at 24 h postinfection can range from 32 to 100% of bacteria phagocytosed in the case of the Crohn's disease-associated strains (4, 51). In our study, the survival of NRG857c in human macrophages at 24 h postinfection was 32.42%, which is consistent with what has been reported for other AIEC strains. The basis of AIEC intramacrophage survival has been associated with several factors, such as the induction of tumor necrosis factor alpha (TNF-α) (4) via an acidification of the phagosome, which contributes to AIEC replication or abnormalities in the autophagy system (54, 55) (such as ATGL16L1 and IRGM [56]). Our results showed that in the absence of IbeA, an impaired intramacrophage survival was observed. The mechanism associated with impaired survival has not been elucidated, but it is plausible to propose that IbeA, which belongs to the Pfam12831 family of FAD-dependent oxidoreductases (19), could play a role in survival to reactive oxygen species (19). Reactive oxygen species are an important part of the macrophage oxidative stress (57, 58), and during the first hours after bacterial invasion, where bacteria encounter stress conditions (59), the role of IbeA as a putative FAD-dependent oxidoreductase might be relevant. The fact that the deletion of the ibeA gene from NRG857c affected its survival within human macrophages supports the role of IbeA in AIEC intracellular survival (data not shown).
In the murine model of infection, we found that AIEC was able to persist within the gut of the mice up to 14 days. This provides additional evidence that AIEC can colonize the guts of conventional mice more efficiently than nonpathogenic E. coli. In our model, AIEC colonization was not directly a result of genetic deficiencies of the host that facilitate AIEC persistence, such as overexpression of CEACAM6 in the gut (5, 7). Colonization of AIEC was not restricted to one exclusive section of the gut, since bacteria were recovered from the small intestine, cecum, and colon, a finding indicating that AIEC has the ability to interact with different intestinal sections.
Although the absence of IbeA did not cause a significant reduction in murine intestinal colonization, this provides an alternative hypothesis indicating that AIEC is equipped with additional adhesins and/or invasins that mediate intestinal interaction. However, we found that IbeA is important for increased pathology in the ilea and ceca of mice, possibly due to increased IFN-γ secretion. The reduction in IFN-γ secretion in the absence of IbeA is possibly a consequence of impaired interaction with specific cells of the innate immune system, such as macrophages, and not a consequence of changes in colonization of the intestinal epithelium. Little difference in pathology was observed in the colon when comparing AIEC-infected animals with the PBS-treated group, suggesting that even though AIEC is present in the colon, greater damage to the epithelium occurs in the ileum and cecum.
We also observed a correlation between colonization by AIEC and development of inflammation with the wt strain, and similar inflammatory responses have been described in mice expressing CEACAM6 (7), as well as conventional mouse strains (8). It is important to clarify that in the conventional mouse strain studies, the host has a depleted microflora due to streptomycin treatment, and the introduction of a high dose (109 CFU) of AIEC might contribute to the inability of the microbiota to fully repopulate the intestine (6). This might be one of the factors associated with the damage reflected by the high pathology score compared to the control in that study. While in general terms, colonization of the gut by bacteria can elicit an inflammatory response, the fact that the response was observed still at 14 days suggests a long-lasting damage to the gut even in the absence of bacteria. It is also possible that in the absence of IbeA, reduced localization to the Peyer's patches (M cells) occurs and accounts for the trend in reduction of IFN-γ levels in the cecum and reduced histopathology observed in the ileum and cecum. If that is the case, future studies should monitor specifically AIEC localized to the Peyer's patches. In conclusion, our study has generated novel information regarding the mechanisms that AIEC has to interact with the host and adds another virulence determinant to the AIEC arsenal that might contribute to the perpetuation of inflammation (6, 44).
Supplementary Material
ACKNOWLEDGMENTS
We thank Vsevolod Popov for his assistance with the electron microscopy experiments.
This study was supported by the University of Texas Medical Branch McLaughlin Endowment predoctoral fellowship to R.J.C., and work in the A.G.T. laboratory was sponsored by National Institutes of Health/National Institute of Allergy and Infectious Disease grant AI079154.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.03003-14.
REFERENCES
- 1.Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. 1999. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn's disease. Infect Immun 67:4499–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eaves-Pyles T, Allen CA, Taormina J, Swidsinski A, Tutt CB, Jezek GE, Islas-Islas M, Torres AG. 2008. Escherichia coli isolated from a Crohn's disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int J Med Microbiol 298:397–409. doi: 10.1016/j.ijmm.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 3.Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, Darfeuille-Michaud A. 2001. Adherent invasive Escherichia coli strains from patients with Crohn's disease survive and replicate within macrophages without inducing host cell death. Infect Immun 69:5529–5537. doi: 10.1128/IAI.69.9.5529-5537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bringer MA, Billard E, Glasser AL, Colombel JF, Darfeuille-Michaud A. 2012. Replication of Crohn's disease-associated AIEC within macrophages is dependent on TNF-alpha secretion. Lab Invest 92:411–419. doi: 10.1038/labinvest.2012.42. [DOI] [PubMed] [Google Scholar]
- 5.Strober W. 2011. Adherent-invasive Escherichia coli in Crohn's disease: bacterial “agent provocateur.” J Clin Invest 121:841–844. doi: 10.1172/JCI46333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT. 2014. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 63:1069–1080. doi: 10.1136/gutjnl-2013-304909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, Darfeuille-Michaud A. 2009. Crohn's disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med 206:2179–2189. doi: 10.1084/jem.20090741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Small CL, Reid-Yu SA, McPhee JB, Coombes BK. 2013. Persistent infection with Crohn's disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Commun 4:1957. doi: 10.1038/ncomms2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menard R, Prevost MC, Gounon P, Sansonetti P, Dehio C. 1996. The secreted Ipa complex of Shigella flexneri promotes entry into mammalian cells. Proc Natl Acad Sci U S A 93:1254–1258. doi: 10.1073/pnas.93.3.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miquel S, Peyretaillade E, Claret L, de Vallee A, Dossat C, Vacherie B, el Zineb H, Segurens B, Barbe V, Sauvanet P, Neut C, Colombel JF, Medigue C, Mojica FJ, Peyret P, Bonnet R, Darfeuille-Michaud A. 2010. Complete genome sequence of Crohn's disease-associated adherent-invasive Escherichia coli strain LF82. PLoS One 5:e12714. doi: 10.1371/journal.pone.0012714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nash JH, Villegas A, Kropinski AM, Aguilar-Valenzuela R, Konczy P, Mascarenhas M, Ziebell K, Torres AG, Karmali MA, Coombes BK. 2010. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics 11:667. doi: 10.1186/1471-2164-11-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krause DO, Little AC, Dowd SE, Bernstein CN. 2011. Complete genome sequence of adherent invasive Escherichia coli UM146 isolated from ileal Crohn's disease biopsy tissue. J Bacteriol 193:583. doi: 10.1128/JB.01290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Homeier T, Semmler T, Wieler LH, Ewers C. 2010. The gimA locus of extraintestinal pathogenic Escherichia coli: does reductive evolution correlate with habitat and pathotype? PLoS One 5:e10877. doi: 10.1371/journal.pone.0010877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang SH, Wan ZS, Chen YH, Jong AY, Kim KS. 2001. Further characterization of Escherichia coli brain microvascular endothelial cell invasion gene ibeA by deletion, complementation, and protein expression. J Infect Dis 183:1071–1078. doi: 10.1086/319290. [DOI] [PubMed] [Google Scholar]
- 15.Huang SH, Stins MF, Kim KS. 2000. Bacterial penetration across the blood-brain barrier during the development of neonatal meningitis. Microbes Infect 2:1237–1244. doi: 10.1016/S1286-4579(00)01277-6. [DOI] [PubMed] [Google Scholar]
- 16.Germon P, Chen YH, He L, Blanco JE, Bree A, Schouler C, Huang SH, Moulin-Schouleur M. 2005. ibeA, a virulence factor of avian pathogenic Escherichia coli. Microbiology 151:1179–1186. doi: 10.1099/mic.0.27809-0. [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Niu C, Shi Z, Xia Y, Yaqoob M, Dai J, Lu C. 2011. Effects of ibeA deletion on virulence and biofilm formation of avian pathogenic Escherichia coli. Infect Immun 79:279–287. doi: 10.1128/IAI.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chouikha I, Bree A, Moulin-Schouleur M, Gilot P, Germon P. 2008. Differential expression of iutA and ibeA in the early stages of infection by extraintestinal pathogenic Escherichia coli. Microbes Infect 10:432–438. doi: 10.1016/j.micinf.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Flechard M, Cortes MA, Reperant M, Germon P. 2012. New role for the ibeA gene in H2O2 stress resistance of Escherichia coli. J Bacteriol 194:4550–4560. doi: 10.1128/JB.00089-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendu DR, Dasari VR, Cai M, Kim KS. 2008. Protein folding intermediates of invasin protein IbeA from Escherichia coli. FEBS J 275:458–469. doi: 10.1111/j.1742-4658.2007.06213.x. [DOI] [PubMed] [Google Scholar]
- 21.Zou Y, He L, Huang SH. 2006. Identification of a surface protein on human brain microvascular endothelial cells as vimentin interacting with Escherichia coli invasion protein IbeA. Biochem Biophys Res Commun 351:625–630. doi: 10.1016/j.bbrc.2006.10.091. [DOI] [PubMed] [Google Scholar]
- 22.Zou Y, He L, Wu CH, Cao H, Xie ZH, Ouyang Y, Wang Y, Jong A, Huang SH. 2007. PSF is an IbeA-binding protein contributing to meningitic Escherichia coli K1 invasion of human brain microvascular endothelial cells. Med Microbiol Immunol 196:135–143. doi: 10.1007/s00430-006-0034-x. [DOI] [PubMed] [Google Scholar]
- 23.Chi F, Wang Y, Gallaher TK, Wu CH, Jong A, Huang SH. 2009. Identification of IbeR as a stationary-phase regulator in meningitic Escherichia coli K1 that carries a loss-of-function mutation in rpoS. J Biomed Biotechnol 2009:520283. doi: 10.1155/2009/520283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou Y, He L, Chi F, Jong A, Huang SH. 2008. Involvement of Escherichia coli K1 ibeT in bacterial adhesion that is associated with the entry into human brain microvascular endothelial cells. Med Microbiol Immunol 197:337–344. doi: 10.1007/s00430-007-0065-y. [DOI] [PubMed] [Google Scholar]
- 25.Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, German AJ, Yates RM, Russell DG, Johnson SE, Berg DE, Harel J, Bruant G, McDonough SP, Schukken YH. 2006. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect Immun 74:4778–4792. doi: 10.1128/IAI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy KC, Campellone KG. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol 4:11. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen CA, Niesel DW, Torres AG. 2008. The effects of low-shear stress on adherent-invasive Escherichia coli. Environ Microbiol 10:1512–1525. doi: 10.1111/j.1462-2920.2008.01567.x. [DOI] [PubMed] [Google Scholar]
- 29.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, Henderson IR, Sperandio V, Ravel J. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gullberg E, Leonard M, Karlsson J, Hopkins AM, Brayden D, Baird AW, Artursson P. 2000. Expression of specific markers and particle transport in a new human intestinal M-cell model. Biochem Biophys Res Commun 279:808–813. doi: 10.1006/bbrc.2000.4038. [DOI] [PubMed] [Google Scholar]
- 31.Kerneis S, Bogdanova A, Kraehenbuhl JP, Pringault E. 1997. Conversion by Peyer's patch lymphocytes of human enterocytes into M cells that transport bacteria. Science 277:949–952. doi: 10.1126/science.277.5328.949. [DOI] [PubMed] [Google Scholar]
- 32.Kerneis S, Caliot E, Stubbe H, Bogdanova A, Kraehenbuhl J, Pringault E. 2000. Molecular studies of the intestinal mucosal barrier physiopathology using cocultures of epithelial and immune cells: a technical update. Microbes Infect 2:1119–1124. doi: 10.1016/S1286-4579(00)01266-1. [DOI] [PubMed] [Google Scholar]
- 33.des Rieux A, Fievez V, Theate I, Mast J, Preat V, Schneider YJ. 2007. An improved in vitro model of human intestinal follicle-associated epithelium to study nanoparticle transport by M cells. Eur J Pharm Sci 30:380–391. doi: 10.1016/j.ejps.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Roberts CL, Keita AV, Duncan SH, O'Kennedy N, Soderholm JD, Rhodes JM, Campbell BJ. 2010. Translocation of Crohn's disease Escherichia coli across M-cells: contrasting effects of soluble plant fibres and emulsifiers. Gut 59:1331–1339. doi: 10.1136/gut.2009.195370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. 2004. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology 127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 36.Rolhion N, Barnich N, Bringer MA, Glasser AL, Ranc J, Hebuterne X, Hofman P, Darfeuille-Michaud A. 2010. Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut 59:1355–1362. doi: 10.1136/gut.2010.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. 2010. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5:e8668. doi: 10.1371/journal.pone.0008668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerquetti M, Serafino A, Sebastianelli A, Mastrantonio P. 2002. Binding of Clostridium difficile to Caco-2 epithelial cell line and to extracellular matrix proteins. FEMS Immunol Med Microbiol 32:211–218. doi: 10.1111/j.1574-695X.2002.tb00556.x. [DOI] [PubMed] [Google Scholar]
- 39.Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. 2013. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol 6:666–677. doi: 10.1038/mi.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tahoun A, Siszler G, Spears K, McAteer S, Tree J, Paxton E, Gillespie TL, Martinez-Argudo I, Jepson MA, Shaw DJ, Koegl M, Haas J, Gally DL, Mahajan A. 2011. Comparative analysis of EspF variants in inhibition of Escherichia coli phagocytosis by macrophages and inhibition of E. coli translocation through human- and bovine-derived M cells. Infect Immun 79:4716–4729. doi: 10.1128/IAI.00023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Etienne-Mesmin L, Chassaing B, Sauvanet P, Denizot J, Blanquet-Diot S, Darfeuille-Michaud A, Pradel N, Livrelli V. 2011. Interactions with M cells and macrophages as key steps in the pathogenesis of enterohemorrhagic Escherichia coli infections. PLoS One 6:e23594. doi: 10.1371/journal.pone.0023594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarra M, Monteleone I, Stolfi C, Fantini MC, Sileri P, Sica G, Tersigni R, Macdonald TT, Pallone F, Monteleone G. 2010. Interferon-γ-expressing cells are a major source of interleukin-21 in inflammatory bowel diseases. Inflamm Bowel Dis 16:1332–1339. doi: 10.1002/ibd.21238. [DOI] [PubMed] [Google Scholar]
- 43.Mimouna S, Goncalves D, Barnich N, Darfeuille-Michaud A, Hofman P, Vouret-Craviari V. 2011. Crohn's disease-associated Escherichia coli promote gastrointestinal inflammatory disorders by activation of HIF-dependent responses. Gut Microbes 2:335–346. doi: 10.4161/gmic.18771. [DOI] [PubMed] [Google Scholar]
- 44.Kostic AD, Xavier RJ, Gevers D. 2014. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146:1489–1499. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaakoush NO, Mitchell HM, Man SM. 2014. Role of emerging Campylobacter species in inflammatory bowel diseases. Inflamm Bowel Dis 20:2189–2197. doi: 10.1097/MIB.0000000000000074. [DOI] [PubMed] [Google Scholar]
- 46.Boudeau J, Barnich N, Darfeuille-Michaud A. 2001. Type 1 pilus-mediated adherence of Escherichia coli strain LF82 isolated from Crohn's disease is involved in bacterial invasion of intestinal epithelial cells. Mol Microbiol 39:1272–1284. doi: 10.1111/j.1365-2958.2001.02315.x. [DOI] [PubMed] [Google Scholar]
- 47.Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. 2003. Regulatory and functional cooperation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn's disease. Mol Microbiol 48:781–794. doi: 10.1046/j.1365-2958.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- 48.Chassaing B, Rolhion N, de Vallee A, Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Soderholm JD, Hugot JP, Colombel JF, Darfeuille-Michaud A. 2011. Crohn's disease-associated adherent-invasive Escherichia coli bacteria target mouse and human Peyer's patches via long polar fimbriae. J Clin Invest 121:966–975. doi: 10.1172/JCI44632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chassaing B, Etienne-Mesmin L, Bonnet R, Darfeuille-Michaud A. 2013. Bile salts induce long polar fimbriae expression favouring Crohn's disease-associated adherent-invasive Escherichia coli interaction with Peyer's patches. Environ Microbiol 15:355–371. doi: 10.1111/j.1462-2920.2012.02824.x. [DOI] [PubMed] [Google Scholar]
- 50.Ratiner YA, Salmenlinna S, Eklund M, Keskimaki M, Siitonen A. 2003. Serology and genetics of the flagellar antigen of Escherichia coli O157:H7a,7c. J Clin Microbiol 41:1033–1040. doi: 10.1128/JCM.41.3.1033-1040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De la Fuente M, Franchi L, Araya D, Diaz-Jimenez D, Olivares M, Alvarez-Lobos M, Golenbock D, Gonzalez MJ, Lopez-Kostner F, Quera R, Nunez G, Vidal R, Hermoso MA. 2014. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int J Med Microbiol 304:384–392. doi: 10.1016/j.ijmm.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vandrangi P, Lo DD, Kozaka R, Ozaki N, Carvajal N, Rodgers VG. 2013. Electrostatic properties of confluent Caco-2 cell layer correlates to their microvilli growth and determines underlying transcellular flow. Biotechnol Bioeng 110:2742–2748. doi: 10.1002/bit.24939. [DOI] [PubMed] [Google Scholar]
- 53.Lertpiriyapong K, Gamazon ER, Feng Y, Park DS, Pang J, Botka G, Graffam ME, Ge Z, Fox JG. 2012. Campylobacter jejuni type VI secretion system: roles in adaptation to deoxycholic acid, host cell adherence, invasion, and in vivo colonization. PLoS One 7:e42842. doi: 10.1371/journal.pone.0042842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lapaquette P, Bringer MA, Darfeuille-Michaud A. 2012. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased proinflammatory response. Cell Microbiol 14:791–807. doi: 10.1111/j.1462-5822.2012.01768.x. [DOI] [PubMed] [Google Scholar]
- 55.Lapaquette P, Darfeuille-Michaud A. 2010. Abnormalities in the handling of intracellular bacteria in Crohn's disease. J Clin Gastroenterol 44:S26–S29. doi: 10.1097/MCG.0b013e3181dd4fa5. [DOI] [PubMed] [Google Scholar]
- 56.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. 2010. Crohn's disease-associated adherent-invasive Escherichia coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol 12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Slauch JM. 2011. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol 80:580–583. doi: 10.1111/j.1365-2958.2011.07612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thi EP, Lambertz U, Reiner NE. 2012. Sleeping with the enemy: how intracellular pathogens cope with a macrophage lifestyle. PLoS Pathog 8:e1002551. doi: 10.1371/journal.ppat.1002551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baptista JM, Justino MC, Melo AM, Teixeira M, Saraiva LM. 2012. Oxidative stress modulates the nitric oxide defense promoted by Escherichia coli flavorubredoxin. J Bacteriol 194:3611–3617. doi: 10.1128/JB.00140-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 61.Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR, Sotman S. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are noninvasive. Lancet i:1119–1122. [DOI] [PubMed] [Google Scholar]
- 62.Rose RE. 1988. The nucleotide sequence of pACYC177. Nucleic Acids Res 16:356. doi: 10.1093/nar/16.1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.