

Abstract
Although modern Chlamydia muridarum has been passaged for decades, there are no reports on the consequences of serial passage with strong selection pressure on its fitness. In order to explore the potential for Pasteurian selection to induce genomic and phenotypic perturbations to C. muridarum, a starter population was passaged in cultured cells for 28 generations without standard infection assistance. The resultant population, designated CMG28, displays markedly reduced in vitro dependence on centrifugation for infection and low incidence and severity of upper genital tract pathology following intravaginal inoculation into mice compared to the parental C. muridarum population, CMG0. Deep sequencing of CMG0 and CMG28 revealed novel protein variants in the hypothetical genes TC0237 (Q117E) and TC0668 (G322R). In vitro attachment assays of isogenic plaque clone pairs with mutations in either TC0237 and TC0668 or only TC0237 reveal that TC0237(Q117E) is solely responsible for enhanced adherence to host cells. Paradoxically, double mutants, but not TC0237(Q117E) single mutants, display severely attenuated in vivo pathogenicity. These findings implicate TC0237 and TC0668 as novel genetic factors involved in chlamydial attachment and pathogenicity, respectively, and show that serial passage under selection pressure remains an effective tool for studying Chlamydia pathogenicity.
INTRODUCTION
Infection with Chlamydia trachomatis in the lower genital tract (LGT) of women can lead to upper genital tract (UGT) inflammatory pathologies, such as hydrosalpinx, resulting in complications including ectopic pregnancy and infertility (1, 2). Hydrosalpinx, which is detectable by laparoscopic examination, has been used as a surrogate marker for tubal factor infertility in women (3, 4). However, the mechanisms by which C. trachomatis induces hydrosalpinges remain unknown. The murine pathogen Chlamydia muridarum, although not known to cause human diseases, has been extensively used for studying the mechanisms of C. trachomatis pathogenesis and immunity (5–8). This is due primarily to the ease of intravaginal infection of mice with C. muridarum organisms and their ability to induce hydrosalpinx in the oviduct, leading to mouse infertility (5, 9).
Both C. trachomatis and C. muridarum share a highly conserved biphasic growth cycle, which begins with the attachment of an infectious elementary body (EB) to a host cell. Multiple putative chlamydial factors, such as the major outer membrane protein (MOMP) (10–13), outer membrane complex (OMC) protein B (OmcB) (14–16), and the polymorphic membrane proteins (Pmps) (17–19), and host-derived factors, such as heparin sulfate (16, 20), epidermal growth factor receptor (EGFR) (21), estrogen receptor (22), and insulin-like growth factor 2 receptor (23), have been proposed to mediate chlamydial interactions with host cells. However, the precise structural basis of the interactions between an EB and a host cell during chlamydial infection in animals and humans remains ill defined. Following attachment to epithelial cells, chlamydiae have been shown to induce endocytosis to aid their own entry into host cells through the release of effectors such as translocated actin-recruiting protein (TARP) (24) and CT694 (25). The internalized EB then differentiates into a noninfectious but metabolically active reticulate body (RB) that is capable of multiplying within a cytoplasmic vacuole, termed an inclusion. To infect new cells, the progeny RBs must differentiate back to infectious EBs that then exit the infected cells. Stimulation of host cells with EBs (26) and chlamydial proteins (27) and intracellular RB replication in cultured cells (28, 29) can lead to the production of inflammatory cytokines, and inflammatory cytokines are also frequently detected in Chlamydia-infected genital tract tissues (30, 31). However, it remains unknown which and how chlamydial proteins contribute to pathogenicity in the genital tracts of animals and humans.
Recently, Sturdevant et al. (32) showed that frameshift mutations in the 360-amino-acid (aa) hypothetical open reading frame (ORF) CT135 of highly passaged C. trachomatis serovar D are responsible for various degrees of infectivity in mouse genital tract. One mutant, isolated 49 days after intravaginal infection of the parental population, acquired a “T” deletion in the 45th codon, resulting in premature termination at the 60th codon. These organisms reproducibly exhibit lower genital tract shedding for infections in excess of 8 weeks, garnering the strain name “Late Clearance” (D-LC). An opposing mutant isolated 10 days after infection carries a “T” insertion at the 182nd codon, terminates at the 194th codon, and can be cleared from the lower genital tract in <4 weeks, giving the strain name “Early Clearance” (D-EC). These findings demonstrate that phenotype-altering mutations can accumulate in genomes of passaged Chlamydia isolates. Although analogous mutations have been discovered in TC0412 (33, 34), the C. muridarum homolog of CT135, it is not known whether these mutations also affect C. muridarum infectivity and pathogenicity in the mouse genital tract.
We have previously shown that both adequate ascension of infection to the upper genital tract and activation of an appropriate tubal inflammatory response are required for the induction of hydrosalpinx (31, 35–37). Defining the virulence factors that contribute to either ascending infection or tubal inflammation has been a priority in Chlamydia research. Recent advances in transforming Chlamydia (38–42) and inducing mutations (43) have provided useful tools for investigating pathogenic mechanisms. However, these approaches rely on either prior discovery of virulence factors encoded on the plasmid and chromosome or functional assays to screen mutant libraries for phenotypes. Serial cell culture passage, an alternative functional assay, has been employed to select for chromosomal mutants of C. trachomatis serovars E and L2; however, this study lacked a novel selection pressure since these strains were historically maintained under the same in vitro conditions as those used during passage (44). Antibiotic resistance adaptations, on the other hand, have successfully been revealed through in vitro selection pressure (45). Despite these advances and prior attempts, no chlamydial genetic factors have been directly associated with the ability of the organism to induce upper genital tract pathology.
In this study, we sought to determine whether Chlamydia can be genetically adapted to an atypical niche, thereby decreasing its pathogenic fitness in vivo. Although this strategy, classically defined as Pasteurian selection, has generated numerous live attenuated microbial vaccines in the past (46, 47), it has not been applied to Chlamydia, which lacks an approved vaccine. To achieve attenuation, C. muridarum was serially passaged in vitro to functionally select for organisms with enhanced infectivity toward cultured host cells. Parental and passaged organisms contain multiple preexisting mutations in TC0412, but the hypothetical ORFs TC0237 and TC0668 acquired novel mutations during passage. The mutation in TC0237 can be attributed solely to an in vitro attachment enhancement phenotype, while TC0237 and TC0668 double mutants, but not TC0237 alone, display severely attenuated in vivo pathogenicity. Unlike CT135, lesions in TC0412 failed to impact either the in vitro attachment or in vivo pathogenicity of the organism. In light of these findings, we propose that TC0237 is a regulator of host adherence and postulate that TC0668 is a significant chlamydial virulence factor.
MATERIALS AND METHODS
Growth and in vitro passage of chlamydial organisms.
Chlamydia muridarum strain Nigg organisms (initially acquired from Robert C. Brunham's laboratory at the University of Manitoba in 1999) were propagated and purified in HeLa cells (human cervical carcinoma epithelial cells; ATCC CCL2), as described previously (48). Prior to C. muridarum infection, host cells were grown in either 24- or 6-well tissue culture plates or tissue culture flasks in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Rockville, MD) supplemented with 10% fetal bovine serum (FBS; Gibco BRL) (D10 medium) at 37°C in an incubator supplied with 5% CO2 (standard incubation conditions). For assisted infections, host cell cultures were preincubated with 30 μg/ml DEAE-dextran (Sigma-Aldrich, St. Louis, MO) in DMEM for 15 min, aspirated to remove the DEAE solution, inoculated with C. muridarum diluted in sucrose-phosphate-glutamate (SPG) medium (218 mM sucrose, 3.76 mM KH2PO4, 7.1 mM K2HPO4, 4.9 mM glutamate [pH 7.2]), and centrifuged at 500 RCF (relative centrifugal force) at room temperature (RT) for 1 h to maximize infection. For unassisted infections, host cell cultures were inoculated with C. muridarum diluted in SPG medium and incubated for 2 h with manual rotation every 15 min to selectively infect organisms with enhanced attachment to and/or entry into host cells. Following infection, inocula were aspirated, replaced with D10 medium supplemented with 2 μg/ml of cycloheximide (Sigma-Aldrich, St. Louis, MO) to make host cells stationary, and incubated for 24 h before being processed.
For in vitro passage, 6-well plates with HeLa cell monolayers were initially infected under the unassisted-infection conditions described above with purified parental C. muridarum EBs, designated CMG0, diluted to a multiplicity of infection (MOI) of 0.5 in a 1-ml inoculum. After incubation for 24 h, the progeny C. muridarum organisms, designated passage generation 1, or CMG1, were harvested and used to infect fresh HeLa cell monolayers under the assisted-infection conditions described above to form passage generation 2, or CMG2. This alternating unassisted/assisted-infection cycle was repeated until CMG28 was reached. As a general rule, odd-passage generations represent the results of an unassisted-infection passage, and even-passage generations represent an assisted-infection passage.
Plaque-forming assay and genotyping.
A plaque-forming assay was used to isolate individual clones from both CMG0 and CMG28. Briefly, purified EBs were inoculated onto confluent monolayers of McCoy cells in 6-well tissue culture plates and treated under the assisted-infection conditions described above. The inoculum was then removed and replaced with agarose overlay medium (1× Dulbecco's modified Eagle's medium, 10% fetal bovine serum, 0.2 μg/ml cycloheximide, and 1% agarose). The cells were then incubated for 5 days to allow plaques to form. Plaques were then picked into SPG medium, and a portion of the plaque stock was used to PCR amplify and Sanger sequence (ABI, Life Technologies, Grand Island, NY) the coding regions of the chromosomal ORFs TC0237, TC0412, and TC0668. Based on the sequence results for the three genes, selected clones were replaqued to ensure monoclonality before large-scale amplification, whole-genome sequencing, and subsequent experiments.
Sequencing and analysis of C. muridarum genomes.
Genomic DNAs from CMG0 and CMG28 populations and their plaque-purified clones were subjected to deep sequencing by using the Illumina HiSeq2000 platform (University of Texas Health Science Center at San Antonio [UTHSCSA] Greehey Children's Cancer Research Institute [GCCRI] Genome Sequencing Facility). Briefly, purified DNA was fragmented by using a Covaris S220 Ultra sonicator, and a DNA sequencing (DNA-Seq) library was prepared by using the TruSeq DNA sample preparation kit (Illumina, Inc.) according to the manufacturer's protocol. DNA-Seq libraries were sequenced with a 50-bp single-end sequencing module on the Illumina HiSeq2000 platform. After demultiplexing with CASAVA, sequence reads and associated qualities were exported in FASTQ format.
Reads in FASTQ format were mapped to the Chlamydia muridarum strain Nigg reference chromosome (GenBank accession number NC_002620.2) and plasmid pMoPn (accession number NC_002182.1) by using the Burrows-Wheeler Aligner mem algorithm (49). The resulting SAM file was converted to BAM format and sorted with Samtools (50). Insertions and deletions (indels) in the mapped reads were realigned by using the Genome Analysis Toolkit (51). A multisample pileup of the processed BAMs for CMG0, CMG28, and CMG0.21.3 was generated by Samtools and pipelined into VarScan (52) for detection and variant call formatted (VCF) output of variants with a minimum average Phred base quality of 20 and a minimum read fraction of 0.05. Low-frequency variants that share read fractions between the two populations and the clonal control (CMG0.21.3) were ruled as sequencing errors, while shared consensus variants against the Nigg reference were determined from parental strain CMG0. All other high-quality variants were mapped to genes, and their protein sequences were predicted using the MathWorks MATLAB r2014a Bioinformatics Toolbox and the GenBank accession numbers listed above. The only variants discovered in passaged organisms lie within the ORFs TC0237, TC0412, and TC0668, with the remaining genome being unaffected.
Based on both the whole-genome sequencing and the targeted-gene sequencing results, three pairs of plaque-purified isogenic clones together with CMG0 and CMG28 organisms were selected for subsequent phenotype analyses.
Centrifugation dependence and attachment assays.
In order to compare dependence on centrifugation, C. muridarum populations and plaque-purified clones were titrated for the number of inclusion-forming units (IFU) per ml under the assisted- and unassisted-infection conditions described above, but without prior incubation with DEAE-dextran. Briefly, purified C. muridarum EBs were serially diluted in SPG medium before being inoculated onto confluent HeLa monolayers grown on glass coverslips in 24-well tissue culture plates. The assisted-infection conditions were modified to include 1 h of preincubation under standard incubation conditions before centrifugation in order to equal the total 2 h of incubation required for unassisted infections. Monolayers were fixed and processed for immunofluorescence detection of C. muridarum inclusions following 24 h of growth. The genome copy number of each organism stock per ml was also quantitated by using quantitative PCR (qPCR), as described previously (41).
The attachment and entry efficiencies of the C. muridarum organisms were further compared via direct observation. Briefly, purified EBs were diluted in SPG medium and inoculated onto HeLa cell monolayers grown on coverslips in 24-well tissue culture plates at 4°C for 1 h. After a brief wash with chilled DMEM to remove unbound EBs, one set of the coverslips was processed for immunofluorescence detection of cell-associated EBs, while 1 ml prewarmed D10 medium was added to the remaining cultures and incubated at 37°C. At different time points after incubation, a set of coverslips was processed for detecting chlamydial organisms inside host cells. Intense fluorescent particles, inclusions, and host cells were counted under the same view across five random views of the coverslip and averaged. The results are expressed as the number of C. muridarum particles or inclusions per host cell.
Mouse infection and live-organism recovery from vaginal swabs and genital tract tissues.
Purified C. muridarum EBs were used to infect 6- to 7-week-old female C3H/HeJ mice (Jackson Laboratories, Inc., Bar Harbor, ME) intravaginally with 2 × 105 IFU in 20 μl of SPG medium. Five days prior to infection, each mouse was injected with 2.5 mg medroxyprogesterone (Depo-Provera; Pharmacia Upjohn, Kalamazoo, MI) subcutaneously to increase mouse susceptibility to infection. After infection, mice were monitored for vaginal live-organism shedding and sacrificed on different days postinfection (as indicated for individual experiments) for quantitating live organisms recovered from different segments of the genital tract and/or for observing gross genital tract pathologies.
For monitoring of live-organism shedding in swab samples, vaginal/cervical swabs were taken every 3 to 4 days for the first week and weekly thereafter until negative shedding was observed for two consecutive time points. To quantitate live chlamydial organisms, each swab was soaked in 0.5 ml of SPG medium and vortexed with glass beads, and the chlamydial organisms released into the supernatant were titrated on HeLa cell monolayers in duplicate. The infected cultures were processed for immunofluorescence assays as described below. Inclusions were counted in five random fields per coverslip under a fluorescence microscope. For coverslips with <1 IFU per field, entire coverslips were counted. Coverslips showing obvious cytotoxicity of HeLa cells were excluded. The total number of IFU per swab was calculated based on the mean number of IFU per view, the ratio of the view area to that of the well, the dilution factor, and inoculation volumes. Where possible, the mean number of IFU/swab was derived from serially diluted and duplicate samples for any given swab. The total number of IFU/swab was converted into a log10 value and used to calculate the means and standard deviations across mice of the same group at each time point.
To monitor ascending infection, mice infected in parallel experiments were sacrificed on day 14 after infection. Whole genital tracts were sterilely harvested, and each tract was divided into three portions, including vagina/cervix (VC), uterus/uterine horn (UH), and oviduct/ovary (OV). VC was defined as the lower genital tract (LGT), while both UH and OV were defined as the upper genital tract (UGT). Tissue segments were homogenized in 0.2 ml cold SPG medium by using a 2-ml tissue grinder (catalog number K885300-0002; Fisher Scientific, Pittsburgh, PA). After brief sonication and centrifugation at 3,000 rpm for 5 min to pellet large debris, the supernatants were titrated for live C. muridarum organisms on HeLa cells, as described above. The results were expressed as log10 IFU per tissue segment.
Evaluation of mouse genital tract pathology.
Mice were sacrificed 60 days after infection to evaluate urogenital tract tissue pathology. Before removal of the genital tract tissues from mice, an in situ gross examination for evidence of hydrosalpinx formation and any other gross abnormalities was performed under a stereoscope (Olympus, Center Valley, PA). The genital tract tissues were then excised in their entirety from the vagina to the ovary and laid onto a blue photography mat for acquisition of digital images. The oviduct hydrosalpinges were visually scored based on their dilation size by using a scoring system described previously (53). No oviduct dilation or swelling observable by stereoscope inspection was defined as no hydrosalpinx and assigned a score 0, hydrosalpinx that was visible only after amplification was assigned a score of 1, hydrosalpinx that was clearly visible with the naked eye but smaller than ovary was assigned a score of 2, hydrosalpinx with a size similar to that of ovary was assigned a score of 3, and hydrosalpinx that was larger than ovary was assigned a score of 4. Both the incidence and severity scores of oviduct hydrosalpinx were analyzed for statistical differences between groups of mice.
For histological scoring and inflammatory cell counting, the excised mouse genital tract tissues, after photography, were fixed in 10% neutral formalin, embedded in paraffin, and serially sectioned longitudinally at 5-μm widths. Efforts were made to include cervix, uterine horns, oviducts, and lumenal structures of each tissue in each section. The sections were stained with hematoxylin and eosin (H&E) as described previously (5). The H&E-stained sections were scored for severity of inflammation and oviduct dilation based on modified schemes established previously (5, 54) by researchers who were blind to mouse group designation. Scores for both sides of the oviducts were added to represent the oviduct pathology for a given mouse, and the scores for individual mice were calculated into medians for each group.
Inflammatory cell infiltrates were scored for oviduct tissue as follows: 0 for no significant infiltration, 1 for infiltration at a single focus, 2 for infiltration at two to four foci, 3 for infiltration at more than four foci, and 4 for confluent infiltration. Dilatation of oviduct was scored as follows: 0 for no significant dilatation, 1 for mild dilation of a single cross section, 2 for one to three dilated cross sections (where the largest diameter is smaller than that of the ovary on the same side), 3 for more than three dilated cross sections (where the largest diameter is equal to that of the ovary on the same side), and 4 for confluent pronounced dilation (where the largest diameter is larger than that of the ovary on the same side).
Immunofluorescence assay.
HeLa cells grown on coverslips were fixed with 4% (wt/vol) paraformaldehyde (Sigma) dissolved in phosphate-buffered saline (PBS) for 30 min at room temperature, followed by permeabilization with 2% (wt/vol) saponin (Sigma) for an additional 30 min. After washing and blocking, the cell samples were subjected to antibody and chemical staining. Hoechst 33342 dye (Sigma) was used to visualize DNA. A rabbit antichlamydial antibody, raised by immunization with purified C. muridarum elementary bodies, plus a goat anti-rabbit IgG secondary antibody conjugated with Cy2 (green; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used to visualize chlamydial organism-containing inclusions. Immunofluorescence images were acquired by using an Olympus AX-70 fluorescence microscope equipped with multiple filter sets and Simple PCI imaging software (Olympus), as described previously (48). The images were processed by using the Adobe Photoshop program (Adobe Systems, San Jose, CA). For titration of live organisms recovered from a given sample, the mean number of inclusions per view was derived by counting inclusions in five random views. The total number of live organisms in a given sample was calculated based on the mean number of inclusions per view, the ratio of the view area to that of the well, the dilution factor, and the inoculum volume and expressed as log10 IFU per sample.
Multiplex array for profiling cytokines in oviduct tissue.
Oviduct/ovary tissues were harvested from mice infected with CMG0 (n = 5) or CMG28 (n = 5) organisms on day 14 after intravaginal inoculation with C. muridarum for generating homogenates, as described previously (35, 54). The homogenates were used for simultaneous measurements of 32 mouse cytokines (23-plex group I [catalog number M60-009RDPD] plus 9-plex group II [catalog number MD0-00000EL]) by using a multiplex bead array assay (Bio-Plex 200 system) (all from Bio-Rad, Hercules, CA) according to the manufacturer's instruction. Levels of all cytokines are expressed as mean pg/ml ± standard deviations. The means from the two mouse groups infected with CMG0 or CMG28 were used for calculating ratios and statistical analysis.
Statistical analyses.
Quantitative data, including the number of live organisms (IFU), IFU ratios, and the number of organisms or IFU per cell, were analyzed by using Student's t test or a Kruskal-Wallis test. Cytokine levels were first subjected to an f test to determine whether the variance of each group is significantly different (P < 0.05), followed by the appropriate t test (two-sample test with either equal or unequal variance). Qualitative data, including incidence rates, were analyzed by using Fisher's exact test. Semiquantitative data, including gross and microscopic pathology scores, were analyzed by using the Wilcoxon rank sum test.
Nucleotide sequence accession numbers.
All reads were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under run accession numbers SRR1531377 for CMG0, SRR1531380 for CMG28, SRR1736634 for CMG0.1.1, SRR1736639 for CMG0.10.1, SRR1531430 for CMG0.21.3, SRR1736644 for CMG28.12.3, SRR1737027 for CMG28.38.1, and SRR1736648 for CMG28.54.1. Select genomes were deposited in GenBank under accession numbers CP009760.1 for the CMG0 parental consensus, CP009608.1 for CMG0.1.1, and CP009609.1 for CMG28.38.1, each being a representative of the population from which they originate.
RESULTS
In vitro passage selects for Chlamydia muridarum with mutations in chromosomal genes TC0237 and TC0668.
C. muridarum organisms were passaged in HeLa cell cultures for 28 generations, as described in Materials and Methods, to generate CMG28 organisms. We compared the genomes of CMG28 organisms to those of the original C. muridarum organisms without passage (CMG0). First, next-generation sequencing (NGS) of the CMG0 and CMG28 populations and a combination of various bioinformatics tools were used to screen for mutations that were either introduced or affected by passage. Most regions of the CMG28 genome were found to be identical to those of CMG0, except for the introduction of novel protein variants in two chromosomal genes: a consensus Q117E substitution in ORF TC0237 and a subconsensus G322R substitution in TC0668. Interestingly, many mutations in TC0412 were detected in both the CMG0 and CMG28 genomes, although no novel TC0412 variants were found in CMG28 compared to CMG0.
We then picked plaques from the CMG0 and CMG28 populations and sequenced their chromosomal regions carrying TC0237, TC0412, and TC0668 using traditional Sanger sequencing. As predicted by NGS, CMG28 clones carry the TC0237(Q117E) mutation, while none of the CMG0 clones do (Table 1). It is likely that TC0412 mutations had accumulated during decades of in vitro maintenance of our C. muridarum stock, as was found for CT135 of C. trachomatis serovar D (32). For this study, we focus on three TC0412 lesions with similarity to the D-LC and D-EC disruptions of CT135 as well as an intermediate mutation that lies between them. These TC0412 variants include a −84T (T insertion at the 84th ORF nucleotide position) frameshift similar to the D-LC disruption, a G262T nonsense intermediate disruption, and a −435T (T insertion at the 435th ORF nucleotide position) frameshift similar to the D-EC disruption. All three lesions are predicted to terminate the ORF at or shortly after their position like those found in D-LC and D-EC.
TABLE 1.
Genotypes of Chlamydia muridarum organisms characterized in this studya
| Chromosomal variant | Affected gene | Gene length (no. of codons) | Nucleotide mutation | Protein mutation | Presence of genotype in Chlamydia muridarum clone |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| CMG0.10.1 | CMG28.54.1 | CMG0.21.3 | CMG28.12.3 | CMG0.1.1 | CMG28.38.1 | |||||
| G277313C | TC0237 | 160 | G349C | Q117E | − | + | − | + | − | + |
| −472842T | TC0412 | 366 | −84T | fs29 | + | + | − | − | − | − |
| G473020T | TC0412 | 366 | G262T | E88 stop | − | − | + | + | − | − |
| −473193T | TC0412 | 366 | −435T | fs146 | − | − | − | − | + | + |
| G797979A | TC0668 | 409 | G964A | G322R | − | + | − | + | − | − |
Isogenic clones were selected by pairing parental control genomes from CMG0 with TC0237(Q117E) single or TC0237(Q117E) and TC0668(G322R) double mutants from CMG28. Aside from carrying either one or both mutations in TC0237 and TC0668, isogenic pairs differ from one another by their unique TC0412 genotypes. Chromosomal variants are numbered according to the parent CMG0 consensus chromosome sequence. “fs” represents a frameshift mutation beginning at the indicated codon. “−” signifies a lack of the genotype, while “+” indicates that the clone carries the genotype.
To correlate the opposing genotypes of TC0237, TC0412, and TC0668 with phenotypes, we selected three pairs of isogenic clones to characterize along with the original CMG0 and CMG28 populations. These six isogenic clones were plaque purified twice and subjected to deep whole-genome sequencing to ensure monoclonality. As shown in Table 1, each of the three pairs possessed a unique TC0412 mutation, and within each pair, the two clones differed in TC0237 and/or TC0668 genotypes. For example, both CMG0.1.1 and CMG28.38.1 have the TC0412 −435T frameshift, but CMG28.38.1 additionally carries the TC0237(Q117E) mutation. The rest of the genomes and plasmids of the two clones are identical. The remaining two CMG28-derived clone pairs have a TC0412 −84T frameshift, G262T nonsense mutations, and both TC0237(Q117E) and TC0668(G322R) mutations. We were unable to isolate plaques with the TC0668 mutation only, as the consensus TC0237(Q117E) mutation is always present in CMG28-derived clones.
TC0237(Q117E) mutants are more efficient in attaching to cultured cells.
As shown in Table 2, three pairs of isogenic clones along with their original CMG0 and CMG28 populations were compared for their in vitro growth in HeLa cells. Each of these eight organisms was amplified, purified, and titrated for determination of the numbers of both genome copies and live organisms per stock volume. Live-organism titration was carried out by using both DEAE-dextran pretreatment of HeLa cells and centrifugation of the infected cultures, which maximizes the in vitro growth of C. muridarum organisms. Indeed, the IFU titers obtained under these maximally assisted infection conditions were closer to the genome copy numbers of the corresponding organisms. We then retitrated the eight organisms under two infection conditions: with or without centrifugation. No DEAE-dextran was used under either condition to prevent confounding charge-related effects. Interestingly, all CMG28 organisms reached significantly higher titers than did CMG0 organisms when titrated without centrifugation. Since the untreated, noncentrifuged conditions were similar to conditions used in alternating cycles during in vitro passage (see Materials and Methods), the above-described observation suggests that the CMG28 organisms were likely selected to be more efficient for infecting HeLa cells in the absence of any assisted-infection conditions. When titrated with versus without centrifugation, the titers of CMG0 plaques increased dramatically, by 19- to 31-fold, compared to the consistent 5-fold increase in the titers of CMG28 organisms. These findings suggest that CMG28 organisms adapted to invade HeLa cells more efficiently in the absence of centrifugation.
TABLE 2.
In vitro growth properties of CMG0 and CMG28 organismsa
| Organism | No. of genome copies/ml | Infection condition | Mean IFU/ml ± SD | Mean IFU ratio | G0 vs G28 IFU ratio P value |
|---|---|---|---|---|---|
| CMG0 | 2.50E+10 | DE + Centri | 9.00E+09 | ||
| Centri | 7.03E+09 ± 1.56E+09 | 43.56 | 0.0115 | ||
| No treat | 1.61E+08 ± 2.22E+07 | ||||
| CMG28 | 6.75E+10 | DE + Centri | 2.70E+10 | ||
| Centri | 2.20E+10 ± 8.79E+09 | 5.80 | |||
| No treat | 3.79E+09 ± 2.27E+09 | ||||
| CMG0.1.1 | 9.43E+10 | DE + Centri | 4.01E+10 | ||
| Centri | 2.41E+10 ± 5.98E+09 | 18.87 | 0.0159 | ||
| No treat | 1.28E+09 ± 7.56E+08 | ||||
| CMG28.38.1 | 4.71E+10 | DE + Centri | 2.35E+10 | ||
| Centri | 1.48E+10 ± 3.57E+09 | 4.87 | |||
| No treat | 3.68E+09 ± 1.86E+09 | ||||
| CMG0.21.3 | 9.26E+09 | DE + Centri | 4.30E+09 | ||
| Centri | 4.29E+09 ± 6.47E+08 | 31.04 | 0.0088 | ||
| No treat | 1.38E+08 ± 4.99E+07 | ||||
| CMG28.12.3 | 2.74E+10 | DE + Centri | 1.33E+10 | ||
| Centri | 1.24E+10 ± 1.68E+09 | 5.11 | |||
| No treat | 2.43E+09 ± 4.25E+08 | ||||
| CMG0.10.1 | 2.67E+10 | DE + Centri | 1.02E+10 | ||
| Centri | 5.20E+09 ± 1.11E+09 | 20.70 | 0.0168 | ||
| No treat | 2.51E+08 ± 6.31E+07 | ||||
| CMG28.54.1 | 6.03E+10 | DE + Centri | 4.63E+10 | ||
| Centri | 4.33E+10 ± 5.03E+09 | 5.23 | |||
| No treat | 8.28E+09 ± 4.10E+09 |
C. muridarum organisms were amplified and purified. Each of the eight purified stock organisms was quantitated for genome copies per ml by using qPCR and titrated for the number of live organisms under three different infection conditions, including DEAE treatment plus centrifugation (DE + Centri), centrifugation alone (Centri), and no treatment (No treat). Numbers of live organisms are expressed as IFU per ml. The ratios of IFU determined by titration under centrifugation conditions to that under the no-treatment condition were calculated for each organism, and the ratios between the CMG0 and CMG28 organisms within each pair were compared. The ratios for the CMG0 organisms were significantly higher than those for the CMG28 organisms.
Although CMG28 organisms display a reduced dependence on centrifugation for infection, these results do not distinguish between more efficient attachment and intracellular growth. To address this question, we compared the attachment efficiencies of CMG0 and CMG28 populations and clones on HeLa cell monolayers under unassisted-infection conditions (Fig. 1). After the organisms were allowed to attach to HeLa cells for 1 h at 4°C, cell samples were rinsed with cold medium three times to remove loosely associated or free-floating EBs and fixed for detection of cell-associated chlamydial particles. We found that even after the numbers of live input organisms were standardized across both organisms, the number of chlamydial particles associated with each cell was significantly higher in cultures inoculated with the CMG28 population and its clones than those in cultures inoculated with CMG0. When the parallel cultures were incubated for another 6 h at 37°C, the number of intracellular chlamydial organisms per cell continued to be significantly higher in cultures inoculated with CMG28 than in those inoculated with CMG0. This trend continued for inclusions inside each cell when observed 22 h after infection. These observations suggest that the CMG28 organisms are more efficient in attaching to HeLa cells during infection in the absence of centrifugation and that enhanced attachment results in more productive infection.
FIG 1.
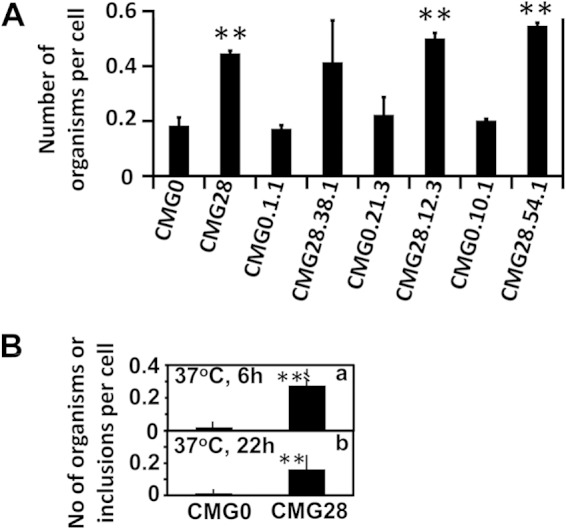
Attachment of CMG0 and CMG28 populations and plaque-purified clones to cultured HeLa cells. The same numbers of CMG0 and CMG28 organisms, as listed along the x axis, were inoculated onto HeLa cell monolayers under unassisted-infection conditions. The infected cells were washed and processed for immunofluorescence detection of the remaining chlamydial organisms 1 h after incubation at 4°C (A) or 6 h (a) or 22 h (b) after incubation at 37°C (B). The cell-associated chlamydial organisms or inclusions were counted and expressed as the number of chlamydial organisms or inclusions per cell, as shown along the y axis. The 37°C incubation conditions were applied only to the CMG0 and CMG28 population cultures (B). All experiments were repeated three times. **, P < 0.01 (Kruskal-Wallis test).
Although their TC0412 and TC0668 genotypes vary, all CMG28-derived clones carry the TC0237(Q117E) mutation. These organisms independently displayed significantly enhanced attachment to HeLa cells compared to their corresponding CMG0 control population or isogenic clones, which strongly suggests that the TC0237(Q117E) mutation is responsible for the enhanced attachment to cultured cells.
TC0668(G322R) mutants are significantly attenuated in inducing hydrosalpinx in mice.
Following in vitro characterization of CMG0 and CMG28 organisms, we evaluated their pathogenicity in the mouse upper genital tract. When oviduct tissues were examined on day 60 after intravaginal infection with 2 × 105 IFU of each of the eight CMG0 or CMG28 organisms (Fig. 2), we found that all mice infected with CMG0 organisms developed significant upper genital tract pathology, with hydrosalpinx incidence rates of 76.9% or higher and severity scores of 3.23 ± 2.24 or higher. However, CMG28 organisms, with the exception of CMG28.38.1 without the TC0668(G322R) mutation, induced significantly reduced levels of hydrosalpinx, with an incidence rate of 30.8% or less and severity scores of <1.5. Although the CMG28.38.1 clone also showed reduced pathogenicity, the attenuation was not as significant, since this clone induced the development of hydrosalpinx in 50% of mice, with a mean severity score of 2.63. Thus, the attenuation of pathogenicity seemed to correlate with the presence of the TC0668(G322R) mutation. The attenuated pathogenicity of the CMG28 population and CMG28 clones was also confirmed by microscopy, which showed significantly reduced oviduct lumenal dilation in mice infected with TC0668(G322R) mutants (Fig. 3).
FIG 2.

Induction of hydrosalpinx in mice by CMG0 and CMG28 populations and plaque-purified clones. C3H/HeJ mice were intravaginally infected with 2 × 105 IFU of the 8 C. muridarum organisms, including the CMG0 (a) and CMG28 (b) population pair (n = 13/group) and three plaque-purified isogenic clone pairs CMG0.1.1 (n = 8) (c) and CMG28.38.1 (n = 8) (d), CMG0.21.3 (n = 8) (e) and CMG28.12.3 (n = 5) (f), and CMG0.10.1 (n = 5) (g) and CMG28.54.1 (n = 5) (h). The mice were sacrificed 60 days after infection for observation of hydrosalpinx. One representative image of whole genital tract from each group of mice is presented in the left columns, with vagina on the left and oviduct/ovary on the right. The areas covering the oviduct/ovary portions were magnified and are shown on the right side of the corresponding images of whole genital tracts. Hydrosalpinges are indicated by white arrows, and hydrosalpinx severity scores are indicated by white numbers. The hydrosalpinx incidence rates and severity scores are listed below the corresponding images. Mice with hydrosalpinx in either or both oviducts were considered positive for hydrosalpinx. The severities of both hydrosalpinges from a given mouse were scored separately and added together for the severity score assigned to that particular mouse. Fisher's exact test was used for comparing incidence rates (&, P < 0.05; &&, P < 0.01) and a Wilcoxon rank sum test was used for comparing severity scores (*, P < 0.05; **, P < 0.01) between the CMG0 and CMG28 organisms. Note that all CMG28 organisms induced more significant hydrosalpinges than did their CMG0 counterparts, with the exception of the CMG0.1.1 and CMG28.38.1 pair.
FIG 3.

Microscopic observation of oviduct dilation induced by CMG0 and CMG28 populations and plaque-purified clones. The oviduct tissues of mice infected with the four pairs of CMG0 and CMG28 organisms were harvested and subjected to H&E staining for microscopic evaluation of oviduct dilation. (A) Representative images for each group were taken under a 4× objective lens from mice infected with CMG0 (a) and CMG28 (b) populations and from the three pairs of plaque-purified clone-infected mice (c to h), as indicated at the top of each image. Uterine horn (UH), normal oviduct, and ovary tissues are marked. Dilated oviducts are indicated with white lines with arrowheads at both ends. Bar, 0.5 mm. (B) Severity of lumenal dilation was scored as described in Materials and Methods and is listed along the y axis. The four pairs of C. muridarum organisms are listed along the x axis. Note that all CMG28 organisms induced more significant oviduct lumenal dilation than did their CMG0 counterparts, with the exception of the CMG0.1.1 and CMG28.38.1 pair. *, P < 0.05; **, P < 0.01 (Wilcoxon rank sum test).
CMG28 organisms are as infectious as CMG0 organisms in mouse genital tract.
Following intravaginal infection with either CMG0 or CMG28 populations or clones as described above, mice were monitored for live-organism shedding from the lower genital tract at different time points (Fig. 4). We found that both the number of live organisms recovered and the percentage of mice remaining positive for shedding of live organisms at each time point were similar regardless of the chlamydial organisms used for infection. This observation suggests that both the CMG0 and CMG28 populations and clones maintain similar levels of infectivity in mouse lower genital tract. Unlike CT135, ORF disruptions within TC0412 have no obvious effect on shedding.
FIG 4.
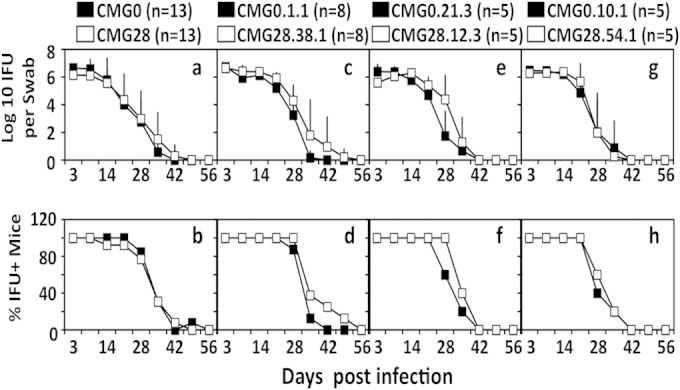
Shedding of live chlamydial organisms from mouse lower genital tract following infection with CMG0 and CMG28 populations and plaque-purified clones. C3H/HeJ mice were intravaginally infected with four pairs of C. muridarum CMG0 and CMG28 organisms, as described in the legend of Fig. 2 and as indicated above the corresponding plots. On different days after infection, as shown along the x axis, vaginal swabs were taken for titration of live organisms on HeLa cell monolayers. The live organisms recovered from each swab are expressed as log10 IFU along the y axis in panels a, c, e, and g. The percentage of mice remaining positive for shedding of live organisms at each time point is plotted along the y axis in panels b, d, f, and h. Note that there is no significant difference in live-organism shedding from the lower genital tracts of mice infected with CMG0 and those infected with CMG28 organisms.
To further compare ascending infection between the CMG0 and CMG28 population organisms, we monitored live-organism recovery from genital tract tissues harvested from mice on day 14 after infection (Fig. 5). There was no significant difference in the numbers of live organisms recovered from mice infected with CMG0 and those infected with CMG28 population organisms. There was also no significant difference in the Chlamydia load detected in the lower and upper genital tracts, including CV, UH, and OV tissues. It appears that both CMG0 and CMG28 organisms comparably ascended to and replicated in the upper genital tract of mice. Although the above-described observation was made on a single day and with the CMG0 and CMG28 populations, we can still conclude that these two types of organisms share similar growth patterns in the mouse genital tract since live organisms from the lower and upper genital tract tissues were surveyed in relation to one another.
FIG 5.
Recovery of live chlamydial organisms from mouse genital tract tissues following infection with CMG0 and CMG28 populations. C3H/HeJ mice intravaginally infected with CMG0 (n = 5) or CMG28 (n = 5) population organisms were sacrificed on day 14 after infection. Vaginal swabs were taken prior to mouse sacrifice. The entire genital tract tissue was harvested from each mouse and divided into lower genital tract (LGT) vagina/cervix (VC) and upper genital tract (UGT) uterus/uterine horn (UH) and oviduct/ovary (OV) sections, as listed along the x axis. Each tissue section was homogenized for titration of live C. muridarum organisms. Vaginal swabs were similarly titrated for live organisms. Log10 IFU values were used to calculate means and standard deviations for each group, as displayed along the y axis. Note that the numbers of live organisms recovered from mice infected with CMG0 and those infected with CMG28 were similar, without any significant difference (Kruskal-Wallis test).
CMG28 organisms are less inflammatory in mouse upper genital tract.
Oviduct tissues collected on day 60 after infection were evaluated for inflammatory cell histopathology under a microscope. As shown in Fig. 6, inflammatory infiltration was significantly reduced in mice infected with the CMG28 population. In order to dissect the immunological mechanisms behind the attenuation of CMG28 organisms, we compared the cytokine levels in oviduct tissue homogenates harvested on day 14 from mice infected with CMG0 to those in mice infected with CMG28 populations (Table 3) and found that the levels of 12 out of the 32 cytokines measured were significantly higher in mice infected with CMG0 than in those infected with CMG28. The remaining cytokines either were not detected in CMG0 or CMG28 homogenates or were not significantly different. The 12 cytokines with decreased concentrations in CMG28-infected mice include the proinflammatory cytokines interleukin-1α (IL-1α) and IL-1β; the Th1-promoting cytokines IL-12 and IL-15; the Th2 cytokines IL-10 and eotaxin; the Th17 cytokine IL-17; the chemokines macrophage inflammatory protein 1α (MIP-1α), monokine induced by gamma interferon (MIG), and MIP-2; as well as the growth factor vascular endothelial growth factor (VEGF). It is clear from these observations that CMG28 organisms are attenuated in inducing inflammation in mouse oviducts despite adequate ascension to the upper genital tract.
FIG 6.

Oviduct inflammatory infiltration induced by CMG0 and CMG28 populations and plaque-purified clones. Oviduct tissues from mice infected with the CMG0 and CMG28 populations and plaque-purified clones, as described in the legend of Fig. 2, were subjected to H&E staining, as described in the legend of Fig. 3, for evaluation of inflammatory histopathology. (A) Representative images from each group taken under 10× (left) and 100× (right) objective lenses. White rectangles in the 10× objective lens images indicate the same areas from which the right images were taken under a 100× objective lens. (B) The severity of inflammatory infiltration in oviduct tissue was scored as described in Materials and Methods and is listed along the y axis. The four pairs of C. muridarum organisms are listed along the x axis. Note that all CMG28 organisms induced more significant oviduct inflammatory infiltration than that induced by their CMG0 counterparts, with the exception of the CMG0.1.1 and CMG28.38.1 pair. *, P < 0.05; **, P < 0.01 (Wilcoxon rank sum test).
TABLE 3.
Cytokines from oviduct tissues of mice infected with C. muridarum CMG0 or CMG28a
| Cytokine | Mean cytokine level (pg/ml) ± SD |
CMG0/CMG28 ratio | P value | |
|---|---|---|---|---|
| CMG0 (n = 5) | CMG28 (n = 5) | |||
| IL-1α | 3,809.7 ± 2,847.6 | 38.2 ± 41.7 | 99.67 | <0.05 |
| IL-1β | 6,248.2 ± 4,272.9 | 122.4 ± 178.1 | 51.05 | <0.05 |
| IL-2 | 7.1 ± 7.8 | 0 | ||
| IL-3 | 5.8 ± 3.5 | 1.7 ± 1.2 | 3.53 | |
| IL-4 | 15.6 ± 10.4 | 2.0 ± 2.3 | 7.89 | |
| IL-5 | 0 | 0 | ||
| IL-6 | 267.1 ± 294.7 | 10.3 ± 12.6 | 25.97 | |
| IL-9 | 304.7 ± 121.4 | 166.0 ± 53.7 | 1.84 | |
| IL-10 | 41.3 ± 27.1 | 6.2 ± 5.0 | 6.70 | <0.05 |
| IL-12(p40) | 312.8 ± 196.9 | 61.2 ± 52.0 | 5.11 | <0.05 |
| IL-12(p70) | 123.0 ± 83.6 | 7.2 ± 6.6 | 17.03 | <0.05 |
| IL-13 | 971.4 ± 553.4 | 400.5 ± 154.2 | 2.43 | |
| IL-17 | 73.8 ± 50.2 | 4.3 ± 6.2 | 17.07 | <0.05 |
| Eotaxin | 1,506.2 ± 1,029.3 | 105.4 ± 124.6 | 14.28 | <0.05 |
| G-CSF | 12,420.0 ± 10,815.5 | 1,552.7 ± 944.3 | 8.00 | |
| GM-CSF | 92.9 ± 62.2 | 24.3 ± 28.1 | 3.83 | |
| IFN-γ | 147.1 ± 121.0 | 21.7 ± 23.5 | 6.76 | |
| MCP-1 | 3,891.9 ± 3,282.3 | 1,356.5 ± 1,558.4 | 2.87 | |
| MIP-1α | 922.4 ± 681.3 | 69.6 ± 77.6 | 13.24 | <0.05 |
| MIP-1β | 221.9 ± 171.3 | 42.0 ± 35.0 | 5.28 | |
| RANTES | 946.6 ± 770.2 | 224.3 ± 233.6 | 4.22 | |
| TNF-α | 163.6 ± 131.3 | 16.9 ± 16.2 | 9.71 | |
| IL-15 | 347.7 ± 233.4 | 18.8 ± 22.0 | 18.46 | <0.05 |
| KC | 498.6 ± 386.0 | 56.1 ± 57.2 | 8.89 | |
| IL-18 | 111.5 ± 103.7 | 0 | ||
| FGF-basic | 1,131.0 ± 536.9 | 703.4 ± 102.6 | 1.61 | |
| LIF | 234.9 ± 203.3 | 11.7 ± 12.9 | 20.03 | |
| M-CSF | 360.9 ± 236.9 | 76.5 ± 44.8 | 4.72 | |
| MIG | 68,208.3 ± 45,450.3 | 7,556.5 ± 9,488.6 | 9.03 | <0.05 |
| MIP-2 | 12,745.6 ± 8,969.6 | 43.4 ± 60.9 | 293.90 | <0.05 |
| PDGF-BB | 0 | 0 | ||
| VEGF | 9,355.2 ± 5,326.1 | 1,668.7 ± 595.6 | 5.61 | <0.05 |
Oviduct tissue homogenates were produced from mice infected with CMG0 (n = 5) or CMG28 (n = 5) on day 14 after intravaginal inoculation for simultaneous measurement of the levels of 32 cytokines using a multiplex bead array assay. The means from the two strains were used for calculating ratios and significance by Student's t test. The corresponding ratios of CMG0 to CMG28 are highlighted in boldface type. G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, gamma interferon; MCP-1, monocyte chemoattractant protein 1; TNF-α, tumor necrosis factor alpha; KC, keratinocyte chemoattractant; FGF-basic, basic fibroblast growth factor; LIF, leukemia inhibitory factor; M-CSF, macrophage-stimulating factor; PDGF-BB, platelet-derived growth factor beta homodimer.
DISCUSSION
In vitro passage of Chlamydia has been used to maintain and acquire pure chlamydial organisms by Chlamydia research laboratories over the years. Normally, assisted-infection conditions such as DEAE-dextran treatment and/or centrifugation are applied to maximize infection rates. As a result, no specific selection pressure, aside from standardized nutrient availability and host cell origin, is applied during routine laboratory passage. Thus, the genomes of the C. muridarum organisms maintained in different laboratories have been relatively stable, although mutations have still accumulated over time. These mutations not only are limited in scope but also lack a tractable association with a definitive phenotype. For example, the TC0412 gene in our CMG0 stock has already accumulated various catastrophic mutations prior to this study, but the functional consequence of the TC0412 mutations remains unknown. Similarly, Miyairi et al. (55) were able to correlate the attenuation of a Chlamydia psittaci 6BC strain in mice with substitution mutations in chromosomal genes, but the lack of isogenic strains and selective pressure context prevents the assignment of individual mutations to the attenuated phenotype.
When the genome of a C. trachomatis L2 population obtained after long-term in vitro propagation under assisted-infection conditions was deep sequenced, nonsynonymous mutations were identified in CTL0084 and CTL0610, encoding a putative transferase and a protein likely implicated in transcription regulation, respectively (44). It is still unclear whether these mutations are advantageous for the replication of L2 organisms. Nevertheless, it is clear that different species or biovars of chlamydial organisms or the same organisms can accumulate different mutations in their genomes during routine passage in different laboratories. Since the impacts of these different mutations on chlamydial pathogenicity are not known, there is an urgent need for all chlamydial research laboratories to begin utilizing standardized clones so that the experimental results from different laboratories can be directly compared.
In the current study, we have used the traditional Pasteurian approach (47) of in vitro passage selection to attenuate pathogenicity in vivo in order to identify chlamydial factors relevant to chlamydial pathogenicity. For every other infection cycle, the chlamydial organisms were applied to cell monolayers without assistance, in the form of DEAE-dextran preincubation and centrifugation. This infection cycle selects for chlamydial organisms with an enhanced affinity for HeLa cells. The successful organisms were then amplified via an assisted-infection cycle. Alternating selection cycles were repeated 28 times, which allowed us to obtain CMG28 organisms with mutations accumulating in the coding regions of TC0237 and TC0668. These genotypes were confirmed by Sanger sequencing of plaque-isolated clones and resequencing of the whole genomes of three pairs of isogenic clones. Although the types of mutations obtained in this study are different from what Borges et al. reported for C. trachomatis serovar L2 or E organisms after passage under fully assisted conditions (44), the general trend in mutability is similar. Only a limited number of ORFs experienced nonsynonymous single nucleotide polymorphisms (SNPs) in both studies, most likely due to the small size of the Chlamydia genome or its relatively high genomic stability following significant reductive evolution.
Following passage, characterization of three pairs of isogenic plaque-purified clones along with the CMG0 and CMG28 populations revealed that all CMG28 organisms developed enhanced attachment to cultured cells. Since TC0237(Q117E) is the only mutation shared by all the CMG28 organisms, this mutation is likely responsible for the in vitro attachment enhancement phenotype. This conclusion is supported by the fact that CMG28.38.1 carrying only the TC0237(Q117E) mutation with a wild-type TC0668 ORF also displayed the enhanced-attachment phenotype. On the contrary, when evaluated in mice, all CMG28 organisms except for the CMG28.38.1 clone were highly attenuated in inducing hydrosalpinx. The attenuated pathogenicity correlates with the presence of the TC0668(G322R) mutation.
It is worth noting that no plaque clones with a wild-type, or annotated, TC0412 ORF sequence were recovered from the CMG0 and CMG28 populations. TC0412 encodes a conserved 365-aa hypothetical protein, although no putative conserved domains have been detected. It shares ∼60% amino acid sequence identity with its homolog CT135 from C. trachomatis serovar D and weaker homology (∼20% amino acid identity) with its homologs in other chlamydial species. Both TC0412 and CT135 are predicted to contain four transmembrane domains. The TC0412 mutants characterized in this study are highly analogous to those previously discovered in CT135 (32), implicating common selective pressures to disrupt the ORF. However, the CT135-associated delayed-clearance (D-LC) or early-clearance (D-EC) phenotype could not be replicated by these TC0412 analogs. TC0412 mutants also did not differ in their abilities to attach to cultured HeLa cells and failed to affect the pathogenicity of C. muridarum in the mouse upper genital tract. Thus, it is unlikely that TC0412 is irrelevant to murine urogenital tract infection, but it is still possible that CT135 is necessary for pathogenesis in the human urogenital tract.
The two ORFs that accumulated mutations during 28 cell culture passages encode two hypothetical proteins that are both conserved and unique to Chlamydiae. TC0237, found on the complementary strand of the C. muridarum strain Nigg reference genome, codes for a 159-aa protein with no known function (56). On the other hand, TC0237 contains a domain of unknown function 720 (DUF720) motif, which is also found in its neighboring ORFs TC0236 (coding for a 172-aa hypothetical protein) and TC0235 (coding for a 170-aa hypothetical protein). These three sequential proteins are paralogous to each other and are predicted to be encoded in an operon (56). TC0237, TC0236, and TC0235 are all highly conserved within the Chlamydia and Chlamydophila genera, an example of such being ∼90% amino acid sequence identity with their C. trachomatis serovar D homologs CT849, CT848, and CT847. Interestingly, Russell et al. previously reported a Q119K substitution mutation in TC0236, which accounted for enhanced infectivity in both cultured cells and mouse lower genital tract tissue (34), but whether this mutation contributes to pathogenicity remains unknown. Since we have correlated a Q117E mutation in TC0237 with enhanced attachment to cultured cells, we hypothesize that this cluster of three proteins may participate in initial chlamydial interactions with host cells. However, we found that the TC0237(Q117E) mutation may not participate in the pathogenicity of C. muridarum in mice. This hypothesis originates from the observation that CMG28.38.1 with only the TC0237(Q117E) mutation was as effective as its isogenic background strain, CMG0.1.1, in inducing hydrosalpinx following intravaginal infection.
TC0668 encodes a highly conserved hypothetical protein of 408 aa and contains a single DUF1207 motif. It shares ∼90% amino acid identity with its homolog CT389 from C. trachomatis serovar D, and its first 24 aa are predicted to constitute a Gram-positive/negative signal sequence. In addition, the region covering residues 360 to 380 of CT389 has weak homology with mammalian phosphatase signatures, suggesting that TC0668 may be a secreted phosphatase. Partially consistent with this prediction and analyses, CT389 was enriched in the outer membrane complex (OMC) fraction of C. trachomatis, suggesting that it is associated with the OMC (15). It appears that the selective advantage of TC0668(G322R) for adaptation to unassisted in vitro infection is less than that of TC0237(Q117E), given that TC0668(G322R) is found at a subconsensus population level and TC0237(Q117E) is found at a consensus population level in CMG28. Although we have not been able to isolate clones that carry the TC0668 mutation alone due to the fact that all CMG28 clones carry the TC0237(Q117E) mutation, the available data have allowed us to associate the TC0668 mutation with the attenuation of in vivo pathogenicity of C. muridarum. However, it is unknown whether TC0668 is solely responsible for pathogenicity or if TC0668 and TC0237 act synergistically to induce upper genital tract disease.
In summary, this study revealed that TC0237 is involved in the chlamydial host attachment process, TC0668 is associated with upper genital tract pathogenesis of C. muridarum, and Chlamydia is tractable to Pasteurian passage and selection.
ACKNOWLEDGMENT
This work was supported in part by grants (to G.Z.) from the U.S. National Institutes of Health.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2009. Sexually transmitted disease surveillance, 2008. US Department of Health and Human Services, Atlanta, GA: http://www.cdc.gov/std/stats08/toc.htm. [Google Scholar]
- 2.Land JA, Van Bergen JE, Morre SA, Postma MJ. 2010. Epidemiology of Chlamydia trachomatis infection in women and the cost-effectiveness of screening. Hum Reprod Update 16:189–204. doi: 10.1093/humupd/dmp035. [DOI] [PubMed] [Google Scholar]
- 3.Strandell A, Waldenstrom U, Nilsson L, Hamberger L. 1994. Hydrosalpinx reduces in-vitro fertilization/embryo transfer pregnancy rates. Hum Reprod 9:861–863. [DOI] [PubMed] [Google Scholar]
- 4.Vasquez G, Boeckx W, Brosens I. 1995. Prospective study of tubal mucosal lesions and fertility in hydrosalpinges. Hum Reprod 10:1075–1078. [DOI] [PubMed] [Google Scholar]
- 5.Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex Transm Dis 32:49–56. doi: 10.1097/01.olq.0000148299.14513.11. [DOI] [PubMed] [Google Scholar]
- 6.Morrison RP, Caldwell HD. 2002. Immunity to murine chlamydial genital infection. Infect Immun 70:2741–2751. doi: 10.1128/IAI.70.6.2741-2751.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Maza LM, Peterson EM. 2002. Vaccines for Chlamydia trachomatis infections. Curr Opin Investig Drugs 3:980–986. [PubMed] [Google Scholar]
- 8.Rockey DD, Wang J, Lei L, Zhong G. 2009. Chlamydia vaccine candidates and tools for chlamydial antigen discovery. Expert Rev Vaccines 8:1365–1377. doi: 10.1586/erv.09.98. [DOI] [PubMed] [Google Scholar]
- 9.de la Maza LM, Pal S, Khamesipour A, Peterson EM. 1994. Intravaginal inoculation of mice with the Chlamydia trachomatis mouse pneumonitis biovar results in infertility. Infect Immun 62:2094–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuo C, Takahashi N, Swanson AF, Ozeki Y, Hakomori S. 1996. An N-linked high-mannose type oligosaccharide, expressed at the major outer membrane protein of Chlamydia trachomatis, mediates attachment and infectivity of the microorganism to HeLa cells. J Clin Invest 98:2813–2818. doi: 10.1172/JCI119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su H, Raymond L, Rockey DD, Fischer E, Hackstadt T, Caldwell HD. 1996. A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells. Proc Natl Acad Sci U S A 93:11143–11148. doi: 10.1073/pnas.93.20.11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swanson AF, Kuo CC. 1994. Binding of the glycan of the major outer membrane protein of Chlamydia trachomatis to HeLa cells. Infect Immun 62:24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su H, Zhang YX, Barrera O, Watkins NG, Caldwell HD. 1988. Differential effect of trypsin on infectivity of Chlamydia trachomatis: loss of infectivity requires cleavage of major outer membrane protein variable domains II and IV. Infect Immun 56:2094–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moelleken K, Hegemann JH. 2008. The Chlamydia outer membrane protein OmcB is required for adhesion and exhibits biovar-specific differences in glycosaminoglycan binding. Mol Microbiol 67:403–419. doi: 10.1111/j.1365-2958.2007.06050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Afrane M, Clemmer DE, Zhong G, Nelson DE. 2010. Identification of Chlamydia trachomatis outer membrane complex proteins by differential proteomics. J Bacteriol 192:2852–2860. doi: 10.1128/JB.01628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stephens RS, Koshiyama K, Lewis E, Kubo A. 2001. Heparin-binding outer membrane protein of chlamydiae. Mol Microbiol 40:691–699. doi: 10.1046/j.1365-2958.2001.02418.x. [DOI] [PubMed] [Google Scholar]
- 17.Becker E, Hegemann JH. 2014. All subtypes of the Pmp adhesin family are implicated in chlamydial virulence and show species-specific function. Microbiologyopen 3:544–556. doi: 10.1002/mbo3.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molleken K, Schmidt E, Hegemann JH. 2010. Members of the Pmp protein family of Chlamydia pneumoniae mediate adhesion to human cells via short repetitive peptide motifs. Mol Microbiol 78:1004–1017. doi: 10.1111/j.1365-2958.2010.07386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crane DD, Carlson JH, Fischer ER, Bavoil P, Hsia RC, Tan C, Kuo CC, Caldwell HD. 2006. Chlamydia trachomatis polymorphic membrane protein D is a species-common pan-neutralizing antigen. Proc Natl Acad Sci U S A 103:1894–1899. doi: 10.1073/pnas.0508983103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang JP, Stephens RS. 1992. Mechanism of C. trachomatis attachment to eukaryotic host cells. Cell 69:861–869. doi: 10.1016/0092-8674(92)90296-O. [DOI] [PubMed] [Google Scholar]
- 21.Molleken K, Becker E, Hegemann JH. 2013. The Chlamydia pneumoniae invasin protein Pmp21 recruits the EGF receptor for host cell entry. PLoS Pathog 9:e1003325. doi: 10.1371/journal.ppat.1003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis CH, Raulston JE, Wyrick PB. 2002. Protein disulfide isomerase, a component of the estrogen receptor complex, is associated with Chlamydia trachomatis serovar E attached to human endometrial epithelial cells. Infect Immun 70:3413–3418. doi: 10.1128/IAI.70.7.3413-3418.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puolakkainen M, Kuo CC, Campbell LA. 2005. Chlamydia pneumoniae uses the mannose 6-phosphate/insulin-like growth factor 2 receptor for infection of endothelial cells. Infect Immun 73:4620–4625. doi: 10.1128/IAI.73.8.4620-4625.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. 2004. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci U S A 101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hower S, Wolf K, Fields KA. 2009. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol 72:1423–1437. doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Lu C, Peng B, Zeng H, Zhou Z, Wu Y, Zhong G. 2012. Induction of protective immunity against Chlamydia muridarum intravaginal infection with a chlamydial glycogen phosphorylase. PLoS One 7:e32997. doi: 10.1371/journal.pone.0032997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Chen D, Zhong Y, Wang S, Zhong G. 2008. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun 76:3415–3428. doi: 10.1128/IAI.01377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang YX, Anderson DJ, Fierer J, Stephens RS, Kagnoff MF. 1997. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J Clin Invest 99:77–87. doi: 10.1172/JCI119136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng W, Shivshankar P, Zhong Y, Chen D, Li Z, Zhong G. 2008. Intracellular interleukin-1alpha mediates interleukin-8 production induced by Chlamydia trachomatis infection via a mechanism independent of type I interleukin-1 receptor. Infect Immun 76:942–951. doi: 10.1128/IAI.01313-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z, Conrad T, Zhou Z, Chen J, Dutow P, Klos A, Zhong G. 2014. Complement factor C5 but not C3 contributes significantly to hydrosalpinx development in mice infected with Chlamydia muridarum. Infect Immun 82:3154–3163. doi: 10.1128/IAI.01833-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Zhou Z, Chen J, Wu G, Yang Z, Zhou Z, Baseman J, Zhang J, Reddick RL, Zhong G. 2014. Lack of long-lasting hydrosalpinx in A/J mice correlates with rapid but transient chlamydial ascension and neutrophil recruitment in the oviduct following intravaginal inoculation with Chlamydia muridarum. Infect Immun 82:2688–2696. doi: 10.1128/IAI.00055-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sturdevant GL, Kari L, Gardner DJ, Olivares-Zavaleta N, Randall LB, Whitmire WM, Carlson JH, Goheen MM, Selleck EM, Martens C, Caldwell HD. 2010. Frameshift mutations in a single novel virulence factor alter the in vivo pathogenicity of Chlamydia trachomatis for the female murine genital tract. Infect Immun 78:3660–3668. doi: 10.1128/IAI.00386-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsey KH, Sigar IM, Schripsema JH, Denman CJ, Bowlin AK, Myers GA, Rank RG. 2009. Strain and virulence diversity in the mouse pathogen Chlamydia muridarum. Infect Immun 77:3284–3293. doi: 10.1128/IAI.00147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell M, Darville T, Chandra-Kuntal K, Smith B, Andrews CW Jr, O'Connell CM. 2011. Infectivity acts as in vivo selection for maintenance of the chlamydial cryptic plasmid. Infect Immun 79:98–107. doi: 10.1128/IAI.01105-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang L, Zhang H, Lei L, Gong S, Zhou Z, Baseman J, Zhong G. 2013. Oviduct infection and hydrosalpinx in DBA1/j mice is induced by intracervical but not intravaginal inoculation with Chlamydia muridarum. PLoS One 8:e71649. doi: 10.1371/journal.pone.0071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lei L, Chen J, Hou S, Ding Y, Yang Z, Zeng H, Baseman J, Zhong G. 2014. Reduced live organism recovery and lack of hydrosalpinx in mice infected with plasmid-free Chlamydia muridarum. Infect Immun 82:983–992. doi: 10.1128/IAI.01543-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Zhang H, Zhou Z, Yang Z, Ding Y, Zhou Z, Zhong E, Arulanandam B, Baseman J, Zhong G. 2014. Chlamydial induction of hydrosalpinx in 11 strains of mice reveals multiple host mechanisms for preventing upper genital tract pathology. PLoS One 9:e95076. doi: 10.1371/journal.pone.0095076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, Watkins H, Zhou B, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 2013. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect Immun 81:636–644. doi: 10.1128/IAI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong S, Yang Z, Lei L, Shen L, Zhong G. 2013. Characterization of Chlamydia trachomatis plasmid-encoded open reading frames. J Bacteriol 195:3819–3826. doi: 10.1128/JB.00511-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Chen C, Gong S, Hou S, Qi M, Liu Q, Baseman J, Zhong G. 2014. Transformation of Chlamydia muridarum reveals a role for Pgp5 in suppression of plasmid-dependent gene expression. J Bacteriol 196:989–998. doi: 10.1128/JB.01161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding H, Gong S, Tian Y, Yang Z, Brunham R, Zhong G. 2013. Transformation of sexually transmitted infection-causing serovars of Chlamydia trachomatis using blasticidin for selection. PLoS One 8:e80534. doi: 10.1371/journal.pone.0080534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci U S A 108:7189–7193. doi: 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borges V, Ferreira R, Nunes A, Sousa-Uva M, Abreu M, Borrego MJ, Gomes JP. 2013. Effect of long-term laboratory propagation on Chlamydia trachomatis genome dynamics. Infect Genet Evol 17:23–32. doi: 10.1016/j.meegid.2013.03.035. [DOI] [PubMed] [Google Scholar]
- 45.Sandoz KM, Rockey DD. 2010. Antibiotic resistance in Chlamydiae. Future Microbiol 5:1427–1442. doi: 10.2217/fmb.10.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hawgood BJ. 1999. Doctor Albert Calmette 1863-1933: founder of antivenomous serotherapy and of antituberculous BCG vaccination. Toxicon 37:1241–1258. doi: 10.1016/S0041-0101(99)00086-0. [DOI] [PubMed] [Google Scholar]
- 47.Hanley KA. 2011. The double-edged sword: how evolution can make or break a live-attenuated virus vaccine. Evolution (N Y) 4:635–643. doi: 10.1007/s12052-011-0365-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Silvachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng B, Lu C, Tang L, Yeh IT, He Z, Wu Y, Zhong G. 2011. Enhanced upper genital tract pathologies by blocking Tim-3 and PD-L1 signaling pathways in mice intravaginally infected with Chlamydia muridarum. BMC Infect Dis 11:347. doi: 10.1186/1471-2334-11-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L, Lei L, Chang X, Li Z, Lu C, Zhang X, Wu Y, Yeh IT, Zhong G. 2010. Mice deficient in MyD88 develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J Immunol 184:2602–2610. doi: 10.4049/jimmunol.0901593. [DOI] [PubMed] [Google Scholar]
- 55.Miyairi I, Laxton JD, Wang X, Obert CA, Tatireddigari Arva VRR, van Rooijen N, Hatch TP, Byrne JI. 2011. Chlamydia psittaci genetic variants differ in virulence by modulation of host immunity. J Infect Dis 204:654–663. doi: 10.1093/infdis/jir333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Utterback T, Berry K, Bass S, Linher K, Weidman J, Khouri H, Craven B, Bowman C, Dodson R, Gwinn M, Nelson W, DeBoy R, Kolonay J, McClarty G, Salzberg SL, Eisen J, Fraser CM. 2000. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res 28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]