

Abstract
BikDD, a phosphorylation-mimic mutant of pro-apoptotic protein Bik, elicits strong apoptosis in cancer cells when introduced via an expression platform termed VP16-GAL4-WPRE integrated systemic amplifier (VISA) under the control of a cancer-specific promoter both in vitro and in vivo. C-VISA-BikDD expression plasmid encapsulated in liposomes is currently in the process to initiate a phase I clinical trial for pancreatic cancer. In this study, we report a potential combination approach of BikDD with proteasome inhibitors on the basis of our findings that exogenously expressed BikDD protein undergoes proteasome-mediated degradation via both ubiquitin-dependent and -independent pathways. Inhibition of proteasome increases the protein stability of BikDD, enhancing the apoptotic effect of BikDD. Hence, high proteasome activity may be a mechanism by which intrinsic and acquired resistance occurs in BikDD gene therapy, and a combination therapy with current clinically approved proteasome inhibitor may overcome resistance.
Keywords: BikDD, proteasome inhibition, apoptosis, combinational therapy
Introduction
Bik, a member of the BH3-only family protein, induces apoptosis by binding to multiple anti-apoptotic molecules, such as Bcl2, Mcl1, and Bcl-xL, and has been reported to function as a tumor suppressor [1-5]. BikDD is a mutant form of Bik in which its Thr 33 and Ser 35 were replaced by aspartate to mimic the constitutively phosphorylated form [6,7]. Compared with the wildtype Bik, BikDD possesses a greater binding affinity to its interacting anti-apoptotic proteins (Bcl-xL, Bcl-2, Bcl-w, and Mcl-1) and induces stronger apoptosis both in vitro and in vivo [6]. Expression of BikDD by the VP16-GAL4-WPRE integrated systemic amplifier (VISA) driven cancer specific promoters demonstrated potent killing effects in pancreatic [8], breast [9,10], lung [11,12], liver [13], and prostate [14] cancer cells and significantly reduced tumor growth in the corresponding tumor xenograft models. Currently, the CCKAR-VISA-BikDD-liposome nanoparticle has been approved by the US Food and Drug Administration (FDA) to initiate a phase I clinical trial for advanced pancreatic cancer (ClinicalTrials.gov identifier NCT00968604). In addition, a Phase I clinical protocol using Claudin4-VISA-BikDD [9] has been approved by the Recombinant DNA Advisory Committee (National Institutes of Health), and we are in the process to file an Investigational New Drug application with the FDA.
Because many cancers eventually develop drug resistance, understanding the mechanisms contributing to BikDD resistance may increase the drug efficacy for intrinsically refractory patients and prevent relapse of cancers with acquired resistance. In this study, we attempted to enhance the clinical benefit of BikDD gene therapy by investigating the mechanisms underlying BikDD degradation and proposed a potential therapeutic strategy in combination with proteasome inhibitors such as bortezomib.
Methods and materials
Cell lines and culture
MDA-MB-231, AsPC-1, MIA PaCa-2 and HEK293T cell lines were purchased from American Type Culture Collection (Manassas, VA). The L3.6pL cell line was kindly provided by Dr. Paul Chiao (UT MD Anderson Cancer Center). Cells were cultured in Dulbecco’s-modified Eagle’s medium mixed 1:1 (v:v) with Ham’s F-12 medium and supplemented with 10% fetal bovine serum, penicillin, and streptomycin.
Plasmids and transient protein expression
N-terminal FLAG-tagged BikDD plasmid (pCDH-CMV-f:BikDD) was constructed by inserting the PCR-amplified BikDD-containing fragment from a DNA template [pLOVE-BikDD] [15] into the pCDH-CMV-MCS-EF1-Puromycin vector (CD510B-1; System Biosciences) between restriction sites EcoRI and BamHI. Sequences of the PCR primers used for cloning are as follows: forward, 5’-ATATATGAATTCCCACCATGGATTACAAGGATGACGACGATAAGATGTCTGAAGTAAGACCCCTCTCC-3’; reverse, 5’-ATATATGGATCCTCACTTGAGCAGCAGGTGCAGGC-3’. BikDD mutants were generated by site-directed mutagenesis following manufacturer’s protocol (QuikChange II; Agilent Technologies) and cloned into pCDH-CMV-MCS-EF1-copGFP (CD511B-1; System Biosciences) or pCDH-CMV-MCS-EF1-Puromycin (contains an N-terminus-fused mCherry tag) vector between restriction sites EcoRI and NotI sites. MSCV-IP N-HAonly WWP2 plasmid was purchased from Addgene (#P6592). BikDD transient expression in different cancer cell lines was achieved by either liposome transfection or lentiviral infection, depending on the feasibility of liposome transfection in specific cell lines. Transfection was carried out in 6-well plates using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. Lentiviral packaging system, pCMV-dR8.2 dvpr (#8455) and pCMV-VSVG (#8454), were purchased from Addgene. For virus packaging, two lentiviral-packaging plasmids and BikDD plasmid were co-transfected into HEK 293T cells using Lipofectamine 2000 (Invitrogen). Virus-containing supernatant was collected 24 to 48 hours after transfection and filtered through 0.45 µm filter. Before adding onto cultured cancer cells, lentivirus-containing medium was diluted by DMEM/Ham’s F-12 fresh medium containing 10% FBS with 10 µg/ml polybrene to facilitate infection. Twenty-four hours after infection, cells were changed into fresh culture medium.
Generation of BikDD-expressing cells
Lentivirus packaging and infection were carried out in the same way as described above for transient lentiviral infection. Twenty-four hours after infection, cells were changed into fresh culture medium. After incubation in fresh culture medium for another 24 hours, puromycin (1 μg/mL) or neomycin (1 mg/mL) were added to select for infected cells.
siRNA experiments
siRNAs targeting autocrine motility factor receptor (AMFR) and BIK were purchased from Sigma-Aldrich (St. Louis, MO). siRNA transfection was performed by electroporation with Amaxa Nucleofector I (Lonza Group Ltd., Basel, Switzerland) according to the manufacturer’s instruction. The efficacy of siRNA transfection was validated by immunoblotting 24 hours post transfection. Subsequent experiments using electroporated cells were analyzed 48 hours post transfection.
shRNA experiments
Lentiviral shRNA constructs targeting AMFR were purchased from MISSIONTM TRC-Hs (human) shRNA Library (Sigma-Aldrich). Lentiviral shRNA targeting HECTD1 and HUWE1 were purchased from GIPZ lentiviral shRNA library (shRNA and OFRome Core, MD Anderson Cancer Center, Houston, TX). GIPZ lentiviral empty vector shRNA was used as a control.
Western blot analysis
Western blot was carried out as described previously [15]. Antibodies against Bik (#4592S), PARP (#9542S), and AMFR (#9590S) were purchased from Cell Signaling Technology (Danvers, MA); α-Tubulin (T5168), β-Actin (A2066), and Flag (F3165) were from Sigma-Aldrich; HECTD1 (A302-908A), HUWE1 (A300-486A), and WWP2 (A302-935A) were from Bethyl Laboratories (Montgomery, TX); Lamin B (Ab16048) was from Abcam (Cambridge, MA).
Flow cytometric analysis
Cells were detached, collected, washed twice with PBS, and resuspended in binding buffer (PBS containing 0.1% FBS) at a concentration of 1×106 cells/ml. Cells (100 µl) were mixed with 5 µl APC-Annexin V for 15 minutes at room temperature in the dark. After incubation, an additional 400 µl binding buffer and 1 µl PI (2 mg/ml) was added to the mixture and evaluated with a BD FACSCanto II cell analyzer (BD Immunocytometry Systems; BD Biosciences, San Jose, CA). CopGFP was used to gate cell populations with a successful transfection.
Mass spectrometry
BikDD-associated protein complexes were immunoprecipitated and eluted from MDA-MB-231 BikDD stable cells with anti-FLAG M2 beads (Sigma Aldrich). Cells were treated with 10 µM of MG132 for 8 hours before harvesting to increase BikDD expression. Proteome analysis was analyzed by nanoelectrospray mass spectrometry using an Ultimate capillary LC system (LC Packings, Amsterdam, The Netherlands) coupled to a QSTARXL quadrupole-time of flight (Q-TOF) mass spectrometer (Applied Biosystem/MDS Sciex, Foster City, CA).
Cell viability assay
The killing effect of BikDD was determined by measuring luciferase activities as previously indicated [9]. Briefly, 100 ng of reporter DNA (pGL3-CMV-Luc plasmid was) was co-transfected with various amount of either vector control or BikDD (pCDH-CMV-FLAG:BikDD) by Lipofectamine 2000 (Invitrogen) in a 6-well plate. Forty-eight hours post-transfection, cells were washed once with PBS. The survived cells were further lysed in RIPA buffer and subjected to luciferase reporter assay (Promega, Madison, WI) according to the commercial protocol.
Results
Establishment of BikDD-tolerant cells
To identify the mechanisms that may counteract the killing effect of BikDD, we established several BikDD-expressing breast (MDA-MB-231 and MDA-MB-468) and pancreatic (L3.6pL and PANC-1) cancer cells by using a lentiviral infection that drives BikDD expression under a strong CMV promoter (Figure 1A). During the selection process, some sensitive populations died, which was likely due to overexpressed BikDD, and those that survived were passaged three times before characterization. These cells expressed BikDD as indicated by Western blot analysis (Figure 1A). The growth rate of BikDD-expressing cells was not significantly different from the control cells, suggesting that well-tolerant cells exist (Figure 1B) and maybe resistant to BikDD.
Figure 1.
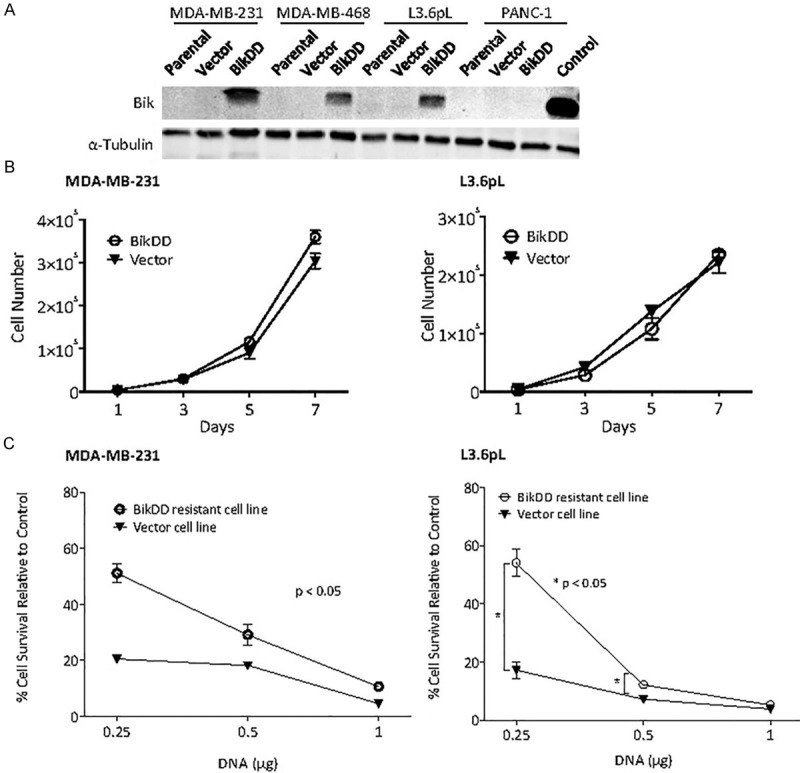
BikDD-expressing stable cells are resistant to BikDD treatment. A. Western blot analysis of BikDD expression in BikDD stable, parental, and vector control breast (MDA-MB-231 and MDA-MB-468) and pancreatic (L3.6pL and Panc-1) cancer cells. 293T cells transfected with BikDD served as positive control (Control). B. Cell proliferation of BikDD-expressing stable and vector control MDA-MB-231 and L3.6pL cells. C. The same dose of BikDD (0.25 µg) results in higher cell viability in BikDD stable cells compared to the vector control cells. Data are shown as mean ± SEM. n = 3. *, p < 0.05. Error bars are present for each point but nominal in some cases.
To further determine whether the selected BikDD-expressing cells are more resistant to the killing effect of BikDD, we re-transfected them with or without BikDD expressing plasmids with a luciferase reporter. The luciferase expression was used as an index for survival cells and control was set as 100%. As shown in Figure 1C, BikDD-expressing cells (MDA-MB-231 and L3.6pL) were more resistant to apoptosis by the same amount of BikDD plasmid DNA compared with the vector control cells, supporting that a resistant phenotype may have emerged from the selection procedure.
BikDD undergoes degradation via the proteasome pathway
Since it has been reported that Bik undergoes degradation through the proteasome-dependent pathway [16], we treated BikDD-expressing breast and pancreatic cancer cells with proteasome inhibitor MG132 and examined its effect on BikDD expression by Western blot analysis. As expected, cells treated with MG132 had substantially higher BikDD protein expression compared with those untreated (Figure 2A). Similar results were observed when we tested the clinically used proteasome inhibitor bortezomib (BZ) for comparison to MG132 in MIA CaPa-2 cells transiently expressing BikDD for 24 hours (Figure 2B) as well as in another pancreatic (L3.6pL; Figure 2C) and a breast (MDA-MB-231; Figure 2D) cancer cell line. We also observed a concomitant increase in the levels of cleaved PARP, indicating that cells underwent apoptosis (Figure 2C and 2D).
Figure 2.
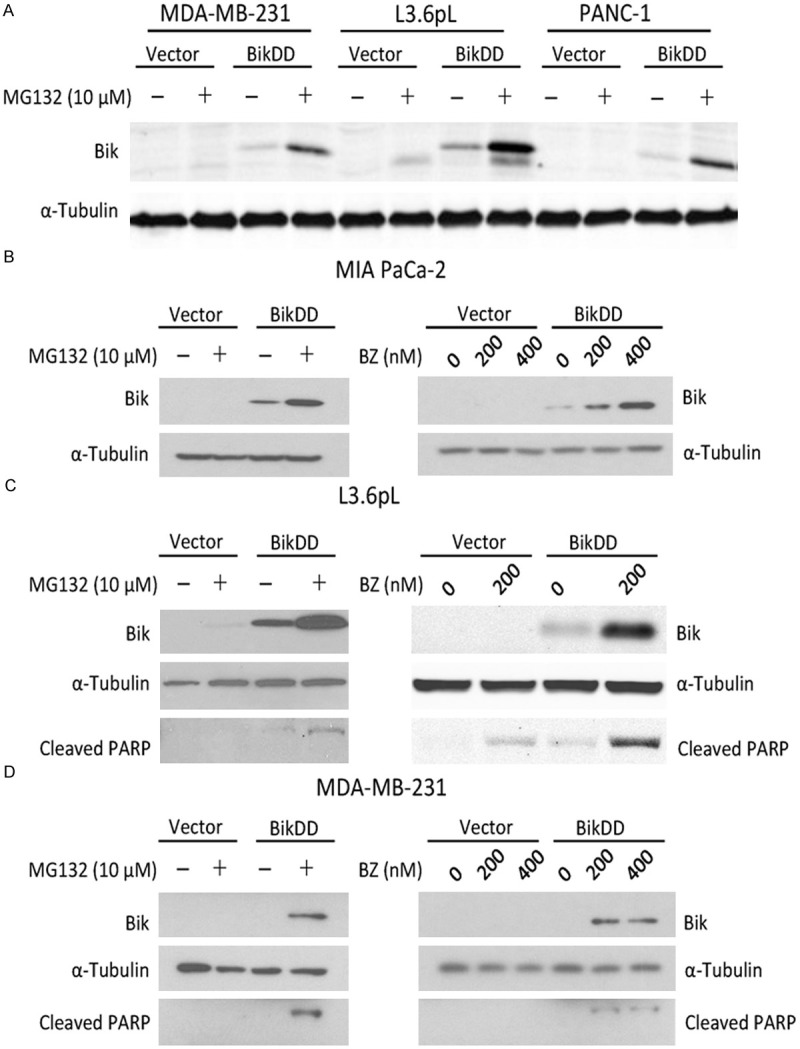
Proteasome inhibition stabilizes exogenously expressed BikDD. A. Three BikDD stable cell lines were treated with MG132 for 8 hours and their BikDD expression levels were analyzed by Western blot. B-D. BikDD was transiently expressed in MIA PaCa-2 and L3.6pL pancreatic and MDA-MB-231 breast cancer cells for 24 hours. Cells were then treated with proteasome inhibitors, MG132 or bortezomib (BZ), at the indicated concentration(s) for 8 hours. Cell lysates were subjected to Western blot analysis to evaluate the effect of proteasome inhibition.
Inhibition of proteasome enhances the killing effect of BikDD in cancer cells
To further validate the increase in apoptosis by BikDD in combination with proteasome inhibition, parental MDA-MB-231 cells with transient BikDD expression with and without BZ (50 nM) treatment were subjected to Annexin V and propidium iodide staining analysis by fluorescence activated cell sorting to compare the cell killing effect of BikDD in the presence of bortezomib. The percentage of cells that underwent apoptosis in the presence of BZ was higher (Figure 3A) in the BikDD group than vector control (BZ vs. control; Figure 3A). Similar results were observed in MIA PaCa-2 pancreatic cancer cells (Figure 3B). Therefore, these results suggest that BZ potentiates BikDD-induced apoptosis.
Figure 3.
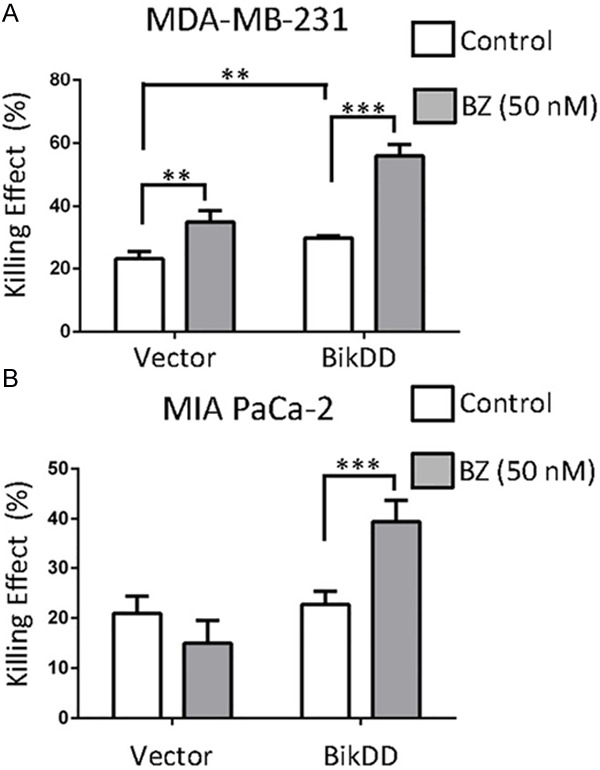
Combination of BikDD with a proteasome inhibitor increases the killing effect compare to monotherapy. BikDD (or control) was transiently expressed in MDA-MB-231 (A) and MIA PaCa-2 (B) cells for 24 hours. Cells were then treated with ethanol or 50 nM bortezomib (BZ) for 16 hours and collected for FACS analysis by staining with APC-Annexin V and propidium iodide (PI). Quantitative results are shown as bar diagrams. Data are shown as mean ± SD. n = 4. **, p < 0.01. ***, p < 0.001. Statistical analysis was performed using Student’s t test.
To determine whether the enhanced killing effect of BikDD when combined with proteasome inhibitor was a result of an accumulation of BikDD protein, we knocked down Bik (also BikDD) by siRNA in BikDD-expressing MDA-MB-231 breast cancer cells (Figure 4A) followed by MG132 treatment. The addition of MG132 to parental MDA-MB-231 cells led to about 50% cell survival compared to that without MG132 (Vector, Figure 4B). However, when BikDD was expressed in the same cells transfected with scrambled siRNA, only about 5% of cells survived. Knocking down Bik restored cell survival from 5% to about 20% to 30% (Figure 4B, right panel). These results suggest that the enhanced killing effect when proteasome inhibitor MG132 was added to BikDD transfection may be at least partially attributed to the accumulation of BikDD protein.
Figure 4.
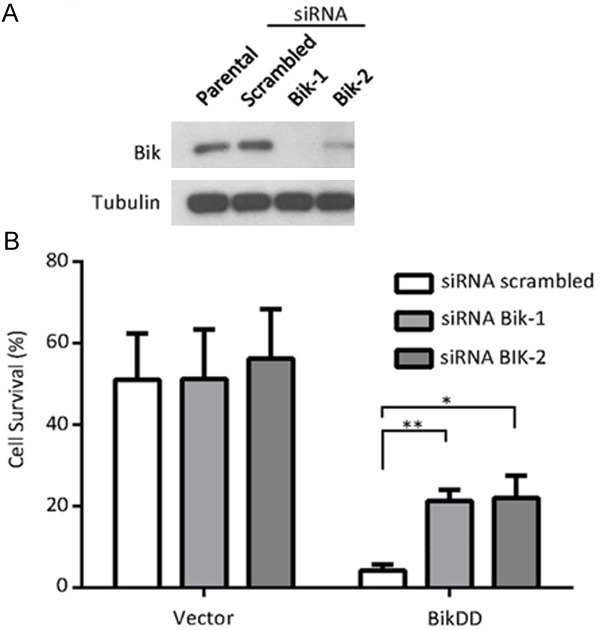
Proteasome inhibition leads to BikDD accumulation and enhanced apoptosis. A. MDA-MB-231 breast cancer cells were infected with vector control or BikDD lentivirus followed by Bik siRNA via electroporation to knock down BikDD expression. Twenty-four hours after electroporation, cells were lysed, and lysates were subjected to Western blot analysis to validate knockdown efficiency. B. Cell survival as indicated by quantitation of cell density after MG132 (2 µM) treatment for 24 hours. Cell density was determined by crystal violet staining, dissolving and measuring absorbance at 590 nm with spectrophotometer. Cell density was normalized to control group without treatment of MG132. Statistical analysis was carried out using Student’s t test. Data are shown as mean ± SD. n = 3. *, p < 0.01. **, p < 0.001.
BikDD is degraded via both ubiquitin-dependent and -independent pathways
Proteasome-mediated protein degradation can occur through an ubiquitin-dependent or -independent pathway. BikDD is a small protein that consists of 160 amino acids with only two lysines at 115 and 160. To determine which pathway mediates BikDD degradation, we created a single and a double Lys to Arg BikDD mutant to block possible ubiquitination. We compared the half-life of these mutants to BikDD and found that those mutations did not significantly prolong the half-life of BikDD (Figure 5A). In addition to Lys residues, N-terminal ubiquitination has been reported to mediate protein degradation, and therefore, we also tagged BikDD with mCherry at its N-terminus. While blocking the N-terminus of BikDD with an mCherry tag did not prolong its protein half-life, it was slightly increased when combined with the double Lys to Arg mutations (Figure 5A). The addition of proteasome inhibitor treatment led to an accumulation of mCherry-K115R/K160R-BikDD as well as WT-BikDD, K115/K160R-BikDD, and mCherry-tagged BikDD (Figure 5B), suggesting that an ubiquitination-independent mechanism is involved in BikDD degradation (Figure 5B). Together, these findings suggest that BikDD undergoes ubiquitination-dependent and -independent degradation pathways.
Figure 5.
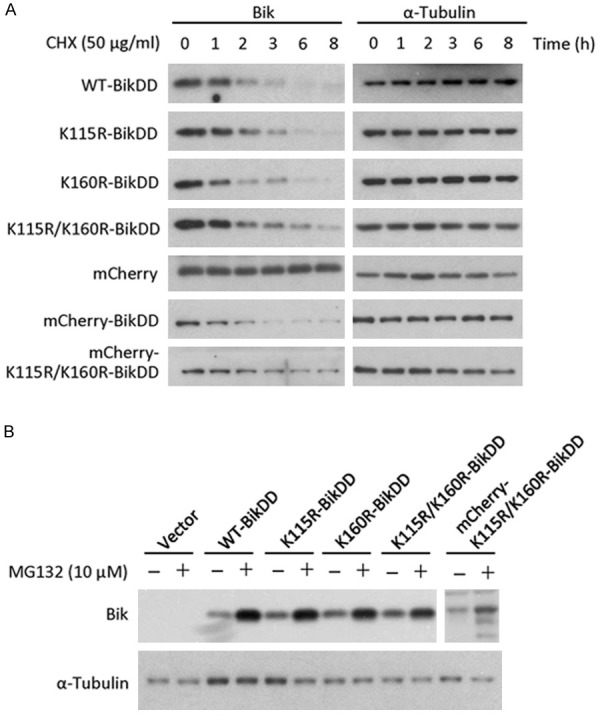
BikDD is degraded via both ubiquitination-dependent and -independent pathways. Lysine mutations and N-terminal tag fusion of BikDD were constructed to block potential ubiquitination sites. A. WT, mutant, and tagged BikDD-expressing MDA-MB-231 cells were treated with cycloheximide (CHX; 50 µg/ml) for the indicated time, and lysates were subjected to Western blot analysis with antibodies against Bik or internal control (α-tubulin). B. MDA-MB-231 cells expressing various forms of BikDD were treated with MG132 (10 µM) for 8 hours. Cell lysates were subjected to Western blot analysis with the indicated antibodies.
Discussion
In normal cells, expression of Bik is tightly regulated to remove damaged or aged cells [1]. A number of reports have indicated that reduced expression of Bik such as through epigenetic silencing, chromosome deletion, or mutation, allows cancer cells to evade apoptosis [17-21]. Moreover, studies have also demonstrated that even though the mRNA of Bik is constitutively transcribed, its protein is actively targeted for degradation by the proteasome system in some cancer cells [16]. Proteasome-inhibitor induced apoptosis appears to be attributed to the accumulation of Bik by preventing its degradation [22,23]. In our study, we found that exogenously expressed BikDD undergoes proteasome degradation as well, and proteasome inhibitors led to significant accumulation of BikDD protein, leading to an enhanced apoptosis. Thus, the addition of proteasome inhibitor to stabilize transfected BikDD, such as in gene therapy, has the potential to further strengthen its therapeutic efficacy. In our current study, we only tested the effect of combining proteasome inhibitor and BikDD transfection by in vitro assays. Further studies in animals will be needed to validate the efficacy and feasibility of the proposed combination therapy.
Based on results from the current study, both ubiquitination-dependent and -independent proteasome degradation pathways contribute to BikDD degradation. Although lysine residues and the N-terminus are the major sites where ubiquitination are known occurs, studies have shown that in rare cases, non-N-terminal ubiquitination can happen on non-lysine residues [24-26]. Thus, the possibility that ubiquitination of other amino acids of BikDD should not be excluded. It would be of interest to further investigate other posttranslational modifications that might also contribute to protein degradation. Xie et al. recently reported a mutation on Ser 124 to block ERK1/2 dependent phosphorylation on BikDD stabilizing its expression [27]. Combining ERK1/2 inhibitors with BikDD, gene therapy should also be considered as a combinational strategy.
Wang et al. previously reported the serine protease RHBDD1 cleaves Bik at the C-terminus, leading to its degradation [28]. However, the proteasome utilizes the N-terminal hydroxyl group of the threonine residue in the catalytic site to catalyze protein degradation, which is structurally different from serine protease. Proteasome inhibitors target threonine catalytic sites with satisfactory high specificity. Thus, the effect of BikDD protein accumulation caused by MG132 and bortezomib is unlikely mediated through the activity of RHBDD1, although RHBDD1 might possess additional effects in BikDD degradation. Furthermore, RHBDD1 is a non-targetable serine protease based on the current protease inhibitor database, thus it may have less value in clinical applications.
It is worthwhile to mention that we intended but failed to identify the E3 ligase responsible for degradation of BikDD. In brief, Flag-BikDD protein stably expressed in MDA-MB-231 cells was immunoprecipitated under conditions with or without proteasome inhibition. BikDD-binding proteins under different conditions were identified by mass spectrum analysis. We identified 4 proteins with published ubiquitin E3 ligase activity, including AMFR, WWP2, HECTD1, and HUWE1. However, we found that overexpression (Supplementary Figure 1A) or knock down (Supplementary Figure 1B) of these candidates individually did not affect BikDD protein levels, suggesting that none of these proteins alone represents the E3 ligases directly responsible for BikDD degradation.
Overall the study shows how combination of BikDD with proteasome inhibitors can increase the killing effect of BikDD in vitro. In addition, a possible explanation for the success of this combination is also shown. Even though in vivo studies are needed, this study opens the door to a possible combination treatment for BikDD that should help in the development of this gene therapy once the phase I clinical trial in pancreatic cancer is completed.
Acknowledgements
This work was supported in part by the following grants: Cancer Prevention and Research Institute of Texas (ETRA RP120727); National Institutes of Health grants (CCSG CA16672); National Breast Cancer Foundation, Inc.; Patel Memorial Breast Cancer Endowment Fund; The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund (to M.-C. Hung); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW104-TDU-B-212-124-002, Taiwan); International Research-Intensive Centers of Excellence in Taiwan (MOST 104-2911-I-002-302, Taiwan); and Center for Biological Pathways. MPS was supported by a fellowship from Sociedad Española de Oncología Médica (SEOM). In memoriam, Mr. Tiong Loi Ang for his courageous battle against cancer.
Disclosure of conflict of interest
Mien-Chie Hung has ownership interest (including patents) for BikDD. All other authors have no conflicts of interest to declare.
Supporting Information
References
- 1.Chinnadurai G, Vijayalingam S, Rashmi R. BIK, the founding member of the BH3-only family proteins: mechanisms of cell death and role in cancer and pathogenic processes. Oncogene. 2008;27(Suppl 1):S20–29. doi: 10.1038/onc.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen JS, Liu JC, Shen L, Rau KM, Kuo HP, Li YM, Shi D, Lee YC, Chang KJ, Hung MC. Cancer-specific activation of the survivin promoter and its potential use in gene therapy. Cancer Gene Ther. 2004;11:740–747. doi: 10.1038/sj.cgt.7700752. [DOI] [PubMed] [Google Scholar]
- 3.Zou Y, Peng H, Zhou B, Wen Y, Wang SC, Tsai EM, Hung MC. Systemic tumor suppression by the proapoptotic gene bik. Cancer Res. 2002;62:8–12. [PubMed] [Google Scholar]
- 4.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 5.Hsu JL, Chao CH, Xie X, Hung MC. Advances in Liposome-Based Targeted Gene Therapy of Cancer. In: Liu XY, Pestka S, Shi YF, editors. Recent Advances in Cancer Research and Therapy. Elsevier; 2012. pp. 113–124. [Google Scholar]
- 6.Li YM, Wen Y, Zhou BP, Kuo HP, Ding Q, Hung MC. Enhancement of Bik antitumor effect by Bik mutants. Cancer Res. 2003;63:7630–7633. [PubMed] [Google Scholar]
- 7.Day CP, Rau KM, Qiu L, Liu CW, Kuo HP, Xie X, Lopez-Berestein G, Hortobagyi GN, Hung MC. Mutant Bik expression mediated by the enhanced minimal topoisomerase IIalpha promoter selectively suppressed breast tumors in an animal model. Cancer Gene Ther. 2006;13:706–719. doi: 10.1038/sj.cgt.7700945. [DOI] [PubMed] [Google Scholar]
- 8.Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, Lee DF, Yang JY, Xie X, Liu JC, Hung MC. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67:4564–4571. doi: 10.1158/0008-5472.CAN-06-1788. [DOI] [PubMed] [Google Scholar]
- 9.Lang JY, Hsu JL, Meric-Bernstam F, Chang CJ, Wang Q, Bao Y, Yamaguchi H, Xie X, Woodward WA, Yu D, Hortobagyi GN, Hung MC. BikDD eliminates breast cancer initiating cells and synergizes with lapatinib for breast cancer treatment. Cancer Cell. 2011;20:341–356. doi: 10.1016/j.ccr.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie X, Li L, Xiao X, Guo J, Kong Y, Wu M, Liu W, Gao G, Hsu JL, Wei W, Hung MC, Xie X. Targeted expression of BikDD eliminates breast cancer with virtually no toxicity in noninvasive imaging models. Mol Cancer Ther. 2012;11:1915–1924. doi: 10.1158/1535-7163.MCT-12-0191. [DOI] [PubMed] [Google Scholar]
- 11.Sher YP, Tzeng TF, Kan SF, Hsu J, Xie X, Han Z, Lin WC, Li LY, Hung MC. Cancer targeted gene therapy of BikDD inhibits orthotopic lung cancer growth and improves long-term survival. Oncogene. 2009;28:3286–3295. doi: 10.1038/onc.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sher YP, Liu SJ, Chang CM, Lien SP, Chen CH, Han Z, Li LY, Chen JS, Wu CW, Hung MC. Cancer-targeted BikDD gene therapy elicits protective antitumor immunity against lung cancer. Mol Cancer Ther. 2011;10:637–647. doi: 10.1158/1535-7163.MCT-10-0827. [DOI] [PubMed] [Google Scholar]
- 13.Li LY, Dai HY, Yeh FL, Kan SF, Lang J, Hsu JL, Jeng LB, Chen YH, Sher YP, Lin WC, Hung MC. Targeted hepatocellular carcinoma proapoptotic BikDD gene therapy. Oncogene. 2011;30:1773–1783. doi: 10.1038/onc.2010.558. [DOI] [PubMed] [Google Scholar]
- 14.Xie X, Kong Y, Tang H, Yang L, Hsu JL, Hung MC. Targeted BikDD expression kills androgen-dependent and castration-resistant prostate cancer cells. Mol Cancer Ther. 2014;13:1813–25. doi: 10.1158/1535-7163.MCT-13-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie X, Xia W, Li Z, Kuo HP, Liu Y, Ding Q, Zhang S, Spohn B, Yang Y, Wei Y, Lang JY, Evans DB, Chiao PJ, Abbruzzese JL, Hung MC. Targeted expression of BikDD eradicates pancreatic tumors in noninvasive imaging models. Cancer Cell. 2007;12:52–65. doi: 10.1016/j.ccr.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Hur J, Bell DW, Dean KL, Coser KR, Hilario PC, Okimoto RA, Tobey EM, Smith SL, Isselbacher KJ, Shioda T. Regulation of expression of BIK proapoptotic protein in human breast cancer cells: p53-dependent induction of BIK mRNA by fulvestrant and proteasomal degradation of BIK protein. Cancer Res. 2006;66:10153–10161. doi: 10.1158/0008-5472.CAN-05-3696. [DOI] [PubMed] [Google Scholar]
- 17.Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, Jung K, Loening S, Dorken B, Daniel PT. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 2006;13:619–627. doi: 10.1038/sj.cdd.4401782. [DOI] [PubMed] [Google Scholar]
- 18.Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI. High-resolution genome-wide mapping of genetic alterations in human glial brain tumors. Cancer Res. 2005;65:4088–4096. doi: 10.1158/0008-5472.CAN-04-4229. [DOI] [PubMed] [Google Scholar]
- 19.Castells A, Ino Y, Louis DN, Ramesh V, Gusella JF, Rustgi AK. Mapping of a target region of allelic loss to a 0.5-cM interval on chromosome 22q13 in human colorectal cancer. Gastroenterology. 1999;117:831–837. doi: 10.1016/s0016-5085(99)70341-0. [DOI] [PubMed] [Google Scholar]
- 20.dos Reis PP, Poli-Frederico RC, dos Santos RM, Nishimoto IN, Kowalski LP, Rogatto SR. Distinct regions of loss of heterozygosity on 22q in different sites of head and neck squamous cell carcinomas. Med Sci Monit. 2002;8:BR89–94. [PubMed] [Google Scholar]
- 21.Arena V, Martini M, Luongo M, Capelli A, Larocca LM. Mutations of the BIK gene in human peripheral B-cell lymphomas. Genes Chromosomes Cancer. 2003;38:91–96. doi: 10.1002/gcc.10245. [DOI] [PubMed] [Google Scholar]
- 22.Zhu H, Zhang L, Dong F, Guo W, Wu S, Teraishi F, Davis JJ, Chiao PJ, Fang B. Bik/NBK accumulation correlates with apoptosis-induction by bortezomib (PS-341, Velcade) and other proteasome inhibitors. Oncogene. 2005;24:4993–4999. doi: 10.1038/sj.onc.1208683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikrad M, Johnson T, Puthalalath H, Coultas L, Adams J, Kraft AS. The proteasome inhibitor bortezomib sensitizes cells to killing by death receptor ligand TRAIL via BH3-only proteins Bik and Bim. Mol Cancer Ther. 2005;4:443–449. doi: 10.1158/1535-7163.MCT-04-0260. [DOI] [PubMed] [Google Scholar]
- 24.Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–130. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Herr RA, Chua WJ, Lybarger L, Wiertz EJ, Hansen TH. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol. 2007;177:613–624. doi: 10.1083/jcb.200611063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tait SW, de Vries E, Maas C, Keller AM, D'Santos CS, Borst J. Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J Cell Biol. 2007;179:1453–1466. doi: 10.1083/jcb.200707063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiao S, Wu M, Ye F, Tang H, Xie X, Xie X. BikDDA, a mutant of Bik with longer half-life expression protein, can be a novel therapeutic gene for triple-negative breast cancer. PLoS One. 2014;9:e92172. doi: 10.1371/journal.pone.0092172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Guan X, Fok KL, Li S, Zhang X, Miao S, Zong S, Koide SS, Chan HC, Wang L. A novel member of the Rhomboid family, RHBDD1, regulates BIK-mediated apoptosis. Cell Mol Life Sci. 2008;65:3822–3829. doi: 10.1007/s00018-008-8452-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.