

Abstract
Autologous chondrocyte implantation (ACI) is a golden treatment for large defects of the knee joint without osteoarthritis or other complications. Despite notable progresses, generation of a stable chondrocyte phenotype using progenitor cells remains a main obstacle for chondrocyte-based cartilage treatment. Monolayer chondrocyte expansion in vitro is accompanied by chondrocyte dedifferentiation, which produces a non-specific mechanically inferior extracellular matrix (ECM) unsuitable for ACI. In-depth understanding of the molecular events during chondrocyte dedifferentiation is required to maintain the capacity of in vitro expanded chondrocytes to produce hyaline cartilage-specific ECM. This review discusses key cytokines and signaling pathways involved in chondrocyte dedifferentiation from the standpoint of catabolism and anabolism. Some potential therapeutic strategies are also presented to counteract chondrocyte dedifferentiation for cell-based cartilage therapy.
Keywords: Chondrocyte dedifferentiation, in vitro, cytokines, signaling pathways, cartilage repair, autologous chondrocyte implantation
Introduction
Articular cartilage is an avascular and aneural load-bearing connective tissue with a very limited capacity for intrinsic repair. Once damaged by trauma or pathology, it is highly susceptible to structural degradation, making it particularly difficult to restore. The chondrocyte is the only cell type that resides within articular cartilage and is solely responsible for the synthesis and turnover of the extracellular matrix (ECM).
For more than decades, autologous chondrocyte implantation (ACI) has been the golden therapy for patients with focal lesions of articular cartilage [1]. ACI procedure consists of three stages (Figure 1). The first stage is to harvest around 100 milligrams (mg) cartilage from a non-weight bearing area of the patient under arthroscopy. The chondrocytes are enzymatically isolated from cartilage and propagated to obtain enough cells in vitro in a specialized laboratory. These cells are re-implanted in combination with a membrane (tibial periosteum or biomembrane) in the damaged area of the articular cartilage when the patient undergoes a second treatment. ACI treatment leads to satisfying clinical results in terms of patient satisfaction, reduction of pain and improvement in knee function [2]. Nevertheless, it is not yet a routine application in clinical practice, with its application being mainly limited to young patients. One of the technical challenges is the isolation and ex vivo expansion of a large enough number of differentiated articular chondrocytes. During this process, the chondrocytes become fibroblast-like cells (Figure 2), characterized by an increase in the expression of type I collagen (COL-1) and a decrease of cartilage-specific markers, such as type II, XI, IX collagens (COL-2, COL-11, COL-9) and aggrecan (ACAN) [3-5]. This process is designated as chondrocyte dedifferentiation. The dedifferentiated chondrocytes produce a non-specific mechanically inferior ECM and are no longer suitable for ACI. Therefore, chondrocyte dedifferentiation is a major obstacle in cell-based cartilage repair. In the past decades, multiple studies have been carried out to explore the molecular mechanisms underlying chondrocyte dedifferentiation.
Figure 1.
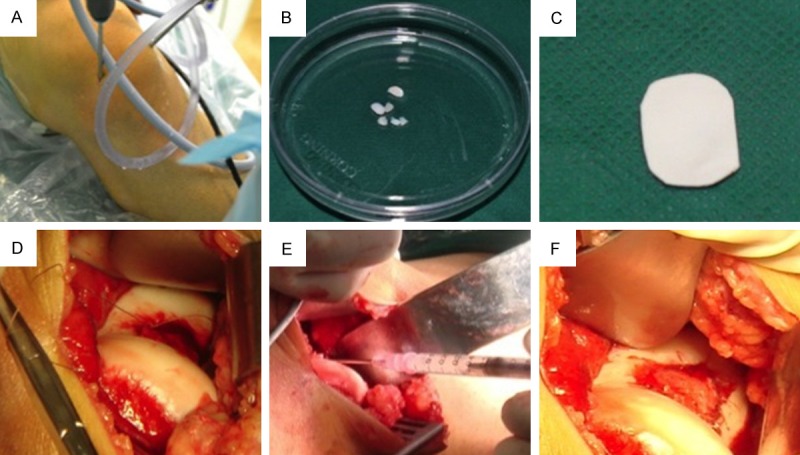
Autologous chondrocyte implantation. A. Cartilage biopsy was harvested from a non-weight bearing area of the knee joint under an arthoscope. B. Around 100 mg cartilage was harvested and enzymatically digested. Chondrocytes were propagated in a GMP lab. C. During the second operation, a collagen membrane was trimmed according to the defect size. D. The periosteum was sutured. E. Chondrocytes were injected into the space between the membrane and the defect. F. After chondrocyte injection, the membrane was sealed with fibrin glue to prevent leaking.
Figure 2.
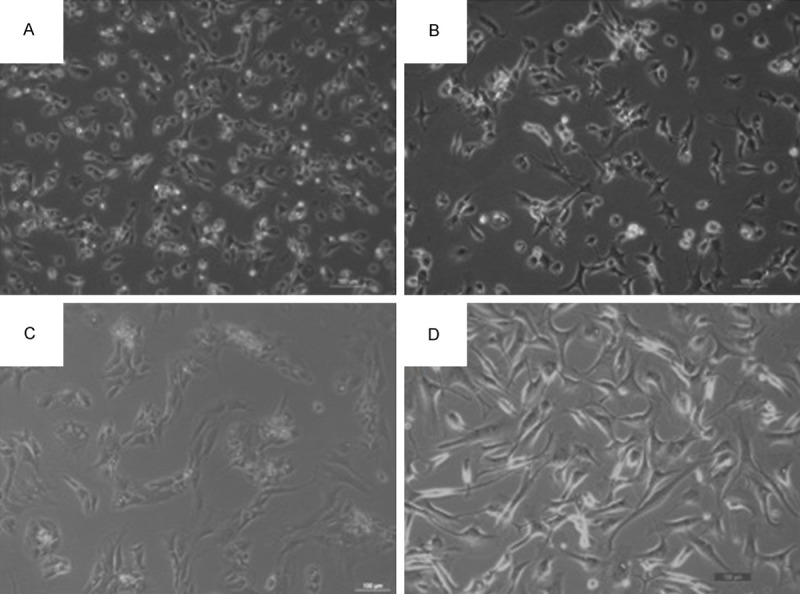
Chondrocyte dedifferentiation during in vitro expansion. Primary human chondrocyte morphology under invert microscope (×100). With passaging, round and polygonal chondrocytes were shifted to fibroblast-like cells. A (P0), B (P1), C (P2), D (P4).
This review focuses on cytokines and signaling pathways of the anabolism and catabolism (summarized in Table 1), which are involved in chondrocyte phenotype maintenance. The purpose of this report is to provide an up-to-date overview of cytokine networking involved in chondrocyte dedifferentiation in vitro and to provide potential targets for improving autologous chondrocyte-based cartilage repair.
Table 1.
Summary of cytokines and signaling pathways involved in chondrocyte dedifferentiation
| Cytokine | Signaling pathway | Effect on chondrocyte phenotype | Reference |
|---|---|---|---|
| IL-1β | p38MAPK, ERK, JNK, PKCα, PKCζ, WNT-3A, WNT-7A | (-) COL-2; | [6,7,10-12,14-16] |
| (+) COL-1; | |||
| (+) COL-3 | |||
| IL-6 | Notch | (-) ACAN; | [18-21] |
| (+) MMP-13 | |||
| IGF-I | PI3K/AKT, ERK/MAPK | (+) ACAN; | [33-41] |
| (+) COL-2; | |||
| (+) COL-1; | |||
| (+) COL-10 | |||
| PDGF | ERK, PI3K/AKT | undefined | [20,42-44] |
| TGF-β1 | ERK/AP-1, SP3/SP1 | (±) COL-2* | [45-52] |
| (+) COL-1 | |||
| BMP-2 | p38MAPK, WNT-7A | (+) COL-2 | [38,62-68] |
| (+) ACAN | |||
| (+) COL-1 | |||
| FGF-18 | ERK, p38MAPK | undefined | [72-74] |
Species - dependent: rabbit/goat/mouse: (+) COL-2; human: (-) COL-2, (-) ACAN.
(+) indicates up-regulation; (-) indicates down-regulation.
Imbalance between catabolism and anabolism
The major function of chondrocytes is to produce cartilage ECM. In turn, the ECM modifies chondrocyte behavior. Metabolic balance of ECM could play a critical role in chondrocyte phenotype maintenance. The failure to maintain the anabolic-catabolic balance within the cartilage matrix may give rise to chondrocyte dedifferentiation. The catabolic genes, such as inflammatory cytokines and proteases, participate in cartilage matrix degradation. In contrast, the anabolic genes including growth factors (e.g. insulin-like growth factor, bone morphogenetic protein) critically contribute to cartilage matrix production and maintenance.
Inflammatory cytokines
Interleukin 1-beta (IL-1β), the major pro-inflammatory cytokine in osteoarthritic synovial fluid, induces the dedifferentiation of cultured articular chondrocytes by decreasing COL-2 while increasing COL-1 and COL-3 expression at mRNA and protein levels [6,7]. During chondrocyte dedifferentiation, IL-1β up-regulates the synthesis of E-series prostaglandins (PGE) to induce chondrocyte differentiation and increase COL-2 expression, thereby counteracting the dedifferentiation-promoting action of IL-1β [8,9]. Thus, the role of IL-1β on chondrocytes appears to be paradoxical. On one hand, IL-1β directly promotes chondrocyte dedifferentiation; on the other hand, it maintains chondrocytes in differentiated status through increasing PGE. To further understand this paradox, several downstream signal molecules have been studied in IL-1β-treated articular chondrocytes and found that IL-1β activates c-Jun-N-terminal kinase (c-JNK), p38 mitogen-activated protein kinase (MAPK) and extracellular-signal-regulated protein kinase (ERK) -1/2 in primary cultures of human articular chondrocytes [10]. ERK-1/2 is the major tyrosine-phosphorylated protein in IL-1β stimulated chondrocytes. In response to IL-1β stimulation, ERK and p38 MAPK are activated at 5 minutes and JNK is activated at 15 minutes [11]. Interestingly, prolonged IL-1β stimulation for 24 hours results in ERK-1/2 inhibition in chondrocytes [12]. Thus, the role of ERK-1/2 in chondrocyte dedifferentiation remains controversial. The dedifferentiated chondrocytes express lower levels of cyclooxygenase-2 (COX-2) mRNA and synthesize less PGE2 than differentiated chondrocytes. The down-regulation of COX-2 expression is associated with a reduction in IL-1β-induced p38 MAPK activation [13]. The WNT family protein (WNT-3A, WNT-7A) activities are also enhanced during IL-1β-induced dedifferentiation of articular chondrocytes [14,15].
Treating lapine articular chondrocytes with human recombinant IL-1β has been shown to boost the production of nitric oxide (NO) and reduce COL-2 synthesis. NO activates ERK-1/2 and p38 MAPK, but inhibits protein kinase C alpha (PKC-α) and PKC zeta (PKC-ζ) in chondrocytes [16]. NO appears to affect the metabolism of cartilage in a stimulus-dependent manner, as NO has been shown to enhance tumor necrosis factor (TNF)-α-induced, but not IL-1β-induced, ACAN degradation in bovine cartilage explants [17]. Taken together, IL-1β regulates chondrocyte dedifferentiation through multiple pathways and the balance between the activities of these pathways may determine the overall effect of IL-1β.
In addition to IL-1β, the role of IL-6 in chondrocyte dedifferentiation has attracted much attention. IL-6 is produced by osteoarthritic synovial lining or expanded chondrocytes rather than by cartilage [18], which exerts catabolic effects on cartilage and chondrocytes [19,20]. According to a recent report, IL-6 induces similar effects as Notch activation in chondrocytes. Furthermore, IL-6 mediates the effects of Notch signaling pathway through ACAN suppression and MMP-13 induction [21].
Proteases
Cartilage matrix components are degraded by various proteases, such as cathepsins [22], matrix metalloproteinases (MMPs) [23] and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) [24]. A microarray analysis revealed that 84 genes are up-regulated and 56 genes are down-regulated in passage 4 (P4) human chondrocytes compared to passage 1 (P1) cells. Among them, cathepsin K (CTSK) expression level is increased 28-fold. Overexpression of CTSK leads to reduced matrix production in cultured human chondrocytes in vitro and poor formation of engineered cartilage in vivo. In contrast, CTSK knockdown increases the capability to maintain the chondrogenic phenotype of in vitro expanded cells with increased mRNA and protein expression of COL-2 and ACAN, in comparison to control cells [25].
To date, the role of MMP-13 in COL-2 degradation [26,27] has been established, which critically contributes to the pathogenesis of experimental osteoarthritis (OA) [28] and human OA [29]. However, there are conflicting results about the expression changes of MMP-13 during chondrocyte expansion. Two studies found that the expression of MMP-13 decreases during cell passaging [30,31] while another in vitro study reported that MMP-13 expression is increased from day 1 up to 21 [32]. Moreover, chitosan-plasmid DNA nanoparticles encoding short hairpin RNA (shRNA) against MMP-13 has been shown to inhibit the expression of dedifferentiation-related genes and help regenerating endurable cartilage [33]. Therefore, genetic modification of MMP-13 or CTSK could be a promising strategy to block chondrocyte dedifferentiation from the standpoint of catabolism.
Insulin-like growth factor-I
Insulin-like growth factor-I (IGF-I), one of the most important anabolic growth factors for articular cartilage, plays a key role in cartilage homeostasis, balancing proteoglycans synthesis and breakdown [34]. Several studies have demonstrated that IGF-I is not only able to extend the chondrogenic potential of dedifferentiated chondrocytes in vitro [35], but also can enhance the chondrogenesis of bovine articular chondrocytes seeded into polymer scaffolds [36]. Interaction of IGF-I and IGF-1R activates both the phosphoinositide-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) and ERK-MAPK pathways, but only the activation of the PI3K/AKT/mTOR pathway is responsible for the ability of IGF-I to increase proteoglycan synthesis of adult human articular chondrocytes [37,38]. The transfection of IGF-I gene (tIGF-I) into chondrocytes stimulates both COL-2 and ACAN expression. Of note, in comparison to the individual transgenes of BMP-2, BMP-7, TGF-β1 and FGF-2, tIGF-I generates the maximal stimulation of COL-2 expression in bovine articular chondrocytes [39].
Monolayer chondrocytes rapidly lose their differentiated phenotype, which is accompanied by a significant decrease in IGF-I expression [30]. IGF-I drives dedifferentiated chondrocytes re-differentiation back to the differentiated phenotype. These re-differentiated chondrocytes still have the capacity to form cartilage-like tissue in a high-density cell culture [35]. Furthermore, IGF-I can block IL-1β or NO-induced rapid dedifferentiation of chondrocytes and help maintaining chondrocyte phenotype [12,40,41]. In addition, IGF-1 blocks NO-induced apoptosis through PI3K pathway [42]. However, it has been shown that IGF-I also increases the synthesis of COL-1 and COL-10 (an indicator of chondrocyte hypertrophy) [39,42]. Therefore, much endeavor has to be directed to explore other factors that may have better re-differentiation potential.
Platelet-derived growth factor
Platelet-derived growth factor (PDGF) is a heat-stable positively charged hydrophilic protein. PDGF activates ERK and up-regulates TGF-β1 and TGF-β3 expression in human articular chondrocytes [20,43]. Growth factor cocktail of TGF-β1, FGF-2, and PDGF-BB has been shown to increase human articular chondrocyte proliferation and maintain their chondrogenic potential [43]. More importantly, different from TGF-β1 and FGF-2, PDGF-BB can promote the proliferation and extracellular matrix sulfation of resting chondrocytes in vitro without inducing a hypertrophic chondrocyte phenotype [44]. Combination of IGF-1 and PDGF-BB inhibits IL-1β-mediated chondrocyte apoptosis, in part through suppression of PI3K/AKT pathway [45]. Therefore, addition of PDGF-BB to a monolayer culture system could be potentially used to maintain chondrocyte phenotype during in vitro chondrocyte expansion.
Transforming growth factor beta
The effects of transforming growth factor beta (TGF-β) on chondrocyte phenotype maintenance appear to be contradictory. For instance, one study demonstrated that TGF-β1 is capable of stimulating COL-2 expression in monolayer culture of rabbit articular chondrocytes after 24 h treatment [46]. In contrast, after 6-day exposure, TGF-β1 exerts an inhibitory effect on collagen synthesis without significant proteoglycan production change [47]. It is postulated that the chondrocyte status may influence the TGF-β1 action on matrix synthesis. Moreover, the action of TGF-β1 on matrix synthesis is species-dependent. Studies have demonstrated that TGF-β1 can stimulate proteoglycan and COL-2 production during either expansion of goat articular chondrocytes or mouse precartilaginous stem cells in monolayer cultures [48,49]. Although human articular chondrocyte dedifferentiation leads to decreasing TGF-β Rece-ptor II (TβRII) expression level and subsequent TGFβ response [50], the addition of TGF-β1 during monolayer expansion of human articular chondrocyte results in the loss of COL-2 and ACAN while increases the level of COL-10 protein [51]. In addition, TGF-β1 transduction pathway is also species-dependent. One study has shown that TGF-β1 induces COL-2A1 expression in rat articular chondrocytes could implicate activation of ERK and subsequent activator protein 1 (AP-1) binding [52]. Another study reported that the COL-2A1 gene transcription is down-regulated by TGF-β1 in rabbit articular chondrocytes through the increase of the Sp3/Sp1 ratio [53].
Recently, the effect of combination of TGF-β with other growth factors or gene intervention on chondrocyte phenotype gets more and more attention. For example, conjunct IGF-I with TGF-β1 stimulates the dedifferentiated human articular chondrocytes to re-differentiate, with an increasing in the expression of the cartilage-specific proteins COL-2 and sex determining region Y box gene 9 (SOX-9) and down-regulation of COX-2 and MMP-13. Thus, they exert anabolic effects on chondrocytes and enhance the chondrogenic potential [12]. Also, adenoviral vector-mediated overexpression of TGF-β3 and RNA interference of COL-1 drive dedifferentiated chondrocytes back to the differentiated phenotype in a three-dimension (3-D) culture system [54].
Bone morphogenetic proteins
Bone morphogenetic proteins (BMPs) are members of the TGF-β superfamily, which regulate a wide range of developmental processes and control the differentiation of several musculoskeletal tissues including bone and cartilage. BMP-2 and -7 have already been clinically used for bone regeneration [55-59]. Besides, several BMPs, including BMP-2, -6, -9, -12 and -13, stimulate cartilage matrix macromolecules synthesis by articular chondrocytes [60-62], revealing their potential to promote cartilage repair. During monolayer culture, BMP-2 expression levels are diminished during passaging. The addition of BMP-2 to monolayer culture could slow down chondrocyte dedifferentiation and reinforce the maintenance of chondrocyte phenotype in long-term culture conditions [31,63]. It was reported that BMP-2 has a stronger capability than TGF-β1 to restore the character of chondrocytes. However, this re-differentiation is limited to a certain degree because COL-1 expression persists after the addition of BMP-2 or TGF-β1 [64]. Combining BMP-2 with COL-1A1 siRNA effectively promotes the assembly of cartilage matrix in agarose hydrogel without production of COL-1 [65]. Simultaneous delivery of BMP-2 and SOX-9, two chondrogenic lineage-determining genes, results in significantly increased expression of chondrogenesis-related markers (COL-2 and ACAN) and glycosaminoglycan (GAG) matrix formation both in vitro and in vivo, compared with individual delivery of BMP-2 or SOX-9 gene [66]. Studies also have suggested that BMP-2 may have a better re-differentiation potential than IGF-I or TGF-β [64,67]. Furthermore, co-transfection of IGF-I and BMP-2 can greatly increase the expression of COL2A1 and ACAN [39].
Although SMAD is the main signaling mechanism of TGF-β, the effect of BMP-2 on COL-2 expression is not mediated by SMAD pathway, as overexpression of SMAD-6 (an inhibitor of the canonical BMP-SMAD signaling pathway) could not block BMP-2-induced COL-2 pro-collagen expression in mouse chondrocytes [64]. Therefore, non-BMP-SMAD signaling pathway could play a key role in BMP-2-induced COL-2 expression in chondrocytes. This hypothesis is consistent with the results from a study that BMP-2 activates p38 MAPK, subsequently inhibits Wnt-7A/β-catenin and eventually up-regulates SOX-9 and COL-2 expression [68]. Taken together, BMP-2 has a stronger potential to prevent dedifferentiation and induce re-differentiation in chondrocytes.
Fibroblast growth factors
Among the members of the fibroblast growth factor (FGF) family, FGF-2 and -18 have been implicated as key regulators in chondrocyte phenotype maintenance in vitro. The role of FGF-2 in chondrocyte dedifferentiation is in discrepancy. A series of studies have determined that FGF-2 functions as an anabolic inducer in animal articular chondrocytes. During monolayer expansion, the addition of FGF-2 reduces expression of fibroblastic molecules in calf articular chondrocytes. Furthermore, FGF-2 en-hances chondrocyte response to BMP-2 in a 3-D culture system on poly (glycolic acid) scaffolds [69]. Of note, the role of FGF-2 in human articular chondrocytes appears to contradict its role in calf articular chondrocytes. Recent studies revealed that FGF-2 stimulates the human articular chondrocyte dedifferentiation in monolayer culture, suggesting it may serve primarily as a catabolic factor [70]. FGF-2-expanded chondrocytes are fully dedifferentiated but retain their potential of re-differentiation to hyaline cartilage in response to environmental changes [71]. Such discrepancies suggest that the function of FGF-2 in articular chondrocytes may differ fundamentally between species.
FGF-18 has been shown to promote early chondrocyte proliferation while inhibit the differentiation and matrix synthesis of chondrocytes [72]. Moreover, FGF-18 regulates chondrocyte proliferation and differentiation via binding FGF receptor 3 (FGFR-3) and activating ERK and p38 MAPK signal pathways [73]. A recent study verified that loss of chondrocyte phenotype is associated with increased FGF-18 expression level, indicating that FGF-18 may serve as a marker of dedifferentiation [74]. In this perspective, strategies to down-regulate the FGF-18 expression may contribute to the chondrocyte phenotype maintenance.
Signaling pathways in chondrocyte dedifferentiation
Extracellular signals evoked by ligand-receptor interaction enter into the intracellular compartment to initiate a series of responses and control multiple fundamental cellular processes such as proliferation, differentiation, survival and apoptosis. There are multiple cellular signaling pathways involved in chondrocyte dedifferentiation, such as MAPK, PI3K/AKT, PKC, WNT and Notch, as shown in Figure 3.
Figure 3.
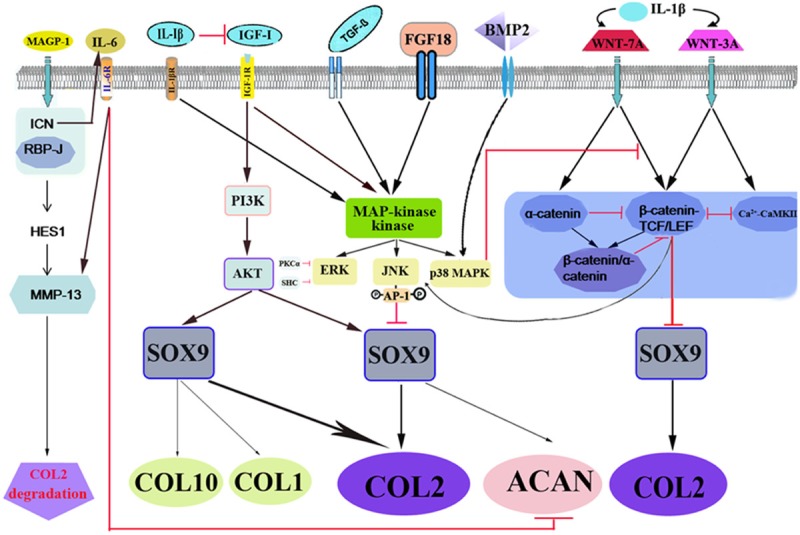
Schematic illustration of molecular events in chondrocyte dedifferentiation. Multiple signaling pathways are evoked during chondrocyte undergoing stimulants, including catabolic factors (IL-1β, IL-6, FGF-18) and anabolic factors (IGF-I, BMP-2, TGF-β). MAPK, PI3K/AKT, PKC, WNT, and Notch proteins are involved in regulating the ratio of COL-2 and COL-1, a key indicator of chondrocyte dedifferentiation. The red dash indicates inhibition and the black arrow indicates promotion.
MAPK signaling
The MAPK pathway (p38MAPK, ERK and JNK) controls multiple fundamental cellular processes such as proliferation, differentiation, survival and apoptosis. p38 MAPK plays a role in the maintenance of differentiated status of articular chondrocytes and induction of chondrocyte apoptosis. p38 MAPK positively regulates ch-ondrogenesis while the ERK pathway functions as a negative regulator in staurosporine-induced re-differentiation [75]. It has been established that ERK signal transduction pathway is involved in the regulation of cell growth and differentiation. ERK-1 and -2 form the central component in the MAPK cascade. ERK-1/2 activation induces chondrocyte dedifferentiation and inhibits NO-induced apoptosis. In contrast, inhibition of ERK-1/2 subsequently blocks dedifferentiation [76]. However, a conflicting report showed that inhibition of ERK pathway can not block chondrocyte dedifferentiation [31]. ERK-1/2 activity in chondrogenesis is negatively regulated by PKCα, whereas ERK-1/2 and PKCα independently regulate dedifferentiation during chondrocyte passaging. Besides PKC, SH2 domain-containing transforming protein (SHC) negatively regulates ERK activation in the absence of growth factors in human cells. The adaptor protein SHC directly binds the ERK, thus preventing its activation in the absence of extracellular stimuli. The SHC-ERK complex restricts ERK nuclear translocation, restraining ERK-dependent transcription of genes [77]. A decrease in SHC-ERK interaction has been suggested as a marker for irreversibly dedifferentiated chondrocytes in tissue engineering [78].
Besides ERK-1/2 and p38 MAPK, the JNK signaling pathway is also reported to play a role in the differentiation of chondrocytes [15,79,80]. However, the results are controversial. JNK phosphorylates c-Jun, leading to the activation of AP-1 to suppress the expression of SOX-9, a major transcription factor regulating COL-2 expression. In addition, c-JNK/AP has been shown to function as downstream of WNT-3A-induced chondrocyte dedifferentiation [15]. A recent study demonstrated that JNK inhibitor suppresses miR-34-targeted Rac1 expression in chick chondroprogenitors, and Rac1 interacts with Rho to negatively modulate reorganization of the actin cytoskeleton, which is one of the essential processes for establishing chondrocyte-specific morphology [80].
PI3K/AKT signaling
IGF-1 binding to a receptor, namely IGF-IR, increases the activity of phosphatidylinositol 3-kinase (PI3K), which up-regulates the levels of the phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] and phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3]. The binding of AKT (also known as protein kinase B, PKB) to PI(4,5)P2 and PI(3,4,5)P3 allows tyrosine phosphorylation of the very important regulatory sites of AKT, resulting in the further phosphorylation of AKT substrates, such as mTOR [81]. PI3K/AKT signaling pathway plays a critical role in cell growth, proliferation and differentiation. Several studies have demonstrated that PI3K/AKT signaling pathway is not only responsible for the ability of IGF-I to increase chondrocyte proteoglycan synthesis [82], but also involved in blockage of the activation of p38 kinase and ERK-1/2 and inhibition of PKCα and PKCζ induced by NO, which in turn suppresses dedifferentiation and apoptosis.
PKC
PKC is a family of protein kinases that control the function of other proteins through the phosphorylation of hydroxyl groups of serine and threonine amino acid residues on these proteins. In humans, the PKC family is divided into three subfamilies: conventional (α, βI, βII, and γ), novel (δ, ε, η, and θ), and atypical (ι and ζ). PKCα promotes chondrogenesis by activating COL-2 expression and blocks dedifferentiation of chondrocytes [83,84]. PKCα expression is down-regulated during chondrocyte passaging, resulting in cellular dedifferentiation [84]. Therefore, PKCα functions as a critical signaling molecule promoting chondrocyte differentiation and maintaining its differentiated phenotype. A decrease in PKCα and -ζ activities is required for both sodium nitroprusside (SNP)-induced dedifferentiation and apoptosis of rabbit knee joint articular chondrocytes. Although the dedifferentiation of chondrocytes during subculturing is not accompanied by any changes in PKCζ expression and activity [76], inhibition of PKCζ activity is required for SNP-induced dedifferentiation of chondrocytes [85].
WNT signaling
WNTs are a family of 19 morphogens that play a fundamental role in developmental processes ranging from embryonic morphogenesis to homeostasis of adult tissues. WNT pathway is divided into at least two branches: the canonical WNT/β-catenin signaling and the β-catenin-independent “non-canonical” pathways.
In the canonical pathway, upon binding of WNTs (such as WNT-3A) to the co-receptor consisting of frizzled (FZD) and lipoprotein receptor-related protein (LRP), the dishevelled (DVL) is activated. Activated DVL inhibits the destruction complex, resulting in nuclear import of β-catenin and its subsequent binding with TCF (T cell factor)/LEF (lymphoid enhancer binding factor) family of transcription factors to initiate the transcription of target genes. The non-canonical WNT pathways include calmodulin-dependent protein kinase (CaMK) II pathway and c-Jun N-terminal kinase-mediated planar cell polarity pathway [86,87].
Most of the cellular effects involved in WNT signaling are through the modulation of β-catenin. For example, WNT-3A and -7A stimulating β-catenin-mediated transcription involved in the regulation of chondrogenesis and dedifferentiation of primary articular chondrocytes [14,15]. Accumulation of β-catenin and subsequent activation of the β-catenin-TCF/LEF complex induces articular chondrocyte dedifferentiation characterized by the suppression of COL-2 expression and the initiation of COL-1 expression. In addition, WNT-3A causes the expression of c-Jun and its phosphorylation by JNK, resulting in activation of AP-1, which suppresses the expression of SOX-9 [15]. Besides β-catenin, α-catenin also accumulates to interact with each other to form a complex in primary monolayer culture during chondrocyte dedifferentiation. This complex inhibits ubiquitin-independent degradation of α-catenin and β-catenin-TCF/LEF’s transcriptional activity to block WNT-7A/β-catenin induced dedifferentiation in chondrocytes [88]. Recent studies have demonstrated that α-catenin recruits the transcriptional repressor Gli3R to β-catenin to form a ternary complex, thus inhibits β-catenin’s transcriptional activity and dedifferentiation of articular chondrocytes [89]. During WNT-3A-induced dedifferentiation of human articular chondrocytes, “non-canonical” CaMKII is also activated and reciprocally inhibits β-catenin-dependent pathways. Therefore, blockade of the canonical β-catenin pathway could induce dedifferentiation through de-repression of the CaMKII pathway [90].
Notch signaling
Notch signaling is a conserved pathway that plays a fundamental role in embryogenesis and maintenance of adult biological processes [91,92]. In mammals, the interaction of Notch ligands (Jagged 1 [JAG-1], JAG-2, Delta-like [DLL] protein, microfibril-associated glycoprotein [MAGP-1], MAGP-2, Notch like EGF-related receptor, and contactin) with its receptors (Notch1, 2, 3 and 4) leads to a series of cleavages mediated by ADAM proteases and γ-secretase complex, resulting in the release of intracellular domain of Notch receptor (ICN) from membrane [93,94]. The active form of ICN fragment translocates into the nucleus and forms a complex with other transcriptional factors (such as recombination signal binding protein J [RBP-J]) and regulates downstream gene expression, such as hairy and enhancer of split 1 (HES1). Besides its important role in carcinogenesis, the Notch pathway has recently been found to be involved in the regulation of articular cartilage development [95], chondrocyte differentiation [96-98] and dedifferentiation during osteoarthritis [99]. Successive passaging of murine articular chondrocytes (MACs) results in chondrocyte dedifferentiation, which is accompanied by increased expression of Notch ligands (DLL-1, JAG-1) and HES1 rather than up-regulation of Notch1 receptor. Sustained activation of Notch1 signaling shows no effect on COL-2 mRNA expression, but decreases COL-2 protein levels in chondrocytes. MMP-13, the main MMP family member involved in COL-2 degradation, is up-regulated in the cells transfected with constitutive active forms of the Notch1 receptor [100]. These results demonstrate that Notch1 contributes to chondrocyte dedifferentiation via enhancing MMP-13-mediated COL-2 degradation. Surprisingly, the Notch inhibitor N-[N-(3,5-diflur- ophenylacetate)-L-alanyl]-(S)-phenylglycine t-butyl ester (DAPT) down-regulates COL-2 mRNA expression [101]. Therefore, besides Notch1, other Notch family members may also participate in the regulation of COL-2 expression.
Unlike the in vitro chondrocyte culture, chondrocytes reside separately in the cartilage lacuna and thus neighboring chondrocytes lack Notch receptor-ligand communication. MAGPs, the components of extracellular microfibrils, are able to dissociate from the Notch1 extracellular domain and activate the receptor [94]. In addition, MAGP-1 has been shown to interact with the chondroitin sulfate proteoglycan decorin in cultured fetal bovine chondrocytes [102], which suggesting that Notch signaling is involved in chondrocyte dedifferentiation and OA. Therefore, we may infer that chondrocyte dedifferentiation and OA share many similarities.
Conclusions
During in vitro monolayer culture, chondrocyte dedifferentiation occurs very early even at the first passage [103]. Although it has been shown that the 3-D culture system can partially help chondrocyte re-differentiation from characteristics of dedifferentiated, it is still quite different from the native matrix in vivo. To date, it remains to be extremely difficult to prevent chondrocyte from dedifferentiation due to the lack of knowledge regarding this process. Given the complexity of growth factors and their complicated intracellular signaling pathways, the regimens of utilizing growth factors to maintain chondrocyte phenotype or using more specific inhibitors to block chondrocyte dedifferentiation need to be optimized. On the basis of crosstalk signaling pathways between anabolism and catabolism, future work should be directed toward identifying the intersection of signaling pathways that functionally impact on chondrocyte dedifferentiation. In addition, to design effective genetic modification-based treatment modalities to control the chondrocyte dedifferentiation for clinical applications, it is important to introduce no deleterious or off-target side effects. With more in-depth understanding of the molecular mechanisms underlying chondrocyte dedifferentiation, better treatment strategies may be on the horizon to combat chondrocyte dedifferentiation and maintain the chondrocyte phenotype and simultaneously enhance hyaline cartilage matrix production. At present, our lab work has begun to investigate the combination of growth factors as well as explore alternative pathways to avoid chondrocyte dedifferentiation for ACI. Although much remains to be explored, we believe that it would greatly contribute to maintain the chondrocyte phenotype if the balance of the anabolic and catabolic signaling pathways can be finetuned.
Acknowledgements
This study was financially supported by the following grants: Natural Science Foundation of China (No.81260161; No.81000460); Natural Science Foundation of Guangdong Province, China (NO.S2012010008129); China Postdoctoral Science Foundation Funded Project (No.2013M530385); The Promotion Program for Shenzhen Key Laboratory, Shenzhen, China (No.ZDSY20120614154551201); Shenzhen Science and Technology Project (No.JSGG2014051905550503; No.JCYJ20140414170821160; No.GJHZ20130412153906740; No.CXZZ20120614160234842). We wish to thank Dr. Wenlan Liu for his valuable comments in improving the quality of the review article.
Disclosure of conflict of interest
None to declare.
Abbreviations
- ACAN
aggrecan
- ACI
autologous chondrocyte implantation
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs
- AP-1
activator protein 1
- BMPs
bone morphogenetic proteins
- CaMK
Ca2+/calmodulin-dependent protein kinase
- c-JNK
c-Jun-N-terminal kinase
- COL-1
type I collagen
- COL-2
type II collagen
- COX-2
cyclooxygenase 2
- CTSK
cathepsin K
- DVL
dishevelled
- ECM
extracellular matrix
- ERK
extracellular-signal-regulated protein kinase
- FGF
fibroblast growth factor
- FGFR-3
FGF receptor 3
- FZD
frizzled
- GAG
glycosaminoglycan
- HES1
hairy and enhancer of split 1
- ICN
intracellular domain of Notch receptor
- IGF-I
insulin-like growth factor-I
- IL-1β
interleukin 1-beta
- JAG-1
Jagged 1
- LEF
lymphoid enhancer binding factor
- MAPK
mitogen-activated protein kinase
- MAGP-1
microfibril-associated glycoprotein
- MMPs
matrix metalloproteinases
- NO
nitric oxide
- OA
osteoarthritis
- PDGF
platelet-derived growth factor
- PGE
E-series prostaglandins
- PKC-α
protein kinase C alpha
- PKC-ζ
protein kinase C zeta
- PI3K
phosphoinositide-3 kinase
- RA
rheumatoid arthritis
- RBP-J
recombination signal binding protein J
- shRNA
short hairpin RNA
- SOX-9
sex determining region Y box gene 9
- SHC
SH2 domain-containing transforming protein
- SNP
sodium nitroprusside
- TGF-β1
transforming growth factor beta 1
- TNF
tumor necrosis factor
- 3-D
three-dimension
- TCF
T cell factor
References
- 1.Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, Peterson L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med. 1994;331:889–895. doi: 10.1056/NEJM199410063311401. [DOI] [PubMed] [Google Scholar]
- 2.Niemeyer P, Porichis S, Steinwachs M, Erggelet C, Kreuz PC, Schmal H, Uhl M, Ghanem N, Südkamp NP, Salzmann G. Long-term outcomes after first-generation autologous chondrocyte implantation for cartilage defects of the knee. Am J Sports Med. 2014;42:150–157. doi: 10.1177/0363546513506593. [DOI] [PubMed] [Google Scholar]
- 3.Malicev E, Kregar-Velikonja N, Barlic A, Alibegović A, Drobnic M. Comparison of articular and auricular cartilage as a cell source for the autologous chondrocyte implantation. J Orthop Res. 2009;27:943–948. doi: 10.1002/jor.20833. [DOI] [PubMed] [Google Scholar]
- 4.Eyre D. Collagen of articular cartilage. Arthritis Res. 2002;4:30–35. doi: 10.1186/ar380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulze-Tanzil G. Activation and dedifferentiation of chondrocytes: implications in cartilage injury and repair. Ann Anat. 2009;191:325–338. doi: 10.1016/j.aanat.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Goldring MB, Birkhead J, Sandell LJ, Kimura T, Krane SM. Interleukin 1 suppresses expression of cartilage-specific types II and IX collagens and increases types I and III collagens in human chondrocytes. J Clin Invest. 1988;82:2026–2037. doi: 10.1172/JCI113823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefebvre V, Peeters-Joris C, Vaes G. Modulation by interleukin 1 and tumor necrosis factor alpha of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochim Biophys Acta. 1990;1052:366–378. doi: 10.1016/0167-4889(90)90145-4. [DOI] [PubMed] [Google Scholar]
- 8.Goldring MB, Sohbat E, Elwell JM, Chang JY. Etodolac preserves cartilage-specific phenotype in human chondrocytes: effects on type II collagen synthesis and associated mRNA levels. Eur J Rheumatol Inflamm. 1990;10:10–21. [PubMed] [Google Scholar]
- 9.Schwartz Z, Gilley RM, Sylvia VL, Dean DD, Boyan BD. The effect of prostaglandin E2 on costochondral chondrocyte differentiation is mediated by cyclic adenosine 3’, 5’-monoph- osphate and protein kinase C. Endocrinology. 1998;139:1825–1834. doi: 10.1210/endo.139.4.5919. [DOI] [PubMed] [Google Scholar]
- 10.Sylvester J, El Mabrouk M, Ahmad R, Chaudry A, Zafarullah M. Interleukin-1 induction of aggrecanase gene expression in human articular chondrocytes is mediated by mitogen-activated protein kinases. Cell Physiol Biochem. 2012;30:563–574. doi: 10.1159/000341438. [DOI] [PubMed] [Google Scholar]
- 11.Geng Y, Valbracht J, Lotz M. Selective activation of the mitogen-activated protein kinase subgroups c-Jun NH2 terminal kinase and p38 by IL-1 and TNF in human articular chondrocytes. J Clin Invest. 1996;98:2425–2430. doi: 10.1172/JCI119056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seifarth C, Csaki C, Shakibaei M. Anabolic actions of IGF-I and TGF-beta1 on Interleukin-1beta-treated human articular chondrocytes: evaluation in two and three dimensional cultures. Histol Histopathol. 2009;24:1245–1262. doi: 10.14670/HH-24.1245. [DOI] [PubMed] [Google Scholar]
- 13.Thomas B, Thirion S, Humbert L, Tan L, Goldring MB, Béréziat G, Berenbaum F. Differentiation regulates interleukin-1beta-induced cyclo-oxygenase-2 in human articular chondrocytes: role of p38 mitogen-activated protein kinase. Biochem J. 2002;362:367–373. doi: 10.1042/0264-6021:3620367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang SG, Ryu JH, Kim IC, Jho EH, Jung HC, Kim K, Kim SJ, Chun JS. Wnt-7a causes loss of differentiated phenotype and inhibits apoptosis of articular chondrocytes via different mechanisms. J Biol Chem. 2004;279:26597–26604. doi: 10.1074/jbc.M401401200. [DOI] [PubMed] [Google Scholar]
- 15.Hwang SG, Yu SS, Lee SW, Chun JS. Wnt-3a regulates chondrocyte differentiation via c-Jun/AP-1 pathway. FEBS Lett. 2005;579:4837–4842. doi: 10.1016/j.febslet.2005.07.067. [DOI] [PubMed] [Google Scholar]
- 16.Yoon JB, Kim SJ, Hwang SG, Chang S, Kang SS, Chun JS. Non-steroidal anti-inflammatory drugs inhibit nitric oxide-induced apoptosis and dedifferentiation of articular chondrocytes independent of cyclooxygenase activity. J Biol Chem. 2003;278:15319–15325. doi: 10.1074/jbc.M212520200. [DOI] [PubMed] [Google Scholar]
- 17.Stevens AL, Wheeler CA, Tannenbaum SR, Grodzinsky AJ. Nitric oxide enhances aggrecan degradation by aggrecanase in response to TNF-alpha but not IL-1beta treatment at a post-transcriptional level in bovine cartilage explants. Osteoarthritis Cartilage. 2008;16:489–497. doi: 10.1016/j.joca.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuchida AI, Beekhuizen M, T Hart MC, Radstake T, Dhert W, Saris D, van Osch G, Creemers LB. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res Ther. 2014;16:441. doi: 10.1186/s13075-014-0441-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldring MB, Otero M, Tsuchimochi K, Ijiri K, Li Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann Rheum Dis. 2008;67(Suppl 3):iii75–82. doi: 10.1136/ard.2008.098764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geng Y, Valbracht J, Lotz M. Selective activation of the mitogen-activated protein kinase subgroups c-Jun NH2 terminal kinase and p38 by IL-1 and TNF in human articular chondrocytes. J Clin Invest. 1996;98:2425–2430. doi: 10.1172/JCI119056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zanotti S, Canalis E. Interleukin 6 mediates selected effects of Notch in chondrocytes. Osteoarthritis Cartilage. 2013;21:1766–1773. doi: 10.1016/j.joca.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dejica VM, Mort JS, Laverty S, Antoniou J, Zukor DJ, Tanzer M, Poole AR. Increased type II collagen cleavage by cathepsin K and collagenase activities with aging and osteoarthritis in human articular cartilage. Arthritis Res Ther. 2012;14:R113. doi: 10.1186/ar3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu W, Zhang W, Li F, Guo F, Chen A. Bortezomib prevents the expression of MMP-13 and the degradation of collagen type 2 in human chondrocytes. Biochem Biophys Res Commun. 2014;452:526–530. doi: 10.1016/j.bbrc.2014.08.102. [DOI] [PubMed] [Google Scholar]
- 24.Ashlin TG, Kwan AP, Ramji DP. Regulation of ADAMTS-1, -4 and -5 expression in human macrophages: differential regulation by key cytokines implicated in atherosclerosis and novel synergism between TL1A and IL-17. Cytokine. 2013;64:234–242. doi: 10.1016/j.cyto.2013.06.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Li J, Zhu J, Zhou G, Zhang WJ, Cao Y, Liu W. Enhanced cartilage formation by inhibiting cathepsin K expression in chondrocytes expanded in vitro. Biomaterials. 2012;33:7394–7404. doi: 10.1016/j.biomaterials.2012.06.070. [DOI] [PubMed] [Google Scholar]
- 26.Billinghurst RC, Dahlberg L, Ionescu M, Reiner A, Bourne R, Rorabeck C, Mitchell P, Hambor J, Diekmann O, Tschesche H, Chen J, Van Wart H, Poole AR. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest. 1997;99:1534–1545. doi: 10.1172/JCI119316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reboul P, Pelletier JP, Tardif G, Cloutier JM, Martel-Pelletier J. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J Clin Invest. 1996;97:2011–2019. doi: 10.1172/JCI118636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huebner JL, Otterness IG, Freund EM, Caterson B, Kraus VB. Collagenase 1 and collagenase 3 expression in a guinea pig model of osteoarthritis. Arthritis Rheum. 1998;41:877–890. doi: 10.1002/1529-0131(199805)41:5<877::AID-ART16>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 29.Bau B, Gebhard PM, Haag J, Knorr T, Bartnik E, Aigner T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002;46:2648–2657. doi: 10.1002/art.10531. [DOI] [PubMed] [Google Scholar]
- 30.Lin Z, Fitzgerald JB, Xu J, Willers C, Wood D, Grodzinsky AJ, Zheng MH. Gene expression profiles of human chondrocytes during passaged monolayer cultivation. J Orthop Res. 2008;26:1230–1237. doi: 10.1002/jor.20523. [DOI] [PubMed] [Google Scholar]
- 31.Ma B, Leijten JC, Wu L, Kip M, van Blitterswijk CA, Post JN, Karperien M. Gene expression profiling of dedifferentiated human articular chondrocytes in monolayer culture. Osteoarthritis Cartilage. 2013;21:599–603. doi: 10.1016/j.joca.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 32.Goessler UR, Bieback K, Bugert P, Naim R, Schafer C, Sadick H, Hormann K, Riedel F. Human chondrocytes differentially express matrix modulators during in vitro expansion for tissue engineering. Int J Mol Med. 2005;16:509–515. [PubMed] [Google Scholar]
- 33.Zhao J, Fan X, Zhang Q, Sun F, Li X, Xiong C, Zhang C, Fan H. Chitosan-plasmid DNA nanoparticles encoding small hairpin RNA targeting MMP-3 and -13 to inhibit the expression of dedifferentiation related genes in expanded chondrocytes. J Biomed Mater Res A. 2014;102:373–380. doi: 10.1002/jbm.a.34711. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt MB, Chen EH, Lynch SE. A review of the effects of insulin-like growth factor and platelet derived growth factor on in vivo cartilage healing and repair. Osteoarthritis Cartilage. 2006;14:403–412. doi: 10.1016/j.joca.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Shakibaei M, Seifarth C, John T, Rahmanzadeh M, Mobasheri A. Igf-I extends the chondrogenic potential of human articular chondrocytes in vitro: molecular association between Sox9 and Erk1/2. Biochem Pharmacol. 2006;72:1382–1395. doi: 10.1016/j.bcp.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 36.Madry H, Padera R, Seidel J, Langer R, Freed LE, Trippel SB, Vunjak-Novakovic G. Gene transfer of a human insulin-like growth factor I cDNA enhances tissue engineering of cartilage. Hum Gene Ther. 2002;13:1621–1630. doi: 10.1089/10430340260201716. [DOI] [PubMed] [Google Scholar]
- 37.Starkman BG, Cravero JD, Delcarlo M, Loeser RF. IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem J. 2005;389:723–729. doi: 10.1042/BJ20041636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies LC, Blain EJ, Gilbert SJ, Caterson B, Duance VC. The potential of IGF-1 and TGF-beta1 for promoting “adult” articular cartilage repair: an in vitro study. Tissue Eng Part A. 2008;14:1251–1261. doi: 10.1089/ten.tea.2007.0211. [DOI] [PubMed] [Google Scholar]
- 39.Shi S, Mercer S, Eckert GJ, Trippel SB. Regulation of articular chondrocyte aggrecan and collagen gene expression by multiple growth factor gene transfer. J Orthop Res. 2012;30:1026–1031. doi: 10.1002/jor.22036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montaseri A, Busch F, Mobasheri A, Buhrmann C, Aldinger C, Rad JS, Shakibaei M. IGF-1 and PDGF-bb suppress IL-1β-induced cartilage degradation through down-regulation of NF-κB signaling: involvement of Src/PI-3K/AKT pathway. PLoS One. 2011;6:e28663. doi: 10.1371/journal.pone.0028663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh CD, Chun JS. Signaling mechanisms leading to the regulation of differentiation and apoptosis of articular chondrocytes by insulin-like growth factor-1. J Biol Chem. 2003;278:36563–36571. doi: 10.1074/jbc.M304857200. [DOI] [PubMed] [Google Scholar]
- 42.Legendre F, Ollitrault D, Hervieu M, Baugé C, Maneix L, Goux D, Chajra H, Mallein-Gerin F, Boumediene K, Galera P, Demoor M. Enhanced hyaline cartilage matrix synthesis in collagen sponge scaffolds by using siRNA to stabilize chondrocytes phenotype cultured with bone morphogenetic protein-2 under hypoxia. Tissue Eng Part C Methods. 2013;19:550–567. doi: 10.1089/ten.TEC.2012.0508. [DOI] [PubMed] [Google Scholar]
- 43.Villiger PM, Lotz M. Differential expression of TGF beta isoforms by human articular chondrocytes in response to growth factors. J Cell Physiol. 1992;151:318–325. doi: 10.1002/jcp.1041510213. [DOI] [PubMed] [Google Scholar]
- 44.Lohmann CH, Schwartz Z, Niederauer GG, Boyan BD. Degree of differentiation of chondrocytes and their pretreatment with platelet-derived-growth factor. Regulating induction of cartilage formation in resorbable tissue carriers in vivo. Orthopade. 2000;29:120–128. doi: 10.1007/s001320050020. [DOI] [PubMed] [Google Scholar]
- 45.Montaseri A, Busch F, Mobasheri A, Buhrmann C, Aldinger C, Rad JS, Shakibaei M. IGF-1 and PDGF-bb suppress IL-1β-induced cartilage degradation through down-regulation of NF-κB signaling: involvement of Src/PI-3K/AKT pathway. PLoS One. 2011;6:e28663. doi: 10.1371/journal.pone.0028663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galéra P, Vivien D, Pronost S, Bonaventure J, Rédini F, Loyau G, Pujol JP. Transforming gro-wth factor-beta 1 (TGF-beta 1) up-regulation of collagen type II in primary cultures of rabbit articular chondrocytes (RAC) involves increased mRNA levels without affecting mRNA stability and procollagen processing. J Cell Physiol. 1992;153:596–606. doi: 10.1002/jcp.1041530322. [DOI] [PubMed] [Google Scholar]
- 47.Galéra P, Rédini F, Vivien D, Bonaventure J, Penfornis H, Loyau G, Pujol JP. Effect of transforming growth factor-beta 1 (TGF-beta 1) on matrix synthesis by monolayer cultures of rabbit articular chondrocytes during the dedifferentiation process. Exp Cell Res. 1992;200:379–392. doi: 10.1016/0014-4827(92)90186-c. [DOI] [PubMed] [Google Scholar]
- 48.Darling EM, Athanasiou KA. Growth factor impact on articular cartilage subpopulations. Cell Tissue Res. 2005;322:463–473. doi: 10.1007/s00441-005-0020-4. [DOI] [PubMed] [Google Scholar]
- 49.Li C, Wang Q, Wang JF. Transforming growth factor-β (TGF-β) induces the expression of chondrogenesis-related genes through TGF-β receptor II (TGFRII)-AKT-mTOR signaling in primary cultured mouse precartilaginous stem cells. Biochem Biophys Res Commun. 2014;450:646–651. doi: 10.1016/j.bbrc.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 50.Baugé C, Duval E, Ollitrault D, Girard N, Leclercq S, Galéra P, Boumédiene K. Type II TGFβ receptor modulates chondrocyte phenotype. Age (Dordr) 2013;35:1105–1116. doi: 10.1007/s11357-012-9433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narcisi R, Quarto R, Ulivi V, Muraglia A, Molfetta L, Giannoni P. TGFβ-1 administration during ex vivo expansion of human articular chondrocytes in a serum-free medium redirects the cell phenotype toward hypertrophy. J Cell Physiol. 2012;227:3282–3290. doi: 10.1002/jcp.24024. [DOI] [PubMed] [Google Scholar]
- 52.Miyazaki Y, Tsukazaki T, Hirota Y, Yonekura A, Osaki M, Shindo H, Yamashita S. Dexamethasone inhibition of TGF beta-induced cell growth and type II collagen mRNA expression through ERK-integrated AP-1 activity in cultured rat articular chondrocytes. Osteoarthritis Cartilage. 2000;8:378–385. doi: 10.1053/joca.1999.0313. [DOI] [PubMed] [Google Scholar]
- 53.Chadjichristos C, Ghayor C, Herrouin JF, Ala-Kokko L, Suske G, Pujol JP, Galéra P. Down-regulation of human type II collagen gene expression by transforming growth factor-beta 1 (TGF-beta 1) in articular chondrocytes involves SP3/SP1 ratio. J Biol Chem. 2002;277:43903–43917. doi: 10.1074/jbc.M206111200. [DOI] [PubMed] [Google Scholar]
- 54.Zhang F, Yao Y, Su K, Pang PX, Zhou R, Wang Y, Wang DA. Redifferentiation of dedifferentiated chondrocytes by adenoviral vector-mediated TGF-β3 and collagen-1 silencing shRNA in 3D culture. Ann Biomed Eng. 2011;39:3042–3054. doi: 10.1007/s10439-011-0398-y. [DOI] [PubMed] [Google Scholar]
- 55.Geesink RG, Hoefnagels NH, Bulstra SK. Osteogenic activity of OP-1 bone morphogenetic protein (BMP-7) in a human fibular defect. J Bone Joint Surg Br. 1999;81:710–718. doi: 10.1302/0301-620x.81b4.9311. [DOI] [PubMed] [Google Scholar]
- 56.Groeneveld EH, Burger EH. Bone morphogenetic proteins in human bone regeneration. Eur J Endocrinol. 2000;142:9–21. doi: 10.1530/eje.0.1420009. [DOI] [PubMed] [Google Scholar]
- 57.Friedlaender GE, Perry CR, Cole JD, Cook SD, Cierny G, Muschler GF, Zych GA, Calhoun JH, LaForte AJ, Yin S. Osteogenic protein-1 (bone morphogenetic protein-7) in the treatment of tibial nonunions. J Bone Joint Surg Am. 2001;83-A:S151–158. [PMC free article] [PubMed] [Google Scholar]
- 58.Boden SD, Kang J, Sandhu H, Heller JG. Use of recombinant human bone morphogenetic protein-2 to achieve posterolateral lumbar spine fusion in humans: a prospective, randomized clinical pilot trial: 2002 Volvo Award in clinical studies. Spine (Phila Pa 1976) 2002;27:2662–2673. doi: 10.1097/00007632-200212010-00005. [DOI] [PubMed] [Google Scholar]
- 59.Granjeiro JM, Oliveira RC, Bustos-Valenzuela JC, Sogayar MC, Taga R. Bone morphogenetic proteins: from structure to clinical use. Braz J Med Biol Res. 2005;38:1463–1473. doi: 10.1590/s0100-879x2005001000003. [DOI] [PubMed] [Google Scholar]
- 60.Gooch KJ, Blunk T, Courter DL, Sieminski AL, Vunjak-Novakovic G, Freed LE. Bone morphogenetic proteins-2, -12, and -13 modulate in vitro development of engineered cartilage. Tissue Eng. 2002;8:591–601. doi: 10.1089/107632702760240517. [DOI] [PubMed] [Google Scholar]
- 61.Blunk T, Sieminski AL, Appel B, Croft C, Courter DL, Chieh JJ, Goepferich A, Khurana JS, Gooch KJ. Bone morphogenetic protein 9: a potent modulator of cartilage development in vitro. Growth Factors. 2003;21:71–77. doi: 10.1080/0897719031000148822. [DOI] [PubMed] [Google Scholar]
- 62.Bobacz K, Gruber R, Soleiman A, Erlacher L, Smolen JS, Graninger WB. Expression of bone morphogenetic protein 6 in healthy and osteoarthritic human articular chondrocytes and stimulation of matrix synthesis in vitro. Arthritis Rheum. 2003;48:2501–2508. doi: 10.1002/art.11248. [DOI] [PubMed] [Google Scholar]
- 63.Claus S, Aubert-Foucher E, Demoor M, Camuzeaux B, Paumier A, Piperno M, Damour O, Duterque-Coquillaud M, Galéra P, Mallein-Gerin F. Chronic exposure of bone morphogenetic protein- 2 favors chondrogenic expression in human articular chondrocytes amplified in monolayer cultures. J Cell Biochem. 2010;111:1642–1651. doi: 10.1002/jcb.22897. [DOI] [PubMed] [Google Scholar]
- 64.Gouttenoire J, Bougault C, Aubert-Foucher E, Perrier E, Ronzière MC, Sandell L, Lundgren-Akerlund E, Mallein-Gerin F. BMP-2 and TGF-beta1 differentially control expression of type II procollagen and alpha 10 and alpha 11 integrins in mouse chondrocytes. Eur J Cell Biol. 2010;89:307–314. doi: 10.1016/j.ejcb.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perrier-Groult E, Pasdeloup M, Malbouyres M, Galéra P, Mallein-Gerin F. Control of collagen production in mouse chondrocytes by using a combination of bone morphogenetic protein-2 and small interfering RNA targeting Col1a1 for hydrogel-based tissue-engineered cartilage. Tissue Eng Part C Methods. 2013;19:652–664. doi: 10.1089/ten.tec.2012.0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cha BH, Kim JH, Kang SW, Do HJ, Jang JW, Choi YR, Park H, Kim BS, Lee SH. Cartilage tissue formation from dedifferentiated chondrocytes by codelivery of BMP-2 and SOX-9 genes encoding bicistronic vector. Cell Transplant. 2013;22:1519–1528. doi: 10.3727/096368912X647261. [DOI] [PubMed] [Google Scholar]
- 67.Liu G, Kawaguchi H, Ogasawara T, Asawa Y, Kishimoto J, Takahashi T, Chung UI, Yamaoka H, Asato H, Nakamura K, Takato T, Hoshi K. Optimal combination of soluble factors for tissue engineering of permanent cartilage from cultured human chondrocytes. J Biol Chem. 2007;282:20407–20415. doi: 10.1074/jbc.M608383200. [DOI] [PubMed] [Google Scholar]
- 68.Jin EJ, Lee SY, Choi YA, Jung JC, Bang OS, Kang SS. BMP-2-enhanced chondrogenesis involves p38 MAPK-mediated down-regulation of Wnt-7a pathway. Mol Cells. 2006;22:353–359. [PubMed] [Google Scholar]
- 69.Martin I, Suetterlin R, Baschong W, Heberer M, Vunjak-Novakovic G, Freed LE. Enhanced cartilage tissue engineering by sequential exposure of chondrocytes to FGF-2 during 2D expansion and BMP-2 during 3D cultivation. J Cell Biochem. 2001;83:121–128. doi: 10.1002/jcb.1203. [DOI] [PubMed] [Google Scholar]
- 70.Claus S, Mayer N, Aubert-Foucher E, Chajra H, Perrier-Groult E, Lafont J, Piperno M, Damour O, Mallein-Gerin F. Cartilage-characteristic matrix reconstruction by sequential addition of soluble factors during expansion of human articular chondrocytes and their cultivation in collagen sponges. Tissue Eng Part C Methods. 2012;18:104–112. doi: 10.1089/ten.tec.2011.0259. [DOI] [PubMed] [Google Scholar]
- 71.Martin I, Vunjak-Novakovic G, Yang J, Langer R, Freed LE. Mammalian chondrocytes expanded in the presence of fibroblast growth factor 2 maintain the ability to differentiate and regenerate three-dimensional cartilaginous tissue. Exp Cell Res. 1999;253:681–688. doi: 10.1006/excr.1999.4708. [DOI] [PubMed] [Google Scholar]
- 72.Liu Z, Lavine KJ, Hung IH, Ornitz DM. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol. 2007;302:80–91. doi: 10.1016/j.ydbio.2006.08.071. [DOI] [PubMed] [Google Scholar]
- 73.Shimoaka T, Ogasawara T, Yonamine A, Chikazu D, Kawano H, Nakamura K, Itoh N, Kawaguchi H. Regulation of osteoblast, chondrocyte, and osteoclast functions by fibroblast growth factor (FGF)-18 in comparison with FGF-2 and FGF-10. J Biol Chem. 2002;277:7493–7500. doi: 10.1074/jbc.M108653200. [DOI] [PubMed] [Google Scholar]
- 74.Yamaoka H, Nishizawa S, Asawa Y, Fujihara Y, Ogasawara T, Yamaoka K, Nagata S, Takato T, Hoshi K. Involvement of fibroblast growth factor 18 in dedifferentiation of cultured human chondrocytes. Cell Prolif. 2010;43:67–76. doi: 10.1111/j.1365-2184.2009.00655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rottmar M, Mhanna R, Guimond-Lischer S, Vogel V, Zenobi-Wong M, Maniura-Weber K. Interference with the contractile machinery of the fibroblastic chondrocyte cytoskeleton induces re-expression of the cartilage phenotype through involvement of PI3K, PKC and MAPKs. Exp Cell Res. 2014;320:175–187. doi: 10.1016/j.yexcr.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 76.Yoon YM, Kim SJ, Oh CD, Ju JW, Song WK, Yoo YJ, Huh TL, Chun JS. Maintenance of differentiated phenotype of articular chondrocytes by protein kinase C and extracellular signal-regulated protein kinase. J Biol Chem. 2002;277:8412–8420. doi: 10.1074/jbc.M110608200. [DOI] [PubMed] [Google Scholar]
- 77.Suen KM, Lin CC, George R, Melo FA, Biggs ER, Ahmed Z, Drake MN, Arur S, Arold ST, Ladbury JE. Interaction with Shc prevents aberrant Erk activation in the absence of extracellular stimuli. Nat Struct Mol Biol. 2013;20:620–627. doi: 10.1038/nsmb.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schulze-Tanzil G, Mobasheri A, de Souza P, John T, Shakibaei M. Loss of chondrogenic potential in dedifferentiated chondrocytes correlates with deficient Shc-Erk interaction and apoptosis. Osteoarthritis Cartilage. 2004;12:448–458. doi: 10.1016/j.joca.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 79.Tuli R, Tuli S, Nandi S, Huang X, Manner PA, Hozack WJ, Dan- ielson KG, Hall DJ, Tuan RS. Transforming growth factor-β-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J Biol Chem. 2003;278:41227–41236. doi: 10.1074/jbc.M305312200. [DOI] [PubMed] [Google Scholar]
- 80.Kim D, Song J, Kim S, Park HM, Chun CH, Sonn J, Jin EJ. MicroRNA-34a modulates cytoskeletal dynamics through regulating RhoA/Rac1 cross-talk in chondroblasts. J Biol Chem. 2012;287:12501–12509. doi: 10.1074/jbc.M111.264382. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 82.Starkman BG, Cravero JD, Delcarlo M, Loeser RF. IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem J. 2005;389:723–729. doi: 10.1042/BJ20041636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang MS, Chang SH, Sonn JK, Lee YS, Kang SS, Park TK, Chun JS. Regulation of chondrogenic differentiation of mesenchymes by protein kinase C alpha. Mol Cells. 1998;8:266–271. [PubMed] [Google Scholar]
- 84.Yoon YM, Kim SJ, Oh CD, Ju JW, Song WK, Yoo YJ, Huh TL, Chun JS. Maintenance of differentiated phenotype of articular chondrocytes by protein kinase C and extracellular signal-regulated protein kinase. J Biol Chem. 2002;277:8412–8420. doi: 10.1074/jbc.M110608200. [DOI] [PubMed] [Google Scholar]
- 85.Kim SJ, Kim HG, Oh CD, Hwang SG, Song WK, Yoo YJ, Kang SS, Chun JS. p38 kinase-dependent and -independent Inhibition of protein kinase C zeta and -alpha regulates nitric oxide-induced apoptosis and dedifferentiation of articular chondrocytes. J Biol Chem. 2002;277:30375–30381. doi: 10.1074/jbc.M205193200. [DOI] [PubMed] [Google Scholar]
- 86.Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009;433:1–7. doi: 10.1016/j.gene.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 87.Rao TP, Kühl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010;106:1798–1806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 88.Hwang SG, Yu SS, Ryu JH, Jeon HB, Yoo YJ, Eom SH, Chun JS. Regulation of beta-catenin signaling and maintenance of chondrocyte differentiation by ubiquitin-independent proteasomal degradation of alpha-catenin. J Biol Chem. 2005;280:12758–12765. doi: 10.1074/jbc.M413367200. [DOI] [PubMed] [Google Scholar]
- 89.Rhee J, Ryu JH, Kim JH, Chun CH, Chun JS. α-Catenin Inhibits β-Catenin-T-cell Factor/Lymphoid Enhancing Factor Transcriptional Activity and Collagen Type II Expression in Articular Chondrocytes through Formation of Gli3R·α-Catenin·β-Catenin Ternary Complex. J Biol Chem. 2012;287:11751–11760. doi: 10.1074/jbc.M111.281014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nalesso G, Sherwood J, Bertrand J, Pap T, Ramachandran M, De Bari C, Pitzalis C, Dell’accio F. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J Cell Biol. 2011;193:551–564. doi: 10.1083/jcb.201011051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138:3593–3612. doi: 10.1242/dev.063610. [DOI] [PubMed] [Google Scholar]
- 92.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 93.Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israël A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci U S A. 1998;95:8108–8112. doi: 10.1073/pnas.95.14.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miyamoto A, Lau R, Hein PW, Shipley JM, Weinmaster G. Microfibrillar proteins MAGP-1 and MAGP-2 induce Notch1 extracellular domain dissociation and receptor activation. J Biol Chem. 2006;281:10089–10097. doi: 10.1074/jbc.M600298200. [DOI] [PubMed] [Google Scholar]
- 95.Hayes AJ, Dowthwaite GP, Webster SV, Archer CW. The distribution of Notch receptors and their ligands during articular cartilage development. J Anat. 2003;202:495–502. doi: 10.1046/j.1469-7580.2003.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Crowe R, Zikherman J, Niswander L. Delta-1 negatively regulates the transition from prehypertrophic to hypertrophic chondrocytes during cartilage formation. Development. 1999;126:987–998. doi: 10.1242/dev.126.5.987. [DOI] [PubMed] [Google Scholar]
- 97.Karlsson C, Jonsson M, Asp J, Brantsing C, Kageyama R, Lindahl A. Notch and HES5 are regulated during human cartilage differentiation. Cell Tissue Res. 2007;327:539–551. doi: 10.1007/s00441-006-0307-0. [DOI] [PubMed] [Google Scholar]
- 98.Watanabe N, Tezuka Y, Matsuno K, Miyatani S, Morimura N, Yasuda M, Fujimaki R, Kuroda K, Hiraki Y, Hozumi N, Tezuka K. Suppression of differentiation and proliferation of early chondrogenic cells by Notch. J Bone Miner Metab. 2003;21:344–352. doi: 10.1007/s00774-003-0428-4. [DOI] [PubMed] [Google Scholar]
- 99.Sassi N, Gadgadi N, Laadhar L, Allouche M, Mourali S, Zandieh-Doulabi B, Hamdoun M, Nulend JK, Makni S, Sellami S. Notch signaling is involved in human articular chondrocytes de-differentiation during osteoarthritis. J Recept Signal Transduct Res. 2014;34:48–57. doi: 10.3109/10799893.2013.856920. [DOI] [PubMed] [Google Scholar]
- 100.Blaise R, Mahjoub M, Salvat C, Barbe U, Brou C, Corvol MT, Savouret JF, Rannou F, Berenbaum F, Bausero P. Involvement of the Notch pathway in the regulation of matrix metalloproteinase 13 and the dedifferentiation of articular chondrocytes in murine cartilage. Arthritis Rheum. 2009;60:428–439. doi: 10.1002/art.24250. [DOI] [PubMed] [Google Scholar]
- 101.Mirando AJ, Liu Z, Moore T, Lang A, Kohn A, Osinski AM, O’Keefe RJ, Mooney RA, Zuscik MJ, Hilton MJ. RBP-Jκ–dependent Notch signaling is required for murine articular cartilage and joint maintenance. Arthritis Rheum. 2013;65:2623–2633. doi: 10.1002/art.38076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Trask BC, Trask TM, Broekelmann T, Mecham RP. The microfibrillar proteins MAGP-1 and fibrillin-1 form a ternary complex with the chondroitin sulfate proteoglycan decorin. Mol Biol Cell. 2000;11:1499–1507. doi: 10.1091/mbc.11.5.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Darling EM, Athanasiou KA. Rapid phenotypic changes in passaged articular chondrocyte subpopulations. J Orthop Res. 2005;23:425–432. doi: 10.1016/j.orthres.2004.08.008. [DOI] [PubMed] [Google Scholar]