

Abstract
Background:
Spectral domain (SD) enhanced depth imaging optical coherence tomography (EDI-OCT) is a useful tool for anatomic, cross-sectional imaging of retinal conditions.
Aims:
The aim was to identify characteristic patterns of retinal and retinal pigment epithelial tumors on EDI-OCT in children and adults.
Settings and Design:
Retrospective review.
Materials and Methods:
Analysis of published reports and personal observations using office-based EDI-OCT for adults and portable hand-held SD OCT for infants and children.
Results:
Using EDI-OCT, retinal tumors such as small retinoblastoma, astrocytic hamartoma, and hemangioblastoma arose abruptly from the retina, immediately adjacent to normal retina. Small exophytic retinoblastoma and retinal hemangioblastoma showed the full-thickness, homogeneous retinal disorganization with surrounding normal retina “draping” over the margins. Retinoblastoma occasionally had intralesional cavities and surrounding subretinal fluid. Hemangioblastoma often had adjacent intraretinal edema and subretinal fluid. Astrocytic hamartoma arose within the nerve fiber layer and sometimes with a “moth-eaten” or cavitary appearance. Retinal pigment epithelial (RPE) lesions such as congenital hypertrophy of RPE appeared flat with shadowing, occasional subretinal cleft, and abrupt photoreceptor loss. Congenital simple hamartoma showed an abrupt elevation from the inner retina with crisp, dark posterior shadowing. Combined hamartoma of the retina/RPE showed vitreoretinal traction causing “sawtooth mini-peak” or gently “maxi-peak” folding of the retina. RPE adenoma often produces remote macular edema or epiretinal membrane and the tumor has an irregular, “rugged” surface with deep shadowing.
Conclusions:
Enhanced depth imaging optical coherence tomography shows characteristic patterns that are suggestive of certain retinal and RPE tumors.
Keywords: Astrocytic hamartoma, enhanced depth imaging optical coherence tomography, hemangioblastoma, retina, retinoblastoma, tumor
Optical coherence tomography (OCT) is a powerful imaging tool for the posterior segment of the eye.[1] Initially, with time-domain OCT (TD-OCT) technology, cross-sectional imaging of the retina was possible with a resolution of approximately 10 microns.[2,3] Further refinements with spectral domain OCT (SD-OCT) has allowed for depiction of the various layers of the neurosensory retina and enhanced depth imaging OCT (EDI-OCT) has recently provided improved resolution to approximately 3-4 microns and the ability to image deeper within the choroid and sclera.[4] Imaging with OCT has become a routine, nearly-essential step of ophthalmology, particularly for diagnosis and therapy of retinal disease.
Standard imaging with OCT has generally been an office-based technique, primarily used in cooperative adults who can maintain ocular alignment for several minutes to enable serial capture of OCT A-scans to produce the final B-scan. In 2004, Shields et al. explored office-based TD-OCT in 44 children and found that cooperative children as young as 4 years of age could be imaged with reliability in the office.[5] New developments in OCT design have allowed for more portable hand-held SD-OCT (HHSD-OCT) for use mainly in the pediatric population under anesthesia. In 2009, Scott et al. published observations on portable HHSD-OCT used under anesthesia on infants with abusive head trauma.[6] They found portable HHSD-OCT produced images comparable to office-based SD-OCT. They could visualize characteristic morphologic retinal features in infants that were previously difficult to visualize by ophthalmoscopy or ultrasonography, such as partial posterior vitreous detachment, epiretinal membrane, macular fold, and lamellar and full-thickness macular hole.
In our practice of ocular oncology,[7,8] office-based EDI-OCT and portable HHSD-OCT is used daily to image tumors within the eye. We have found this modality to provide exquisitely reliable, high-resolution anatomic information on tumors, much of which is not clinically visible to the examiner. Herein, we review the most salient features of retinal and retinal pigment epithelial (RPE) tumors on EDI-OCT.[9]
Retina
Retinoblastoma
Worldwide, retinoblastoma is the most common primary intraocular malignancy.[7,8,10,11] When detected at an early stage, OCT can be useful in delineating the relationship of the tumor to the surrounding retina and foveola. Shields et al. studied TD-OCT in 10 eyes with retinoblastoma and observed the retinal mass with disorganization at the site of the tumor with occasional intralesional cavities.[5] Rootman et al. evaluated 16 patients with retinoblastoma at median age 1.5 years with HHSD-OCT and found in 5 cases, in which the tumor was small and could be evaluated, that it was located in the middle retinal layers.[12] In other cases, they noted the retinoblastoma mass in the outer retinal layers, elevating the normal-appearing inner retinal layers inward, a feature that we have termed “retinal draping.” This feature is common with small retinoblastoma.
Cao et al. analyzed 3 infants with small retinoblastomas (mean thickness 4.8 mm) using HHSD-OCT, in the operating room and noted smooth tumor surface in 2 and full-thickness retinal involvement and low optical density in all 3 cases.[13] In each case, the foveola was buried within the tumor at presentation, and there was an abrupt transition from normal to tumor-involved retina. Subretinal fluid was noted in each case. Following therapy, the tumor surface was more irregular, optical density higher, and subretinal fluid resolved. In one case, the foveola was intact, despite being inapparent pretreatment.
More recently, Shields et al. imaged 74 eyes of 57 infants with retinoblastoma using portable HHSD-OCT.[14] The median patient age was 9 months. The authors commented that this technique required precise positioning of the infant's head and rotation of the eye so that the foveola, tumor, and other sites could be appropriately seen. Anesthesia-related supraduction or infraduction of the eye could lead to difficulty with HHSD-OCT. They noted that the tumor arose abruptly within the retina, often with full-thickness disorganization of the retina [Fig. 1]. In exophytic tumors, an important finding was normal retina draped over an exophytic mass. This “retinal draping” implied that the normal tissue could be inadvertently damaged by thermotherapy or other treatments during tumor consolidation as it was overlying the retinal malignancy. Other findings on HHSD-OCT included intratumor cavities, intratumor calcification, subretinal seeds, epiretinal vitreous seeds, photoreceptor loss in areas of chronic subretinal fluid, traction retinal detachment, retinal edema, epiretinal membrane, and retinal thinning following thermotherapy or laser photocoagulation. HHSD-OCT was useful in identifying foveolar anatomy and estimating visual potential.
Figure 1.
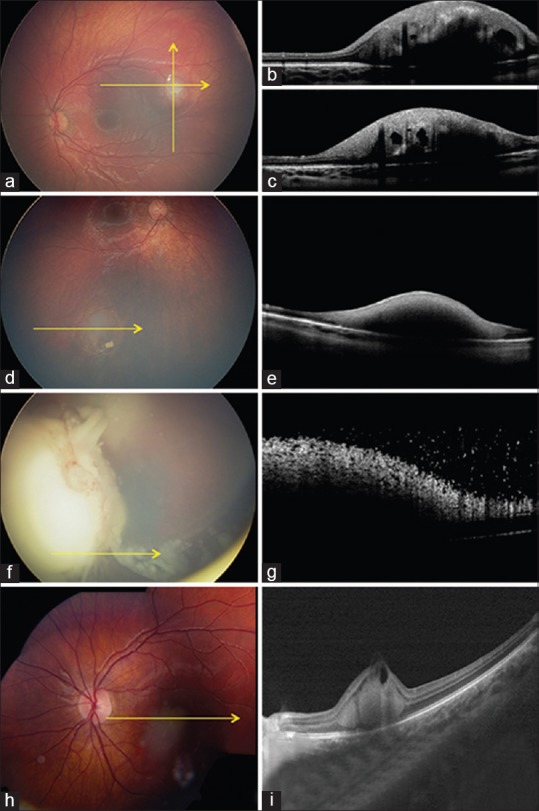
Retinoblastoma. (a-c) Infant with small macular retinoblastoma (a) with spectral domain optical coherence tomography (SD-OCT) (b and c) showing smooth anterior tumor surface, full-thickness retinal involvement, optically empty cavities, shadowing from calcification, and retinal draping over margins. (d and e) Small retinoblastoma (d) with SD-OCT (e) showing smooth tumor surface and full-thickness retinal involvement. (f and g) Large endophytic retinoblastoma (f) with SD-OCT (g) showing irregular, “frosty” tumor surface with overlying fine vitreous seeds. (h and i) Macular retinoblastoma (h) with SD-OCT (i) showing outer retinal involvement, anterior cavity, and notable inner retinal draping
In summary, EDI-OCT of small retinoblastoma shows an abrupt transition from normal retina to the tumor, with tumor involving middle retinal layers or full-thickness retina and occasional “retina draping” of normal retina over the tumor.
Retina astrocytic hamartoma
Retinal astrocytic hamartoma is a benign tumor that appears as a yellow white inner retinal mass, often with calcification and minimally dilated retinal vessels. TD-OCT has characterized this tumor with retinal elevation and “moth-eaten” lucent areas.[15] Serafino et al. described EDI-OCT features in 86 eyes of 47 patients with retinal astrocytic hamartoma and classified the P = tumors into type I (42%), type II (26%), type III (20%), and type IV (12%).[16] They described type I as flat and generally in the nerve fiber layer; type II with slight elevation of the nerve fiber layer and retinal traction; type III with “moth-eaten” lucent areas suggestive of calcification involving inner and outer retina; and type IV with optically empty intralesional cavities [Fig. 2]. They found that type II correlated with cutaneous forehead plaques (P < 0.001) and type III correlated with brain astrocytoma (P < 0.001). Veronese et al. illustrated two cases of retinal astrocytic hamartoma with OCT-evidence of intralesional cavities.[17]
Figure 2.
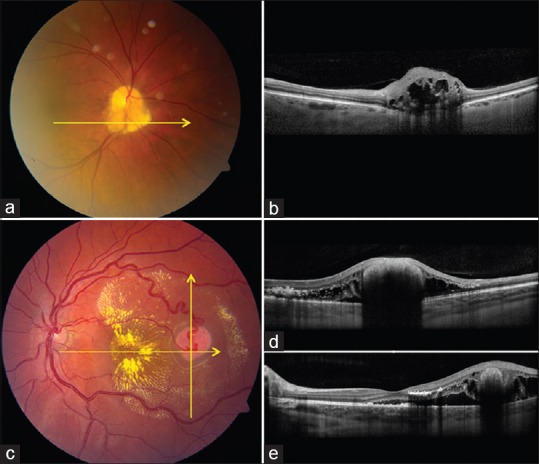
Retinal astrocytic hamartoma and hemangioblastoma. (a and b) Retinal astrocytic hamartoma (a) with enhanced depth imaging optical coherence tomography (EDI-OCT) (b) showing the inner retinal mass compressing the outer retina and with central full-thickness involvement and “moth-eaten” lucencies. (c-e) Retinal hemangioblastoma (c) with EDI-OCT (d and e) showing exophytic mass pushing inner retina, outer retinal edema, subretinal debris and fluid
In summary, we believe that retinal astrocytic hamartoma typically begins as a flat tumor in the nerve fiber layer with gradual enlargement to nodular full-thickness retinal mass, occasionally with “moth-eaten” calcified nummular lucent areas causing optical showing or degenerative intralesional cavitary findings.
Retina hemangioblastoma
Retinal hemangioblastoma is a benign retinal vascular tumor, often associated with von Hippel–Lindau disease.[7,18] This tumor commonly produces subretinal fluid, exudation, and vitreoretinal traction. There are no published case series studying OCT findings in this condition. By personal experience, EDI-OCT shows an abruptly elevated full-thickness retinal mass with smooth domed surface, retinal disorganization, and deep optical shadowing [Fig. 2]. The surrounding normal retina is commonly draped over the exophytic mass. Surrounding subretinal fluid with subretinal exudation and macrophages are noted. Intraretinal edema with noncystoid and cystoid features, as well as intraretinal exudation, can be found.
Retinal Pigment Epithelium
Congenital hypertrophy of the retinal pigment epithelial
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is a fairly common, benign, pigmented RPE lesion, classically located in the retinal periphery.[19] The peripheral location of this tumor causes difficulty in obtaining EDI-OCT; however, much has been learned about this tumor in the few cases in which the tumor was postequatorial, in the region suitable for imaging.
Shields et al. reported on TD-OCT of CHRPE in 10 consecutive cases and observed a flat lesion of the RPE with minimal, barely perceptible increased thickness by 52% compared to normal RPE and with overlying retinal thinning and overlying photoreceptor loss in every case.[20] They commented that the photoreceptor loss began precisely at the margin of the CHRPE, with normal photoreceptors in the immediately adjacent surrounding normal retinal tissue.
Fung et al. described EDI-OCT of 18 eyes with CHRPE and found all lesions were flat.[21] In the region of the CHRPE, they noted that the RPE was absent (11%), thickened (89%) or irregular (83%) [Fig. 3]. Intralesional lacunae typically showed absent RPE with bright transmission of OCT through the defect. The overlying retina displayed marked photoreceptor loss in all cases immediately at the site of the CHRPE. Another interesting feature was subretinal cleft (33%), which was speculated to represent thinned excavated posterior retina with the photoreceptor atrophy, leaving an empty space. The underlying choroid appeared normal.
Figure 3.
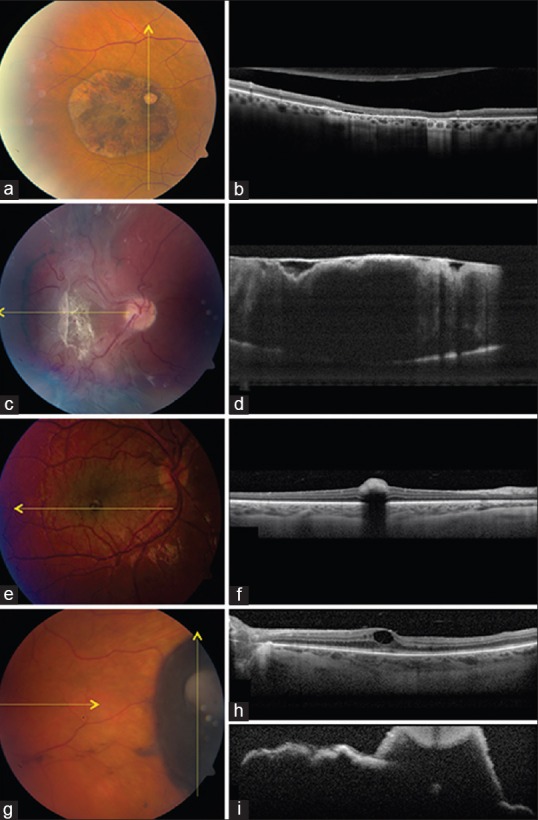
Retinal pigment epithelial (RPE) tumors. (a and b) Congenital hypertrophy of the retinal pigment epithelium (a) with enhanced depth imaging optical coherence tomography (EDI-OCT) (b) showing abrupt photoreceptor loss over the flat lesion, subretinal cleft, and transmission of light through lacunae. (c and d) Combined hamartoma of the retina and RPE (c) with EDI-OCT (d) showing thickened retina with disorganization and “folded” appearance with overlying dense epiretinal membrane. (e and f) Congenital simple hamartoma of the RPE (e) with EDI-OCT (f) showing an abrupt dense inner retinal mass with absolute shadowing and normal choroidal thickness. (g-i) RPE adenoma (g) with EDI-OCT showing remote cystoid macular edema (h) and irregular surface, tumor nodule, and deep shadowing (i)
In summary, EDI-OCT of CHRPE classically shows flat, slightly irregular RPE with an abrupt photoreceptor atrophy, occasional subretinal cleft, and normal underlying choroid.
Congenital simple hamartoma of the retinal pigment epithelial
Congenital simple hamartoma of the RPE is a rare pigmented hamartoma believed to arise from RPE and typically located in the parafoveal region. This tumor generally appears at approximately 1 mm in diameter, involves full-thickness retina, and generally remains stable with minimal effect on visual acuity.[22]
TD-OCT shows this mass to be a modest dome-shaped elevation of the inner retinal surface with abrupt, absolute shadowing of the deeper retinal tissue.[22,23,24,25] The small size and marked shadowing of this tumor are somewhat characteristic and suggestive of the diagnosis. Personal experience with EDI-OCT of this tumor reveals similar elevation if the inner retina, marked shadowing, no adjacent retinal tissue disturbance and apparently normal underlying choroid [Fig. 3].
Combined hamartoma of the retina and retinal pigment epithelial
Combined hamartoma of the retina and retinal pigment epithelium is a presumed-congenital intraocular mass characterized by an ill-defined disorganized glial, vascular, and melanocytic tissue involving the neurosensory retina and retinal vessels as well as the RPE.[25,26] Overlying vitreoretinal traction in both a horizontal and vertical direction can lead to retinal dragging, foveal ectopia, and vision loss.[26] Persistent traction can produce retinal folds or tractional retinal detachment. In an analysis of 77 eyes with a combined hamartoma, clinical evidence of retinal traction was documented in 81%, and poor visual acuity of 20/200 or worse in 47%.[26]
Shields et al. reported on TD-OCT of combined hamartoma in 11 eyes and noted remarkable vitreoretinal traction, epiretinal membrane, retinal striae, retinal disorganization, and photoreceptor attenuation.[27] No case demonstrated posterior vitreous detachment or RPE detachment. One case showed subretinal fluid. They concluded that TD-OCT could provide important information on this tumor and could influence surgical decisions, for example, if the retina appeared relatively intact, then better surgical result could be anticipated. Huot et al. noted that SD-OCT allows higher resolution imaging with delineation of the vitreoretinal interface abnormalities and visualization of the transition from normal retina to tumor.[28]
Arepalli et al. analyzed the EDI-OCT features of 8 patients with a combined hamartoma at a median age of 4 years. They found striking vitreoretinal traction causing irregularities in the inner retina (100%) and/or all retinal layers (38%). This traction lead to a pattern of mini-peaks (sawtooth appearance) (25%), large maxi-peaks of full-thickness retinal folds (38%), or both (38%)[29] [Fig. 3]. The mean number of mini-peaks per tumor scan was 5, most of which were sharp, pointed and hyperacute and others less pointed. The mean number of maxi-peaks per scan was 3, and all were gently folded retina. The tumor thickness was mean of 608 μm compared to 244 μm in the unaffected eye (P = 0.0004). Interestingly, the underlying choroidal thickness was slightly decreased to a mean of 210 μm compared to 328 μm in the corresponding area of the unaffected eye (P = 0.009).
In summary, EDI-OCT of combined hamartoma shows dramatic vitreoretinal traction in the inner or full-thickness retina leading to patterns of sawtooth (mini-peaks) and/or full-thickness retinal folds (maxi-peaks).
Retinal pigment epithelial adenoma/adenocarcinoma
Retinal pigment epithelial adenoma/adenocarcinoma is a rare intraocular tumor. Shields et al. reviewed the clinical and histopathologic features of this tumor in 13 cases and found that this lesion classically appears as a black (85%), full-thickness retinal mass, often with enlarged feeding vessels (62%), surrounding exudative retinopathy (38%), vitreous hemorrhage (15%), and remote epiretinal membrane and macular edema.[30] By personal experience, EDI-OCT shows the tumor surface as irregular or “rugged” with full-thickness retinal tumor and dense posterior optical shadowing. Occasional vitreous seeds can be seen. Remote macular findings of epiretinal membrane, cystoid and noncystoid edema, and occasionally macular hole can be found.
Summary
Enhanced depth imaging optical coherence tomography is an important tool for the imaging of retinal and RPE tumors in adults and children. Office-based EDI-OCT for adults and portable HHSD-OCT for young infants are employed. New information regarding tumor anatomy and related findings allow a better understanding of ocular conditions. Retinoblastoma and retinal hemangioblastoma show “retinal draping” over tumor, retinal astrocytic hamartoma originates in the nerve fiber layer and can eventually develop intralesional cavities, CHRPE shows flat tumor with overlying cleft and lacunar transmission, congenital simple hamartoma of RPE shows slightly elevated retinal mass with abrupt, absolute shadowing, and combined hamartoma of retina and RPE shows vitreoretinal traction in “sawtooth (mini-peaks)” or folded (maxi-peaks) appearance. RPE adenoma often produces remote macular edema or epiretinal membrane and the tumor has an irregular, “rugged” surface with deep shadowing.
Precis
Spectral domain enhanced depth imaging optical coherence tomography shows smooth tumor surface with retinal “draping” with small retinoblastoma and hemangioblastoma and intralesional cavities with retinoblastoma and astrocytic hamartoma. Congenital hypertrophy of the retinal pigment epithelium shows flat mass with photoreceptor loss and combined hamartoma shows occasionally surface “sawtooth” traction.
Footnotes
Source of Support: Support provided by Eye Tumor Research Foundation, Philadelphia, PA (CLS, JAS), Lift for a Cure, Morrisdale, PA (CLS, JAS), and the Lucille Wiedman Fund for Pediatric Eye Cancer, Philadelphia, PA (JAS, CLS). The funders had no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, and in the preparation, review or approval of the manuscript. Carol L. Shields, M. D. has had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis
Conflict of Interest: None declared.
References
- 1.Huang D, Swanson EA, Lin CP, Schuman JS, Stinson WG, Chang W, et al. Optical coherence tomography. Science. 1991;254:1178–81. doi: 10.1126/science.1957169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shields CL, Materin MA, Shields JA. Review of optical coherence tomography for intraocular tumors. Curr Opin Ophthalmol. 2005;16:141–54. doi: 10.1097/01.icu.0000158258.01681.40. [DOI] [PubMed] [Google Scholar]
- 3.Say EA, Shah SU, Ferenczy S, Shields CL. Optical coherence tomography of retinal and choroidal tumors. J Ophthalmol 2012. 2012 doi: 10.1155/2012/385058. 385058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spaide RF, Koizumi H, Pozzoni MC. Enhanced depth imaging spectral-domain optical coherence tomography. Am J Ophthalmol. 2008;146:496–500. doi: 10.1016/j.ajo.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 5.Shields CL, Mashayekhi A, Luo CK, Materin MA, Shields JA. Optical coherence tomography in children: Analysis of 44 eyes with intraocular tumors and simulating conditions. J Pediatr Ophthalmol Strabismus. 2004;41:338–44. doi: 10.3928/01913913-20041101-04. [DOI] [PubMed] [Google Scholar]
- 6.Scott AW, Farsiu S, Enyedi LB, Wallace DK, Toth CA. Imaging the infant retina with a hand-held spectral-domain optical coherence tomography device. Am J Ophthalmol. 2009;147:364–373.e2. doi: 10.1016/j.ajo.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Shields JA, Shields CL. 2nd ed. Philadelphia, PA: Lippincott, Williams and Wilkins; 2008. Intraocular Tumors: An Atlas and Textbook. [Google Scholar]
- 8.Ramasubramanian A, Shields CL, editors. New Delhi, India: Jaypee Brothers Medical Publishers; 2012. Retinoblastoma. [Google Scholar]
- 9.Shields CL, Pellegrini M, Ferenczy SR, Shields JA. Enhanced depth imaging optical coherence tomography of intraocular tumors: From placid to seasick to rock and rolling topography – the 2013 Francesco Orzalesi Lecture. Retina. 2014;34:1495–512. doi: 10.1097/IAE.0000000000000288. [DOI] [PubMed] [Google Scholar]
- 10.Shields CL, Fulco EM, Arias JD, Alarcon C, Pellegrini M, Rishi P, et al. Retinoblastoma frontiers with intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Eye (Lond) 2013;27:253–64. doi: 10.1038/eye.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shields CL, Lally SE, Leahey AM, Jabbour PM, Caywood EH, Schwendeman R, et al. Targeted retinoblastoma management: When to use intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Curr Opin Ophthalmol. 2014;25:374–85. doi: 10.1097/ICU.0000000000000091. [DOI] [PubMed] [Google Scholar]
- 12.Rootman DB, Gonzalez E, Mallipatna A, Vandenhoven C, Hampton L, Dimaras H, et al. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: Clinical and morphologic considerations. Br J Ophthalmol. 2013;97:59–65. doi: 10.1136/bjophthalmol-2012-302133. [DOI] [PubMed] [Google Scholar]
- 13.Cao C, Markovitz M, Shields CL. Handheld enhanced depth imaging optical coherence tomography of foveal and parafoveal retinoblastoma in infants and young children under anesthesia in 13 cases. Submitted for publication [Google Scholar]
- 14.Shields CL, Schonbach E, Nickerson S, Weiner J, Schwendemann R, Ferenczy S, et al. Hand-held spectral domain optical coherence tomography of retinoblastoma in infants. Submitted for publication [Google Scholar]
- 15.Shields CL, Benevides R, Materin MA, Shields JA. Optical coherence tomography of retinal astrocytic hamartoma in 15 cases. Ophthalmology. 2006;113:1553–7. doi: 10.1016/j.ophtha.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Serafino M, Pichi F, Giuliari GP, Shields CL, Ciardella A, Nucci P. Retinal astrocytic hamartoma: Spectral domain optical coherence tomography classification and correlation with tuberous sclerosis complex. J AAPOS. 2012;17:e27. doi: 10.1097/IAE.0000000000000829. [DOI] [PubMed] [Google Scholar]
- 17.Veronese C, Pichi F, Shields CL, Ciardella A. Cystoid changes within astrocytic hamartomas of the retina in tuberous sclerosis. Retin Cases Brief Rep. 2011;5:113–6. doi: 10.1097/ICB.0b013e3181c59959. [DOI] [PubMed] [Google Scholar]
- 18.Singh AD, Shields CL, Shields JA. Major review: Von Hippel-Lindau disease. Surv Ophthalmol. 2001;46:117–42. doi: 10.1016/s0039-6257(01)00245-4. [DOI] [PubMed] [Google Scholar]
- 19.Shields CL, Mashayekhi A, Ho T, Cater J, Shields JA. Solitary congenital hypertrophy of the retinal pigment epithelium: Clinical features and frequency of enlargement in 330 patients. Ophthalmology. 2003;110:1968–76. doi: 10.1016/S0161-6420(03)00618-3. [DOI] [PubMed] [Google Scholar]
- 20.Shields CL, Materin MA, Walker C, Marr BP, Shields JA. Photoreceptor loss overlying congenital hypertrophy of the retinal pigment epithelium by optical coherence tomography. Ophthalmology. 2006;113:661–5. doi: 10.1016/j.ophtha.2005.10.057. [DOI] [PubMed] [Google Scholar]
- 21.Fung AT, Pellegrini M, Shields CL. Congenital hypertrophy of the retinal pigment epithelium: Enhanced-depth imaging optical coherence tomography in 18 cases. Ophthalmology. 2014;121:251–6. doi: 10.1016/j.ophtha.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Shields CL, Shields JA, Marr BP, Sperber DE, Gass JD. Congenital simple hamartoma of the retinal pigment epithelium: A study of five cases. Ophthalmology. 2003;110:1005–11. doi: 10.1016/S0161-6420(03)00087-3. [DOI] [PubMed] [Google Scholar]
- 23.Shields CL, Materin MA, Karatza EC, Shields JA. Optical coherence tomography of congenital simple hamartoma of the retinal pigment epithelium. Retina. 2004;24:327–8. doi: 10.1097/00006982-200404000-00031. [DOI] [PubMed] [Google Scholar]
- 24.Shukla D, Ambatkar S, Jethani J, Kim R. Optical coherence tomography in presumed congenital simple hamartoma of retinal pigment epithelium. Am J Ophthalmol. 2005;139:945–7. doi: 10.1016/j.ajo.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 25.Schachat AP, Shields JA, Fine SL, Sanborn GE, Weingeist TA, Valenzuela RE, et al. Combined hamartomas of the retina and retinal pigment epithelium. Ophthalmology. 1984;91:1609–15. doi: 10.1016/s0161-6420(84)34094-5. [DOI] [PubMed] [Google Scholar]
- 26.Shields CL, Thangappan A, Hartzell K, Valente P, Pirondini C, Shields JA. Combined hamartoma of the retina and retinal pigment epithelium in 77 consecutive patients visual outcome based on macular versus extramacular tumor location. Ophthalmology. 2008;115:2246–2252.e3. doi: 10.1016/j.ophtha.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Shields CL, Mashayekhi A, Dai VV, Materin MA, Shields JA. Optical coherence tomographic findings of combined hamartoma of the retina and retinal pigment epithelium in 11 patients. Arch Ophthalmol. 2005;123:1746–50. doi: 10.1001/archopht.123.12.1746. [DOI] [PubMed] [Google Scholar]
- 28.Huot CS, Desai KB, Shah VA. Spectral domain optical coherence tomography of combined hamartoma of the retina and retinal pigment epithelium. Ophthalmic Surg Lasers Imaging. 2009;40:322–4. doi: 10.3928/15428877-20090430-19. [DOI] [PubMed] [Google Scholar]
- 29.Arepalli S, Pellegrini M, Shields CL, Shields JA. Combined hamartoma of the retina and retinal pigment epithelium. Findings on enhanced depth imaging optical coherence tomography (EDI-OCT) in 9 eyes. Retina. 2014 doi: 10.1097/IAE.0000000000000220. In press. [DOI] [PubMed] [Google Scholar]
- 30.Shields JA, Shields CL, Gündüz K, Eagle RC., Jr Neoplasms of the retinal pigment epithelium: The 1998 Albert Ruedemann, Sr, memorial lecture, Part 2. Arch Ophthalmol. 1999;117:601–8. doi: 10.1001/archopht.117.5.601. [DOI] [PubMed] [Google Scholar]