

Abstract
Defects in the Lhx4, Lhx3, and Pitx2 genes can cause combined pituitary hormone deficiency and pituitary hypoplasia in both humans and mice. Not much is known about the mechanism underlying hypoplasia in these mutants beyond generally increased cell death and poorly maintained proliferation. We identified both common and unique abnormalities in developmental regulation of key cell cycle regulator gene expression in each of these three mutants. All three mutants exhibit reduced expression of the proliferative marker Ki67 and the transitional marker p57. We discovered that expression of the cyclin-dependent kinase inhibitor 1a (Cdkn1a or p21) is expanded dorsally in the pituitary primordium of both Lhx3 and Lhx4 mutants. Uniquely, Lhx4 mutants exhibit reduced cyclin D1 expression and have auxiliary pouch-like structures. We show evidence for indirect and direct effects of LHX4 on p21 expression in αT3-1 pituitary cells. In summary, Lhx4 is necessary for efficient pituitary progenitor cell proliferation and restriction of p21 expression.
Regulation of the eukaryotic cell cycle is studied in numerous fields of molecular and cellular biology. The basic aspects of cell division and growth control are fundamental not only for the physiological development of a multicellular organism but also for tissue maintenance and pathological situations, such as tumorigenesis. Improper development of the major endocrine center can affect the survival of the organism, even in very ancient forms of life (1). The pituitary is a critical endocrine gland in vertebrates because it is responsible for the production of critical hormones for growth, reproduction, homeostasis, and stress response, which include GH, prolactin, LH, FSH, TSH, and ACTH, and it integrates complex signaling and feedback pathways (2).
Height is a product of environmental and genetic factors, and early-onset abnormal height is a common reason for referral to pediatric endocrinologists (3). Sporadic GH deficiency is the most frequently diagnosed pituitary hormone deficiency, and it has an estimated prevalence up to 1 of 4000 live births in humans (4, 5). Up to 4% of the these sporadic GH deficiency cases arise from identified genetic defects (6). In combined or multiple pituitary hormone deficiencies the percentage of known genetic causes is proportionally higher, yet most individual patients, including early-onset and familial cases, are of unknown cause (7).
Many genetic cases of combined and isolated pituitary hormone deficiency in humans and mice are caused by defects in transcription factor (TF) genes. Pitx2, Pitx1, Sox2, Sox3, Hesx1, Otx2, Lhx4, Lhx3, Tpit, Gli2, Prop1, and Pou1f1 are all critical for the formation of a fully functional pituitary gland (2, 8). Combined and isolated pituitary hormone deficiencies are frequently coupled with hypoplasia of the gland (7, 9–13). The hypoplasia in global knockout mouse models of Pitx2, Lhx3, and Lhx4 is attributed to both increased apoptosis and reduced proliferation detected by TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling staining) and BrdU (5-bromo-2′-deoxyuridine labeling), respectively (9, 11, 13–16). Little is known about how these transcription factors regulate proliferation and/or the cell cycle.
There are many examples of organ hypoplasia that are caused by TF mutations in mouse and man. For example, renal hypoplasia results from Trp53 and Pax2 deficiencies (17, 18), and numerous TFs are required for the growth of midline structures in the central nervous system, eg, zinc finger protein 423 (Zfp423) for the cerebellum (19) and empty spriacles homeobox protein-1 (Emx1), empty spriacles homeobox protein-2 (Emx2), homeobox gene expressed in ES cells 1 (Hesx1), nuclear factor 1 A type (Nfia), paired box transcription factor 6 (Pax6), ventral anterior homeobox 1 (Vax1), and tailless (Tlx) for the corpus callosum (reviewed in reference 20). Similarly, hypoplasia of the anterior segment of the eye can be caused by deficiency in Pax6, paired-like homeodomain transcription factor 2 (Pitx2), and forkhead box C1 (Foxc1) (reviewed in reference 21). The same is true for endocrine organs. For example, complex pharyngeal, thymus, and parathyroid gland hypoplasia can originate from the loss of T-box TF genes (Tbx1, Tbx2, or Tbx3) (22). Adrenal hypoplasia congenita occurs in patients with nuclear receptor subfamily 0, group B, member 1 (Nrb0b1 also known as Dax1) mutations (23, 24), and pancreas hypoplasia is caused by mutations in pancreas specific transcription factor, 1a (Ptf1a), hairy and enhancer of split 1 (Hes1), and motor neuron and pancreas homeobox 1 (Mnx1) (reviewed in reference 25). Thyroid, pituitary, and lung hypoplasia are profound when the NK2 homeobox 1 (Nkx2.1) gene is defective (26, 27), and Nkx2.5, Pax8, Foxe1, and hematopoietically expressed homeobox (Hhex) mutations can cause thyroid hypoplasia through apoptotic progenitor cell death (reviewed in reference 28). The downstream target genes that are responsible for organ growth are largely unknown. Understanding how transcription factor deficiencies cause organ hypoplasia is a fundamental question in organogenesis.
The hypoplasia characteristic of Pitx2−/−, Lhx4−/−, and Lhx3−/− global knockout mice is evident at early stages of embryonic pituitary development, consistent with the normal temporal and spatial expression of these genes (15, 16, 29, 30). Pitx2 is expressed as early as mouse embryonic day (E) 8.5 in the area of the oral ectoderm that invaginates to produce Rathke's pouch (RP), and it remains expressed through postnatal life (31). Lhx4 is expressed from E9.5 with peak expression between E10.5-E12.5, followed by a lower level until approximately E15.5 (15, 30). Between E9.5 and E12.5, Lhx3 expression follows a pattern similar to Lhx4, but it is expressed at a higher level during postnatal life (15). The absence of each of these transcription factors leads to lethality caused by developmental failure of vital organs. Pitx2−/− die due to heart defects at E12.5, and there are many other anomalies including ventral body wall closure defects, lung isomerism, gastric malrotation, and eye defects (29). Death within 24 hours after birth in Lhx3−/− and Lhx4−/− mice is attributed to ventral motor neuron defects that impair respiratory movements, and Lhx4−/− mice also have lung hypoplasia (16, 30).
Traditionally the cell cycle is divided into four major parts in which the gap phases (G1 and G2) separate the mitotic (M) phase from the phase in which DNA duplication/synthesis occurs (S phase) (32). The expression of specific cyclin (Ccn) and cyclin-dependent kinase (Cdk) genes regulates progression through the phases of the cell cycle. At the transition from one phase to the next, checkpoint mechanisms safeguard the completion of the phase, eg, the proper duplication of the DNA after the S phase. The decision to progress through the cell cycle to produce two daughter cells is made at the G1/S phase. At this checkpoint the retinoblastoma (Rb) protein becomes phosphorylated, which releases it from a complex with the E2F transcription factor, and it remains in this state until the next G1 phase (33). During early to mid-G1, the predominant cyclins are the D-type cyclins, which form complexes with cyclin-dependent kinase (CDK)-4 and CDK6 (34). These Cdks are inhibited by their Cdk inhibitors (Cdkns) as Cdkn1a (p21WAF1/CIP1), Cdkn1b (p27KIP1), Cdkn1c (p57KIP2), Cdkn2a (INK4A, p16), Cdkn2b (INK4B, p15), Cdkn2c (INK4C, p18), Cdkn2d (INK4D, p19) and Cdkn3 (35, 36). In late G1 the E-type cyclins and CDK2 cause Rb phosphorylation and stimulate the transition to S phase (37). Many signaling pathways were shown to affect cell cycle regulation (38).
The cell cycle is dynamically regulated during anterior pituitary gland development (39). As progenitors leave the cell cycle, they express CCNE (also known as cyclin E) and the cell cycle inhibitor p57. Subsequently, cells differentiate and are inhibited from proliferation by expression of p27. Some transcription factor mutations disrupt this aspect of cell cycle regulation. For example, pituitaries deficient in the T-box transcription factor Tpit exit the cell cycle and are arrested at the transition between p57 and p27 expression, and corticotrope differentiation fails. This example provides a precedent that supports the hypothesis that transcription factor mutations cause pituitary hypoplasia through the misregulation of the cell cycle.
We tested this idea in mice deficient in the LIM (Lin-11, Isl-1, Mec-3)-type homeodomain TF proteins LHX4 and LHX3 and the paired-like homeodomain TF PITX2 by examining the temporal and spatial expression of cell cycle markers. Lhx3, Lhx4, and Pitx2 are critical for the early growth of RP, and we found altered cell cycle expression in each of these mutants. Our findings have advanced our understanding of the link between transcription factor deficiencies, abnormal regulation of the cell cycle, and reduced proliferation that underlie clinical causes of growth insufficiency in humans.
Materials and Methods
Experimental animals, sample collection, and tissue processing
The University of Michigan University Committee on Use and Care of Animals approved the animal care and use protocols. Mice were housed in specific pathogen-free conditions in ventilated cages with automatic watering. Mice were fed 5020 chow (PMI Nutrition International). Pitx2 mice were from our own stock (29), and the Lhx4 and Lhx3 mice were originally provided by Steven Potter (Cincinnati Children's Hospital, Cincinnati, Ohio) (30) and Heiner Westphal (National Institutes of Health, Bethesda, Maryland) (16), respectively. We maintained these stocks with occasional backcrosses to C57BL/6J mice. The original genetic backgrounds were as follows: Pitx2tm2Sac: (129×1/SvJ × 129S1/Sv)F1-Kitl+; for Lhx4tm1Ssp: 129S2/SvPas CF1; and for Lhx3tm1Lmgd: 129S4/SvJae.
For each genotype, heterozygous mutant mice were mated and checked for copulation plugs in the morning (E0.5). Embryos were collected on each day of the pregnancy from E9.5 until E15.5. At least three embryos of each genotype were analyzed at each time point. Mice were genotyped from a tail tissue sample or yolk sac DNA as described previously (13, 30).
Dissected embryos were fixed in 4% formaldehyde for 0.5–2 hours, depending on the size of the embryo and embedded in paraffin. Five-micrometer-thick sections were placed onto frosted slides. At least two midsagittal sections from two different embryos were selected/processed for immunohistochemistry or in situ hybridization at each representative embryonic day. Parasagittal sections from the same embryos containing RP or more developed pituitary structures were used as negative controls.
Immunohistochemistry (IHC) and in situ hybridization
For IHC, samples were boiled for 10–15 minutes in 0.01 M citric acid (pH 6.0) for antigen retrieval. The Tyramide Signal Amplification fluorescein kit was used for blocking and signal amplification according to the manufacturer's instructions (PerkinElmer). Endogenous peroxidase activity was blocked with a solution of 3% hydrogen peroxide, 47% phosphate buffered saline, and 50% methanol for 20 minutes. The following antibodies were used: antibodies raised in mouse: anti-cyclin D1 (dilution of 1:200; Santa Cruz Biotechnology; number sc-8396), anti-p21 (1:200; BD Pharmingen; number 556431), anti-LHX3 (1:1000; Developmental Studies Hybridoma Bank at the University of Iowa, Iowa City, Iowa; number 67.4E12), anti-ISL1 (1:600; Developmental Studies Hybridoma Bank; number 40.2D6); rabbit antibodies: anti-cyclin D2 (1:200; Santa Cruz Biotechnology; number sc-593), anti-cyclin E (1:100; Santa Cruz Biotechnology; number sc-481), anti-Ki67 (1:200; Novocastra Laboratories-Leica Microsystems; number NCL-Ki67p), anti-p57 (1:200; Thermo Fischer Scientific; number RB-1637-P0), anti-phospho-histone H2A.X (Ser139) (1:250; Cell Signaling Technology; number 9718), antichoriogonadotropin-α (CGA) (1:300; National Institute of Diabetes and Digestive and Kidney Diseases, Torrance, California; number AFP-6619986). Negative control slides did not have any primary antibody added during the IHC procedure. Five percent of normal donkey serum was used in blocking reagent with p21 staining procedure (40). Secondary, biotinylated, antimouse or antirabbit antibodies and normal donkey serum were purchased from Jackson ImmunoResearch Laboratories (1:100).
For antibodies generated in mouse the M.O.M. immunodetection kit was used according to the company's recommendations (Vector Laboratories). DAPI (Diamidino-2-phenylindole) was used for nuclear counterstaining. The Leica DMRB microscope (Leica Microsystems) was used with a Leica EL6000 light source, a Retiga 2000R digital camera, and Q Capture Pro 6.0 software (QImaging). Slides were photographed using two different light filter cubes: for DAPI (Leica Microsystems A4) and for fluorescein (Leica Microsystems L5). Slides were also checked for autofluorescence with Leica Microsystems N3 filter cube and captured when a signal was seen. Acquired images were overlaid. Composite figures in the paper were compiled with Adobe Illustrator CS6 (Adobe Systems).
For in situ hybridization, we used digoxigenin-labeled antisense Pitx2 riboprobes described elsewhere (41) with a standard alkaline phosphatase activity detection technique. We used the Pitx2 sense riboprobe for negative control slides. Hybridization temperature was 51°C–54°C. The detailed protocol can be found in the Supplemental Materials and Methods.
Computational analysis of the murine p21 gene
We used Vista Genome Browser 2.0 for the analysis of genomic DNA sequence homology searches with default settings (100 bp window and 75% sequence identity for conservation cutoff level; sequence releases for mouse July 2007, for human February 2009, for rhesus January 2006, for dog May 2005, for horse January 2007, for rat November 2004, and for chicken May 2006) (42). The clusters of conserved elements (CEs) were cloned from mouse genomic DNA (gDNA) in three segments, and the nucleotide sequence position was defined relative to the transcription start site initiating in exon 1a [ENSMUST00000023829 transcript at www.ensembl.org or NM_007669.4 at GenBank at the National Center for Biotechnology Information (NCBI), Bethesda, Maryland]. The coordinates and sizes of the three cloned segments of conserved elements are: CE 1, −3224/−1696 (1528 bp); CE 2, +595/+1906 (1311 bp); CE 3, +3,283/+4,495 (1212 bp). We designed primers to amplify these CEs together with approximately 100 bp of upstream and downstream sequences using the NCBI-Primer BLAST software (http://www.ncbi.nlm.nih.gov/tools/primer-blast/), with special attention to avoid repeat rich regions (CE1, forward, TCCTGACCCTCGTGCTTAGACCAT, reverse, ATCCCGGCACTCAGGAGACAGA; CE2, forward, AGCGCAGAGCGGTTCTCCGA, reverse, GCCTAGCCGGCCTTGCAGTC; CE3, forward, GCCACTGGGGCTCACCTTGC, reverse, GCACCCCAAGGTCACGGGTG). We used the RestrictionMapper version 3.0 (http://www.restrictionmapper.org/) for a restriction enzyme cleavage site analysis.
Cloning
The mouse Lhx4 cDNA in the pcDNA3.1/myc-His C was a kind gift of Simon Rhodes (Indiana University, Indianapolis, Indiana) (43). The Cdkn1a conserved noncoding DNA sequence elements (CEs) were PCR amplified with GoTaq DNA polymerase (Promega) from Lhx4+/+ mouse gDNA. The CEs were sequenced with the Sanger method using multiple primers, compared with the Ensembl sequence database, TA cloned into the TOPO2.1 vector, and transformed into Escherichia coli DH5α-E (both from Life Technologies-Invitrogen). Inserts were subcloned with the InFusion HD cloning system (Clontech) into the pGL3-promoter firefly luciferase reporter plasmid (Promega) between the restriction enzyme cut sites KpnI-XhoI (CEs 1 and 3) or KpnI-NheI (CE2). Restriction enzymes were purchased from New England Biolabs, and high-fidelity enzymes were used where applicable. Final subclones were transformed into E coli Stellar chemically competent cells (Clontech). Bacteria were plated on Luria-Bertani agar plates containing the selective drug ampicillin (Sigma-Aldrich). The plasmid DNAs were purified using the Miniprep and Plasmid Maxi Kits (QIAGEN). The final plasmid DNA sequences were confirmed by the Sanger method.
Cell culture and transfection
We used the mouse αT3-1 pituitary pregonadotrope cell line, provided by Pamela Mellon (University of California, San Diego, San Diego, California) for functional studies (44). Cells were plated at 3.5 × 105 cells/well in 12-well cluster plates 24 hours before transfection in DMEM supplemented with 584 mg/L glutamate, 4.5 g/L D-glucose, 110 mg/L sodium pyruvate, and 10% heat-inactivated fetal bovine serum (Life Technologies Gibco; number 11995-065 and number 16140-071). The Promega FuGene6 transfection reagent was used with a 3:1 Fugene6 to DNA ratio (FuGene6 amount in microliters, DNA in micrograms) according to the manufacturer's instructions. The cells were cotransfected with the pRL-SV40 renilla luciferase plasmid (Promega) as an internal control. The mass ratio of renilla to total DNA was 1:25. The −480/+43 choriogonadotropin-α (Cga)-pA3-luc and pA3 plasmids were used as positive controls for showing the activation effect of LHX4 (43, 45). Eight hundred nanograms per well of DNA were used. The firefly luciferase reporter plasmid amount was 512 ng in all applicable cases, and 256 ng of the Lhx4 plasmid was used where indicated. The pcDNA3.1− plasmid was added to keep the total DNA amount constant in all wells used for transfection. The cells were incubated for 48 hours at 37°C in 5% CO2-supplemented air. The experiment was performed in triplicate and repeated on 3 separate days.
Ten 100-mm culture dishes were plated with 6.0 × 106 αT3-1 cells each. Five dishes were transfected with 11 μg Lhx4 plasmid using FuGene6, and five were not transfected. Four plates were used for preparing nuclear extracts and one plate was used for the confirmation of Lhx4 expression.
Reporter activity assay and statistical analysis
The transfected cells were assayed using the dual-luciferase reporter assay system and read on a GloMax-96 microplate luminometer (Promega). The firefly to renilla luminescence ratios were averaged for triplicate wells of each experiment. We considered the background activity of the firefly reporter on its own and the effect of the LHX4 expression vector on the pGL3-promoter plasmid backbone without the conserved element (46). The results from independent experiments were tested for normality and homogeneity with Shapiro-Wilk's and Levene's tests, respectively. One-way ANOVA was used with a Scheffe post hoc test (IBM SPSS Statistics 19.0; IBM SPSS Statistics). The significance level was set to P ≤ .05. Graphs were prepared with MS-Excel 2010 (Microsoft).
Semiquantitative detection of p21 transcript variants with PCR
A confluent, 100-mm culture dish of αT3-1 cells (passage 10) was treated with 0.05% trypsin-EDTA (Life Technologies-Invitrogen) and centrifuged at 100 × g for 5 minutes. After removing excess media, cells were resuspended in RNAlater (Life Technologies-Ambion) and frozen at −80°C. Whole pituitary and large intestine tissues were dissected from a wild-type (C57BL/6J), 10-week-old female mouse after a cervical dislocation. Tissues were preserved in RNAlater at −80°C until processed. RNA was isolated with the RNAqueous-4PCR kit including subsequent deoxyribonuclease treatment (Life Technologies-Ambion). The SuperScript II first-strand synthesis system was used for reverse transcription with oligo dT12–18 primers (Life Technologies-Invitrogen). RNA and DNA were measured with NanoDrop (Thermo Fischer Scientific).
Primers were designed to amplify only one transcript variant. Primer sequences are as follows: v1_forward, CGGTGTCAGAGTCTAGGGGA, v1_reverse, AGGATTGGACATGGTGCCTG; v2_forward, TGGGGTAAACAGGACGGTGA, v2_reverse, CAGGTGCTTTTCCACCACAC; and v3_forward, ACTACCAGCTGTGGGGTGAG, v3_reverse, TCGGACATCACCAGGATTGG (Integrated DNA Technologies). Product sizes were 88, 116, and 125 nucleotides, respectively. Twenty nanograms of cDNA were used in each reaction with GoTaq (Promega). The thermocycle program was as follows: 95°C for 2 minutes and then 30 cycles of 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 30 seconds followed by a final extension step of 72°C for 5:00 minutes. PCR products were visualized on a 1.5% agarose gel stained with ethidium bromide and were verified by DNA sequence analysis. The 1-kb Plus DNA ladder was used as a reference for molecular weight (Life Technologies-Invitrogen). The gel images were captured using the UVP BioDocIt system (UVP).
Electrophoretic mobility shift assay
For the detection of LHX4 protein binding to the described conserved elements in and around the p21 gene, we used the LightShift chemiluminescent EMSA kit (Thermo Pierce Biotechnology) according to the manufacturer's instructions. We designed primer pairs internal to p21 CE1 and CE2. CE2 fragments were as follows: number 1 (forward-reverse), CAGAGCGGTTCTCCGATCC-TGTCACAATGAGTCACCTCCTC;number 2, CGAGGAGGTGACTCATTGTGAC-AACATACTGTGCCCGCCAAATA; number 3, TATTTGGCGGGCACAGTATGTT-GGTGGGTGGGACCCTTTG; number 4, CAAAGGGTCCCACCCACC-CTGGTCACCTTCCTACACTGG;number 5, CACCCAGTGTAGGAAGGTGAC-TGAGTGTCCTCTCTGAAACGC; number 6, CTACGTCGCGTTTCAGAGAGG-GAAAGTGCTCTTAGCTCTGGC; number 7, CGTGGAGATCAAGGTGGAGG-GGCTTCCTAAATTCCCGCCTA; number 8, ATAGGCGGGAATTTAGGAAGCC-AAGGAGTGGTGAGTCAGTTTCC; and number 9, TCCGGTGCCCAAGCAGTTTT-GCCTAGCCGGCCTTGCAGTC. DNA fragment sizes were 182, 183, 159, 180, 191, 142, 196, 165, and 154 bp, respectively. We used PCR for generating the DNA pieces with wild-type mouse gDNA, applying the same thermal parameters as described in the previous paragraph. Unlabeled primer pairs were used for generating the competitor DNA fragments, whereas labeled DNA was generated using 5-prime biotinylated forward and unlabeled reverse oligonucleotides (Integrated DNA Technologies). All PCR products were gel purified with QIAquick gel extraction kit (QIAGEN) and quantified with Nanodrop 2000 (Thermo Fischer Scientific). All fragments were confirmed with bidirectional Sanger sequencing.
Nuclear extracts were prepared from the transfected and untransfected αT3-1 cells after 48 hours of incubation using the NE-PER kit (Thermo Pierce Biotechnology), aliquoted, and stored at −80ºC until use. The bicinchoninic acid (BCA) protein assay kit (Thermo Pierce Biotechnology) was used with an Eppendorf biophotometer for measuring the protein concentration.
Modified parameters for performing the EMSA with the LightShift kit (Thermo Pierce Biotechnology) are briefly outlined here. Twenty femtomoles of labeled DNA, 8 pM of unlabeled competitor DNA (400-fold molar excess), and 2.82 μg of LHX4-transfected or untransfected nuclear extracts (NEs) was used. The DNA strand length corrected molarity of the DNAs was calculated with a web tool (http://molbiol.edu.ru/eng/scripts/01_07.html). Ten microliters of each EMSA sample were run on nondenaturing 5% polyacrylamide, mini-Protean Tris borate-EDTA gels with 15-well comb and 15 μl sample space in Mini-Protean II gel electrophoresis apparatus (Bio-Rad Laboratories, Hercules, CA) for 40 minutes at 160 V in filtered 0.5× Tris borate-EDTA buffer at +4ºC. Gels were blotted to positively charged Amersham Hybond-N+ nylon membrane (GE Healthcare) with Bio-Rad Laboratories Transblot SD semidry transfer cell for 1 hour at 20 V and then UV cross-linked with Stratalinker 2400 (Stratagene) with default settings (120 mJ/cm2, 1 min). Chemiluminescent detection was performed in Slimline autoradiography cassette (Denville Scientific) with Kodak BioMax XAR film (Carestream).
Results
Altered cell cycle marker expression in Pitx2−/−, Lhx3−/−, and Lhx4−/− pituitaries
Detachment of RP from the oral ectoderm occurs around E11.5 between somite stages 16 and 18 (47). We initiated an assessment of cell cycle marker expression patterns at this time and included cyclin D1, p21, cyclin D2 (Figure 1), p57, and Ki67 (Figure 2). D-type cyclins have similar functions, but their tissue specific expression is thought to permit regulation of proliferation in different cell types (48). Cyclin D1 expression is normally enriched in the dorsal aspect of RP and absent from the closure site and oral ectoderm, and the expression pattern is similar in Pitx2−/− and Lhx3−/− mutants (Figure 1 A, D, and G). The most striking change is the near absence of cyclin D1 expression throughout most of RP in Lhx4−/− mice (Figure 1J). Cyclin D1 is detected in abnormal, additional RP invaginations in the Lhx4−/− mice and only a few cells within the pouch (Figure 1J, white arrows). These extra invaginations were not described before. Normal p21 expression contrasts with cyclin D1 in that it is enriched in the ventral aspect of RP. This mutually exclusive pattern of cyclin D1 and p21 expression is present in wild-type, Pitx2−/−, Lhx3−/−, and Lhx4−/− mutants, but the border between the expression domains is shifted dorsally in Pitx2−/−, Lhx3−/− mutants (compare Figure 1, A and B, D and E, and G and H). In Lhx4−/− mice, p21 is expressed in all parts of RP except the abnormal invaginations, which express cyclin D1 (Figure 1, K and J, and their inserts). Thus, the mutants all preserve the mutually exclusive pattern of cyclin D1 and p21 expression, but the boundary shifts dorsally in the mutants. Changes in cyclin D1 and p21 expression are more extreme in Lhx4−/− mutants than in Pitx2−/− or Lhx3−/−. Cyclin D1 expression is slightly reduced throughout the small RP in Pitx2−/−. Cyclin D2 is expressed throughout RP and in the oral ectoderm, and its expression is not obviously changed in any of the three mutants (Figure 1, C, F, I, and L).
Figure 1.

Abnormal cell cycle marker gene expression in mutants with pituitary hypoplasia. Midsagittal sections from wild-type (+/+), Pitx2−/−, Lhx3−/−, and Lhx4−/− embryos (E11.5) were immunostained for the cell cycle regulators Cyclin D1, p21, and cyclin D2. Arrows and insets show clusters of cells (additional invaginations) extruded from the main RP of Lhx4 mutants (panels J–L). Brackets indicate immunopositive areas. Original magnification is the same for all panels. Scale bar, 100 μm. Embryos are facing toward the right.
Figure 2.
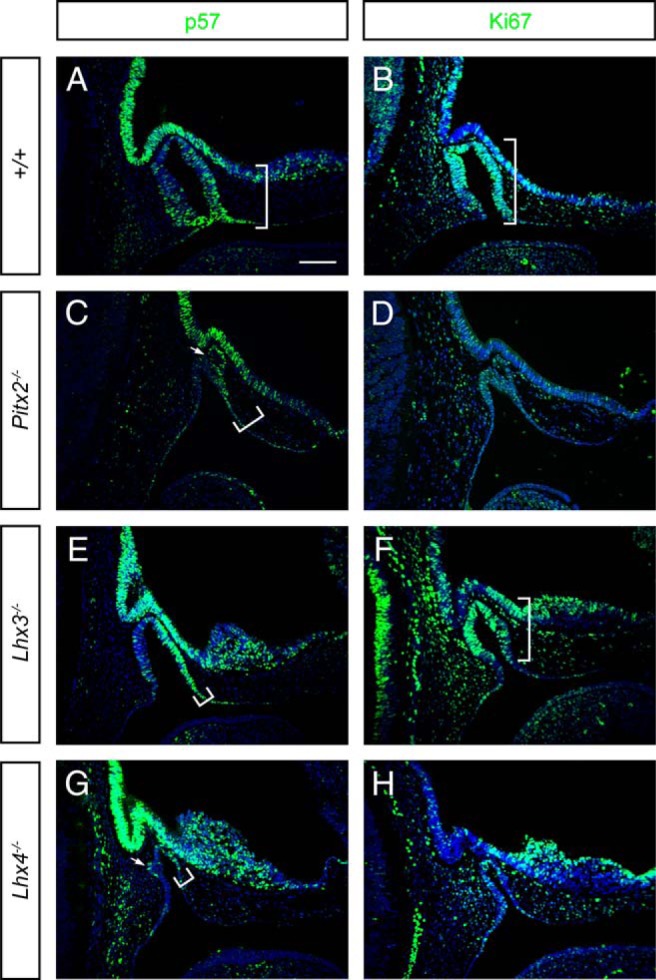
Altered p57 and Ki67 expression in embryonic pituitaries from mice with pituitary hypoplasia. Mouse embryonic pituitaries collected at E11.5 from normal and mutant Pitx2, Lhx3, and Lhx4 mouse embryos were fixed, embedded, sectioned in the midsagittal plane, and immunostained for p57 and the proliferation marker Ki67. The white arrow marks an abnormal invagination at the side of Rathke's pouch. White brackets show the extent of the representative immunostaining. Scale bar, 100 μm. Embryos are facing to the right.
p57 is an inhibitor in the G1 phase of the cell cycle. Mice missing p57 display anterior lobe hyperplasia, midline defects (cleft palate, omphalocele), and eye lens, bone, kidney, adrenal cortex abnormalities with proliferation changes and increased cell death (39, 49). p57 expression is similar to p21 in that it is enriched at the ventral aspect of RP in which the first differentiated cells emerge. p57 expression shifts dorsally and rostrally in all three mutants (Figure 2). It is intriguing that p57 is asymmetrically expressed in Pitx2, Lhx3, and Lhx4 mutants. Expression is well balanced on the rostral and caudal sides of the pouch in normal mice, but it is enriched rostrally in the mutants and present in the additional invaginations of Lhx4−/− mutants.
Ki67, which marks cells active in the cell cycle, is reduced in all mutants, particularly in Pitx2−/− and Lhx4−/− mice (Figure 2). Thus, all three mutants have fewer cycling cells and alterations in expression of cell cycle regulators cyclin D1, p21, and p57. We focused a more detailed analysis on Lhx4−/− mice because they exhibited the most extreme changes in cell cycle marker expression patterns.
p21 is expressed in a mutually exclusive pattern with cyclin D1 throughout early pituitary development
At E11.5 Lhx4 mutants already exhibited RP hypoplasia and abnormal morphology. We undertook a study of p21 and cyclin D1 expression from early development, E9.5 through E14.5. In wild-type mice, p21 expression is detected at E9.5 at the dorsal epithelial layer of the first pharyngeal arch, and at E10.5 it is present at the caudal lip of the pouch closure site. It remains restricted to this thin margin and the adjacent area of the oral cavity until E11.5 (Figure 3, first row). There is little or no expression at E12.5 or E13.5. The initial onset and location of initial p21 expression in Lhx4−/− mice is the same, but the expression persists abnormally throughout the whole RP at Ell.5 rather than being restricted to the ventral aspect. In addition, p21 expression persists through E13.5, 2 days longer than in wild-type mice (Figure 3 second row). p21 is not detectable at E14.5 (data not shown). Cyclin D1 is readily detectable in wild-type mice at E10.5 and persists through E13.5. It is initially enriched on the rostal lip of the pouch and becomes predominant in the dorsal aspect of the pouch and throughout the prospective intermediate lobe at E13.5, coincident with the most active sites of cell proliferation. The Lhx4 mutant expresses cyclin D1 at E9.5, a day earlier than wild type. From E10.5 through E13.5 the overall cyclin D1 expression is significantly reduced relative to normal mice; it is expressed only in the small, ectopic invaginations that extend from the major aspect of the mutant pouch. Cyclin D1 and p21 expression are normally mutually exclusive in RP at E10.5 and E11.5 (Figure 3). Although this exclusivity exists to a certain extent in Lhx4 mutants, they obviously lack dorsoventral patterning. Thus, the absence of Lhx4 causes multiple defects in cell cycle regulation.
Figure 3.

p21 and cyclin D1 expression are mutually exclusive in developing pituitary gland. Midsagittal sections of wild-type and Lhx4 mutant embryos collected at E9.5-E13.5 were immunostained for p21 and cyclin D1. White arrows indicate ectopic invaginations from RP. Magenta arrows indicate onset of p21 expression along the oral ectoderm at E10.5. Scale bar, 100 μm. Embryos are facing to the right. PL, posterior lobe.
Transitional cell marker cyclin E altered in Lhx4−/−
Cyclin E expression marks cells that progress to the late G1 phase and pass the G1 to S transition point (50). In the pituitary gland, the cyclin E expressing cells are located between the zone of most active proliferation and the zone of differentiating cells at E14.5 (39). At E13.5 the anterior lobe (AL) and the adjacent intermediate lobe (IL) structures express cyclin E at a higher level than at E14.5. The Lhx4−/− mice have reduced cyclin E expression at all ages examined (E13.5-E14.5), consistent with the presence of fewer transitional cells and differentiating cells (Supplemental Figure 1). Cyclin E is detected in the additional invaginations. Cyclin E was either undetectable or showed cytoplasmic staining between E9.5 and E12.5 (data not shown). A few cells in the IL stain with cyclin E staining in wild-type RP at E13.5, a time when cyclin D1 and cyclin D2 staining are both strong (Supplemental Figure 1). This is consistent with the later differentiation of IL cells than AL cells in normal mice.
Rathke's pouch cells undergo DNA fragmentation, apoptosis, and delayed cell specification
In Lhx4−/− embryos, cells throughout the rudimentary RP undergo programmed cell death (9). We examined phospho-histone H2A.X (Ser139) (pH2A.X) staining to determine whether the extensive expression of the G1 phase inhibitor p21 is a sign of senescence and/or premature aging during pituitary progenitor proliferation (51). pH2A.X marks only a few cells at the closure site of RP in normal mice, but it marks cells throughout RP in the Lhx4 mutants (Supplemental Figure 2). No pH2A.X staining is detectable in the extra invaginations unique to the mutants. The pH2A.X staining is similar to the previous reports of apoptotic cells at the normal point of RP separation at E10.5-E11.5 in normal mice and throughout the pouch in the mutants (9). To support or reject earlier terminal differentiation in Lhx4 mutants, we stained for the first pituitary hormone marker, CGA, the α-subunit common to LH, FSH, TSH, and choriogonadotropin (52, 53). We detected one to two CGA-positive cells at E11.5 in the wild-type pouch, an increasing number at E12.5, and robust, widespread staining at E13.5–14.5. In contrast, the first CGA-positive cells were detected at E14.5 in the ectopic invaginations in Lhx4 mutants, several days later than in normal mice (Supplemental Figure 3). Thus, Lhx4 deficiency promotes expression of the senescence marker pH2A.X, but it is associated with cell death, not premature cell differentiation.
Lhx4 deficiency affects LHX3 and ISL1 expression during pituitary development
We analyzed the expression of LIM and paired-homeodomain family transcription factors to explore whether expression of any of these genes were altered by LHX4 deficiency (11, 15). LHX4, LHX3, and ISL1 are members of the broader protein group of the LIM-type homeodomain transcription factors (54, 55). There is overlapping expression and function of Lhx3 and Lhx4 in Rathke's pouch and the anterior lobe, suggesting that LHX3 might be up-regulated in a compensation for the LHX4 deficiency. The hypocellular pituitaries of Lhx3−/− and Lhx4−/− are similar to those of Isl1−/− and Pitx2−/− (11, 31, 56). We did not observe any increase in LHX3 expression in LHX4 mutant RP. Instead, LHX3 expression was delayed and confined to the additional invaginations (Figure 4). In wild-type mice, LHX3 is readily detectable throughout RP and the developing AL and IL from E9.5 through E14.5. In contrast, LHX3 was detected in only one to two cells at E10.5, and a few cells at the dorsal aspect of RP at E11.5. At later stages, E12.5-E14.5, LHX3 expression increased in Lhx4 mutants, but it was never observed throughout RP.
Figure 4.

LIM-homeodomain genes Lhx3 and Isl1 do not compensate for Lhx4 deficiency. Midsagittal sections of wild-type and Lhx4−/− embryos collected from E9.5 to E14.5 were immunostained for ISL1 and LHX3. Arrow indicates the presence of ISL1-immunopositive cells in Lhx4 mutants at E14.5. Scale bar, 100 μm. Embryos are facing right.
Normally ISL1 expression initiates at E9.5 throughout RP, but from E11.5 onward, it is confined to cells in the ventral aspect during which cells are differentiating to form the AL. The initiation of ISL1 expression is indistinguishable in Lhx4−/− RP, but it does not become ventrally localized at E11.5, and it is detected in only a few cells at later stages (Figure 4). This is consistent with the paucity of differentiating cells in LHX4 mutants, and it suggests that LHX4 expression is not necessary for initial activation of ISL1.
Pitx2 mRNA expression is evident at E9.5 throughout RP and is maintained throughout the IL and AL through E14.5. The onset of Pitx2 expression is similar in Lhx4 mutant pituitaries at E9.5-E10.5 (Figure 5). In the Lhx4 mutants, Pitx2 mRNA levels appear lower than normal from E11.5 through E13.5. Expression was mainly in the additional invaginations (Figure 5). This suggests that LHX4 is not necessary for activation of Pitx2 transcription, but it may have a role in maintenance. In summary, the expression of LHX3, ISL1, and Pitx2 was reduced at some developmental times in Lhx4 mutants, and there were no examples of elevated expression.
Figure 5.

Pitx2 is initially expressed appropriately in the Lhx4 mutants but is not maintained. Midsagittal sections from wild-type and Lhx4 mutant embryos collected at E9.5-E14.5 were analyzed for Pitx2 expression by in situ hybridization. Pitx2 is expressed at a robust level on the epithelial surface of the first pharyngeal arch from E9.5 through E11.5 in mutant and wild-type embryos (dark purple). In situ hybridization signal is detected in RP of wild-type embryos from E9.5 through 11.5, but only E9.5 through 10.5 in mutant embryos. Scale bar, 100 μm. Embryos are facing to the right.
LHX4 can repress p21 expression via two conserved noncoding sequences
Three different transcription start sites have been reported for the p21 gene, resulting in unique 5′ untranslated regions that are encoded by exons 1a, 1b, and 2. To test whether LHX4 could repress p21 directly, we first determined which p21 transcript variant predominates in the pituitary gland and the pregonadotrope αT3-1 cell line by semiquantitative RT-PCR with primers designed to each unique 5′ end (Figure 6, A–C). The p21 transcript variant initiating in exon 1b (1 ENSMUST00000023829 or NM_007669.4) is clearly the predominant variant in both the αT3-1 cell line and whole pituitary tissue from normal 10-week-old mice. We detected a lower level of transcripts that initiated in exon 1a and a trace of transcripts initiating in exon 2 in αT3-1 cells, but transcripts from exons 1a or 2 were barely detected in whole pituitary tissue.
Figure 6.

Molecular genetics of p21 gene regulation. A, Multiple transcription start sites in p21 produce transcripts with different 5′ and 3′ untranslated regions, but they all encode the identical protein. Boxes represent exons; white is noncoding and black indicates protein coding. Exons are identified by numbers and letters. Exons 1a, 1b, and 2 contain transcription start sites (horizontal arrows), and exon 3 has three termination sites (vertical arrows). Transcripts initiating in exons 1a and 1b are spliced into exon 2 at the splice acceptor site (SA), just downstream of the transcription start site. The dominant transcript in the pituitary is transcribed from exon 1b. B, Three transcripts are generated from the p21 gene. Isoforms labeled 1–3 correspond to the following: 1, initiation at exon 1b (ENSMUST00000023829 or NM_007669.4, NCBI); 2, initiation at exon 2 (ENSMUST00000122348, no NCBI correspondent); or 3, initiating at exon 1a (ENSMUST00000119901 or NM_001111099.1). C, The p21 transcript isoform 1 predominates in the pituitary gland. RNA from αT3-1 cells, intestine, and adult pituitary gland was used to prepare the cDNA, which was amplified using p21 isoform-specific primers and separated on an agarose gel with size markers. The negative, no template control served as a screen for contaminating gDNA amplification. Expected PCR product sizes are as follows: isoform 1 = 88 bp, 2 = 116 bp, and 3 = 125 bp. D, Conserved noncoding elements in p21. Vista plot analysis revealed putative regulatory elements conserved 75% or greater (gray boxes) between murine p21 and the human, rhesus, canine, and equine genes. These were cloned in three groups clustered around the different transcription start sites: CE1, CE2, and CE3. Number 6 and number 9 indicate sites of specific LHX4 binding in EMSA. E, LHX4 inhibits p21 expression in transfected αT3-1 pituitary cell lines. A typical result is shown from transfecting the pregonadotrope cell line αT3-1 in triplicate with conserved element clusters (CE1, CE2, CE3) driving the expression of a minimal promoter and luciferase reporter gene, normalized to cotransfected renilla luciferase values. Results are presented as relative arbitrary units (RAU) for samples from untransfected cells (white, set to 1.0 RAU) or cotransfected LHX4 expression vector (black). The level of significance is indicated with two asterisks for P < .01 and one for P < .05. **, CE1, P = .008; *, CE2, P = .029; CE3, P = .832, not significant. The Cga promoter sequence serves as a positive control for the LHX4-mediated activation. *, P = .041. F, LHX4 protein binds specifically to two sequences in p21. Representative results from an EMSA are shown for the elements in CE2. NEs were prepared from αT3-1 cells transiently transfected with an Lhx4 expression plasmid (LHX4+) or from untransfected cells (LHX4−). Two fragments of CE2 (number 6 and number 9) exhibited specific binding (lanes 2 and 8, marked with one and two asterisk symbols, respectively). Both shifts are effectively competed with unlabeled DNA of the same sequence (lane 3 and 9) C, competitor DNA. The same settings are repeated with NEs from untransfected cells in which the shifted bands (lanes 5 and 11) are not competed with unlabeled competitor (lanes 6 and 12, respectively), indicating they are not specific. The upper bands in lanes 5, 6, 8, 9, 11, and 12 are all nonspecific bands.
Next we identified noncoding regions in and around the p21 gene that are evolutionarily conserved across higher-order vertebrates. We found 10 highly conserved noncoding elements (Figure 6D). Five of these elements cluster around transcription start site 1a (CE1). Four elements are located just downstream of the major pituitary transcription start site at exon 1b (CE2), and one is near the transcription start site at exon 2 (CE3). To test function, we cloned each of these three CE clusters into a separate luciferase reporter plasmid and cotransfected them with or without a mouse Lhx4 expression vector into αT3-1 cells (46). The results were normalized to exclude any effects of LHX4 on the plasmid backbone, and the experiment was performed in triplicate wells and repeated on 3 separate days. LHX4 repressed luciferase reporter activity modestly but significantly through CE1 and CE2 (P = .008 and P = .029, respectively) (Figure 6E). There was no effect on CE3 (P = .832). LHX4 modestly activated the CGA promoter-luciferase construct, which was normalized to its own empty backbone reporter plasmid (P = .041) (43, 45). The activation we observed for LHX4 on the Cga promoter in αT3-1 cells is similar to that reported in human embryonic kidney-293T and HeLa cells (43, 57).
LHX4 binds to conserved noncoding elements in intron 1 of p21
To detect protein-DNA binding, we performed EMSA using NEs from αT3-1 cells that were untransfected or transiently transfected with an Lhx4 expression vector. We tested 20 DNA fragments spanning CE1 and CE2. We did not detect any specific binding in CE1 (data not shown). Two of the nine fragments tested from CE2, numbers 6 and 9, exhibited specific DNA-protein binding by LHX4. These two fragments are within the four conserved elements in CE2. Fragment number 6 exhibits strong LHX4 binding that is competed off by unlabeled competitor DNA of the same sequence, and there was no specific binding by NEs from nontransfected cells, indicating that LHX4 expression was necessary to cause the specific mobility shift (Figure 6F). No LHX4 antibodies were available for supershift experiments. Fragment number 9 also exhibits specific LHX4 binding, although it is weaker than that observed with fragment number 6. We did not detect specific binding with any of the other fragments (data not shown).
Discussion
Defects in Pitx2, Lhx3, and Lhx4 cause pituitary hypoplasia early in development of Rathke's pouch due to reduced proliferation and enhanced cell death (14, 58). We report changes in multiple cell cycle markers in the developing pituitaries of these mutants (summarized in Figure 7). There are mutually exclusive expression domains of p21 and cyclin D1 in wild-type and Pitx2−/−, Lhx3−/−, and Lhx4−/− pituitaries. The border between these domains is shifted somewhat dorsally in the pouch of Lhx3 mutants and more dramatically in Lhx4 mutants. Cyclin D2 has a broader pattern of expression in RP than cyclin D1, and its expression is similar in each mutant. Although cell culture studies suggest that PITX2 directly activates both cyclin D1 and cyclin D2 in muscle cells (14, 58), it is clear that PITX2, LHX3, and LHX4 are not necessary for initiating cyclin D2 expression in the developing pituitary gland. Pitx2 is necessary for maintaining cyclin D1 but not D2 at E14.5 in the pituitary (data not shown). The distinct expression patterns of cyclin D1 and cyclin D2 imply that these genes are regulated by different mechanisms in pituitary organogenesis (14, 58). Consistent with this idea, both cyclin D1 and cyclin D2 exhibit decreased expression in Prop1 mutants (M.L.B., unpublished observations and reference 59). The hypoplasia and cell death characteristic of Prop1 mutants manifests much later than that of Pitx2, Lhx3, and Lhx4 mutants.
Figure 7.

Aberrant cell cycle regulation and dorsal-ventral boundaries in Lhx4 mutants underlies pituitary hypoplasia. Normal pituitary development is characterized by a well-orchestrated regulation of cell proliferation, cell cycle exit, and transition to cell differentiation. In cases of pituitary hypoplasia, progenitor proliferation is reduced, cells exit the cell cycle prematurely, differentiation is delayed or blocked, and cells undergo apoptosis. We discovered aberrant dorsal-ventral boundaries in gene expression, particularly for p21, in Lhx4 mutant pituitaries. Several transcription factors normally initiate expression in the oral ectoderm of mice at E9.5. These include PITX1, PITX2, ISL1, LHX4, and LHX3, which are all important for normal pituitary development. The cell cycle marker CCND1 is coexpressed throughout this area at E9.5. Lhx4 mutants fail to activate LHX3 expression at this time. Normal progression from E9.5 to E11.5 is characterized by activating CCND2 and developing oppositional expression of CCND1 and p21 in dorsal and ventral aspects of Rathke's pouch, respectively. Ki67 and p57 also exhibit oppositional expression in dorsal and ventral regions, respectively. The Lhx4 mutants exhibit a dramatic disruption of these oppositional expression patterns, with CCND1 and p57 only appearing in clusters of cells abnormally extruded from the pouch, and p21 expanding dorsally throughout the pouch. p57 exhibits an abnormal spatial pattern of expression in Pitx2 and Lhx3 mutants at E11.5, with expression prominent on the rostral side, instead of being restricted ventrally as observed in normal mice. The factors regulating rostral and caudal expression are not known but could involve inhibitors of bone morphogenetic protein signaling (111). Ki67 expression is obviously reduced in Pitx2 and Lhx4 mutants but not Lhx3 mutants. Multiple pieces of evidence reveal altered differentiation in Lhx4 mutants. LHX3 exhibits delayed expression and abnormal restriction to dorsal aspects of the pouch, and PITX2 expression wanes prematurely at E11.5. ISL1 persists in the dorsal region of mutant pituitaries and wanes early at E12.5. CCNE1 expression is reduced (E14.5). The differentiation marker CGA is delayed by 2 days in Lhx4 mutants. Excessive cell death occurs in Pitx2, Lhx3, and Lhx4 mutants (9, 11, 13), and Lhx4 mutants also exhibit elevated pH2A.X from E10.5 through E12.5. Thus, the mechanism underlying pituitary hypoplasia is complex, with some unique features in Pitx2, Lhx3, and Lhx4 mutants. In particular, dorsal ventral patterning is disrupted in Lhx4 mutants, and there is excessive dorsal expansion of the cell cycle inhibitor p21. Proliferation is reduced, markers of transitional cells are reduced, differentiation markers are delayed or blocked, and excessive cell death ensues. Cells are indicated as small circles and the colors indicate expression of p21 (green), CCND1 (blue), and CGA (yellow).
We noted clusters of cells that appear to be extruded from or invaginated from the main RP at E11.5 in the Lhx4−/− mice. They were cyclin D1 positive and p21 negative, suggesting active participation in the cell cycle. These clusters start to form at E10.5 and separate entirely from the original RP over time. A reexamination of the Lhx4−/−, Prop1df/df mice revealed the same structures (9). These structures likely give rise to the few differentiated cells detectable in more mature Lhx3 and Lhx4 mutant pituitaries (9, 13, 15, 16).
Redundancy in cell cycle regulation and the role of genetic background
Pituitary cell cycle regulation has been investigated in connection with sporadic and familial pituitary tumors and with developmental abnormalities (60–62). D-type cyclins can substitute for each other in some but not all of their molecular roles (48). In addition, the Ccnd1−/− phenotype can be rescued either by a Ccne knock-in to the Ccnd1 locus or by disrupting p27 (63, 64). There are a few known cell cycle regulators that cause pituitary hyperplasia when disrupted. p57−/− mice have AL hyperplasia during embryogenesis (39), and p27−/−, p18−/−, and Rb+/− mice develop IL hyperplasia or tumors (33, 39, 65–68). Given this, it is intriguing that the failure to express p57 on the caudal side of Rathke's pouch was not sufficient to promote proliferation in Pitx2, Lhx3, or Lhx4 mutants.
Pituitary hypoplasia occurs in Pttg1−/− (69, 70) and in Cdk4−/− mice (71, 72), but mutations in most cyclins and their regulators do not cause obvious abnormalities. These include Cdk6 (73), Cdk2 (74), Cdk1 (75), Cyclin A (76), Cyclin B (77), Cyclin D (34, 78), Cyclin E (50), p16 (79), p15 (80), p19 (36), E2f (81), and Trp53 (82). Most of the critical cell cycle genes control passage through the G1 phase, during which the ultimate decision is made to exit the cycle, undergo at least one more cycle, or remain in G1 and progress to differentiation (32). We observed reduced expression of cyclin E and cyclin D1 in Lhx4 mutants, but these changes are not likely to be drivers of the hypoplasia in Lhx4 mutants since systemic deletion of cyclin E and cyclin D1 had no effect on pituitary function. Thus, the elevated expression of p21 may be the most critical change with regard to the hypoplasia in Lhx4 mutants.
In some contexts p21 deficiency is associated with pituitary hyperplasia. The p21tm1Tyj mice are grossly normal and have an increased incidence of radiation-induced tumors in various tissues including the AL and IL of the pituitary on a mixed C57BL/6J × 129S1/SvImJ genetic background (83). In contrast, p21tm1Led mice, on the 129S6/SvEvTac genetic background, develop normal pituitary glands by birth, exhibit autoimmune glomerulonephritis, and have no increase in spontaneous tumor incidence (47, 84, 85). Thus, there may be redundancy for cell cycle regulation in mouse pituitary development that supports normal growth without p21, at least on some genetic backgrounds.
Variable phenotypes among humans and mice with LHX4 deficiency
Patients with heterozygous LHX4 gene mutation represent a phenotypic spectrum with variable penetrance even within the same family (86). Lhx4 heterozygous mice are normal, and homozygous Lhx4−/− mice have severe combined pituitary hormone deficiency and die at birth from lung and ventral motor neuron developmental failure (15, 30). We observed earlier lethality of Lhx4 embryos than previously reported. Expected Mendelian ratios of all genotypes were present until E14.5 [E9.5 to E14.5 (n = 255, P = .19)], but there were no homozygous mutants among 16 embryos at E15.5-E16.5 (n = 16, P = .02). This suggests that Lhx4 mutants are more severely affected on this stock, which is enriched for C57BL/6J relative to the original mixed background (15, 30, 87). The genomic variation that influences the viability among homozygous Lhx4−/− mice may correspond to the effects of human genomic variation on the clinical presentation of human patients heterozygous for LHX4 mutations.
Lack of LHX4 affects expression of multiple transcription factors and delays differentiation
The expression of LHX3 and CGA is delayed in Rathke's pouch of Lhx4 mutants. ISL1 expression is activated normally, but it is not maintained or spatially refined appropriately as development ensues. This is consistent with the observation that Lhx3-deficient mice also fail to activate Isl1 appropriately (88). Pitx2 expression is initiated normally in Lhx4 mutants, but it is not maintained. These observations suggest an important early role of Lhx4 in the regulatory cascade. Pitx2 and Pitx1 are expressed in the unusual clustered pituitary cells in Lhx4 mutants at later time points (9). There is no evidence for elevated expression of LHX3, ISL1, or PITX2 in compensation for the lack of LHX4, despite the similarities between the genes and evidence for overlapping functions in other tissues such as the retina or the ventral motor neurons (11, 15, 89–92).
p21 has multiple upstream regulators
LHX4 can modestly repress transcription of p21 in transfected αT3-1 cells through two clusters of conserved elements, CE1 and CE2, located upstream and downstream of the primary pituitary transcription start site. LHX4 can bind two areas of CE2 specifically, but no direct binding sites are evident in CE1. Thus, LHX4 influences the activity of CE1 indirectly. LHX4 may interact with other proteins through its LIM domains and induce repression as a component of a protein complex that binds DNA. Thus, regulation of p21 expression is complex and cannot be explained by LHX4 DNA binding activity alone.
Many factors are known to regulate p21 transcription and protein levels in various tissues (93, 94). These include transformation related protein 53 (TRP53), trans-acting transcription factor 1 (SP1), trans-acting transcription factor 2 (SP2), myelocytomatosis oncogene (MYC), E1A binding protein p300 (EP300), pituitary tumor-transforming gene (PTTG), HES1, and PITX2 (95, 96). Although TRP53 is the major effector of the G1 cell cycle checkpoint arrest, the embryonic expression of p21 is TRP53 independent and is associated with terminal differentiation (reviewed in references 62, 97, and 98). PTTG, a known regulator of the M phase of the cell cycle, represses p21 transcription. Pttg−/− and p21 mice both exhibit pituitary hypoplasia, and Pttg restrains pituitary tumor growth (69, 70, 99). In the absence of the Notch signaling pathway target Hes1, ectopic p21 expression is detected in the dorsal compartment of the RP in the pituitary progenitor cells, coincident with areas of increased cell death (40). This is similar to the dorsal activation of p21 and cell death in Lhx4 mutants, suggesting that Lhx4 and Notch are both involved in restricting p21 expression to the ventral zone of RP. Because multiple factors regulate p21 expression, and LHX4 deficiency affects expression of several important pituitary transcription factors, it is not surprising that the direct repression of p21 by LHX4 is modest in cell culture. The ectopic, excess p21 expression and pituitary hypoplasia in Lhx4 mutants are attributable to the pleiotropic effects of LHX4 deficiency.
LHX4 may regulate cell cycle in other tissues
The clinical features associated with the few known LHX4 mutations are variable, ranging from combined pituitary hormone deficiency to isolated GH deficiency, even within a family with the same genetic defect (86, 100). Pituitary stalk interruption syndrome or other extrapituitary findings like corpus callosum hypoplasia, respiratory distress, and Chiari malformation may occur (100–104). Interestingly, the affected individuals are heterozygous for LHX4 mutations, suggesting that haploinsufficiency for LHX4 is sufficient to affect pituitary and brain development in humans, whereas Lhx4+/− mice are normal. LHX4 may affect cell proliferation in hepatocellular carcinoma and primary lung cancer cell lines (105, 106). LHX4 is located on human 1q, a chromosomal region that is prone to genomic instability and has multiple roles in neurogenic differentiation and senescence of human embryonic stem cell lines, in poor outcome pediatric malignant brain tumors, and in certain types of hepatic and pulmonary malignancies (107–110). Some of these examples of genomic instability and altered differentiation and growth may involve the loss of LHX4 expression. Our demonstration that LHX4 deficiency affects the expression of multiple cell cycle regulators could provide insight into the mechanism of LHX4 action in the brain and other tissues.
In summary, Lhx4 has an important role in stimulating the rapid proliferation of undifferentiated pituitary progenitors, activating Lhx3, and maintaining expression of Isl1 and Pitx2 (Figure 7). If Lhx4 is missing, the launch of pituitary organogenesis is delayed, the potent cell cycle inhibitor p21 is overexpressed, and the pituitary progenitors undergo senescence and die. Suppression of p21 expression may be an important aspect of early pituitary development.
Acknowledgments
We thank the following individuals for their expert advice: Audrey Seasholtz and Christopher Krebs (University of Michigan), Lori Raetzman (University of Illinois, Urbana-Champaign), the University of Michigan DNA Sequencing Core, and members of the Camper Laboratory. Peter Gergics is a participant of the International Endocrine Scholars Program of The Endocrine Society.
This work was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant R01HD34283 (to S.A.C.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AL
- anterior lobe
- CCN
- cyclin
- CDK
- cyclin-dependent kinase
- CDKN
- CDK inhibitor
- CE
- conserved element
- CGA
- choriogonadotropin-α
- E
- embryonic day
- gDNA
- genomic DNA
- IHC
- immunohistochemistry
- IL
- intermediate lobe
- ISL1
- islet-1
- LHX
- LIM homeobox protein
- LIM
- Lin-11, Isl-1, and Mec-3
- NCBI
- National Center for Biotechnology Information
- NE
- nuclear extract
- PAX
- paired box transcription factor
- PITX2
- paired-type homeodomain transcription factor 2
- PTTG
- pituitary tumor-transforming gene
- RB
- retinoblastoma
- RP
- Rathke's pouch
- TF
- transcription factor
- TRP53
- transformation related protein 53.
References
- 1. De Loof A, Lindemans M, Liu F, De Groef B, Schoofs L. Endocrine archeology: do insects retain ancestrally inherited counterparts of the vertebrate releasing hormones GnRH, GHRH, TRH, and CRF? Gen Comp Endocrinol. 2012;177:18–27. [DOI] [PubMed] [Google Scholar]
- 2. Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. 2009;30:790–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pfaffle R. Genetics of growth in the normal child. Eur J Endocrinol. 2006;155:S27–S33. [Google Scholar]
- 4. Vimpani GV, Vimpani AF, Lidgard GP, Cameron EH, Farquhar JW. Prevalence of severe growth hormone deficiency. Br Med J. 1977;2:427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Regal M, Paramo C, Sierra SM, Garcia-Mayor RV. Prevalence and incidence of hypopituitarism in an adult Caucasian population in northwestern Spain. Clin Endocrinol (Oxf). 2001;55:735–740. [DOI] [PubMed] [Google Scholar]
- 6. Alatzoglou KS, Dattani MT. Genetic causes and treatment of isolated growth hormone deficiency-an update. Nat Rev Endocrinol. 2010;6:562–576. [DOI] [PubMed] [Google Scholar]
- 7. Reynaud R, Gueydan M, Saveanu A, et al. Genetic screening of combined pituitary hormone deficiency: experience in 195 patients. J Clin Endocrinol Metab. 2006;91:3329–3336. [DOI] [PubMed] [Google Scholar]
- 8. Davis SW, Potok MA, Brinkmeier ML, et al. Genetics, gene expression and bioinformatics of the pituitary gland. Horm Res. 2009;71:101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raetzman LT, Ward R, Camper SA. Lhx4 and Prop1 are required for cell survival and expansion of the pituitary primordia. Development. 2002;129:4229–4239. [DOI] [PubMed] [Google Scholar]
- 10. Nasonkin IO, Ward RD, Raetzman LT, et al. Pituitary hypoplasia and respiratory distress syndrome in Prop1 knockout mice. Hum Mol Genet. 2004;13:2727–2735. [DOI] [PubMed] [Google Scholar]
- 11. Charles MA, Suh H, Hjalt TA, Drouin J, Camper SA, Gage PJ. PITX genes are required for cell survival and Lhx3 activation. Mol Endocrinol. 2005;19:1893–1903. [DOI] [PubMed] [Google Scholar]
- 12. Raetzman LT, Cai JX, Camper SA. Hes1 is required for pituitary growth and melanotrope specification. Dev Biol. 2007;304:455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ellsworth BS, Butts DL, Camper SA. Mechanisms underlying pituitary hypoplasia and failed cell specification in Lhx3-deficient mice. Dev Biol. 2008;313:118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kioussi C, Briata P, Baek SH, et al. Identification of a Wnt/Dvl/β-catenin–> Pitx2 pathway mediating cell-type-specific proliferation during development. Cell. 2002;111:673–685. [DOI] [PubMed] [Google Scholar]
- 15. Sheng HZ, Moriyama K, Yamashita T, et al. Multistep control of pituitary organogenesis. Science. 1997;278:1809–1812. [DOI] [PubMed] [Google Scholar]
- 16. Sheng HZ, Zhadanov AB, Mosinger B, Jr, et al. Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science. 1996;272:1004–1007. [DOI] [PubMed] [Google Scholar]
- 17. Harshman LA, Brophy PD. PAX2 in human kidney malformations and disease. Pediatr Nephrol. 2012;27:1265–1275. [DOI] [PubMed] [Google Scholar]
- 18. Saifudeen Z, Liu J, Dipp S, et al. A p53-Pax2 pathway in kidney development: implications for nephrogenesis. PloS One. 2012;7(9):e44869 http://www.ncbi.nlm.nih.gov/pubmed/22984579 Accessed November 15, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng LE, Zhang J, Reed RR. The transcription factor Zfp423/OAZ is required for cerebellar development and CNS midline patterning. Dev Biol. 2007;307:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richards LJ, Plachez C, Ren T. Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet. 2004;66:276–289. [DOI] [PubMed] [Google Scholar]
- 21. Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011;22:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mesbah K, Rana MS, Francou A, et al. Identification of a Tbx1/Tbx2/Tbx3 genetic pathway governing pharyngeal and arterial pole morphogenesis. Hum Mol Genet. 2012;21:1217–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El-Khairi R, Martinez-Aguayo A, Ferraz-de-Souza B, Lin L, Achermann JC. Role of DAX-1 (NR0B1) and steroidogenic factor-1 (NR5A1) in human adrenal function. Endocr Dev. 2011;20:38–46. [DOI] [PubMed] [Google Scholar]
- 24. McCabe ER. DAX1: increasing complexity in the roles of this novel nuclear receptor. Mol Cell Endocrinol. 2007;265–266:179–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cano DA, Hebrok M, Zenker M. Pancreatic development and disease. Gastroenterology. 2007;132:745–762. [DOI] [PubMed] [Google Scholar]
- 26. Krude H, Schutz B, Biebermann H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2–1 haploinsufficiency. J Clin Invest. 2002;109:475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kimura S, Hara Y, Pineau T, et al. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. [DOI] [PubMed] [Google Scholar]
- 28. Fagman H, Nilsson M. Morphogenetics of early thyroid development. J Mol Endocrinol. 2011;46:R33–R42. [DOI] [PubMed] [Google Scholar]
- 29. Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–4651. [DOI] [PubMed] [Google Scholar]
- 30. Li H, Witte DP, Branford WW, et al. Gsh-4 encodes a LIM-type homeodomain, is expressed in the developing central nervous system and is required for early postnatal survival. EMBO J. 1994;13:2876–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suh H, Gage PJ, Drouin J, Camper SA. Pitx2 is required at multiple stages of pituitary organogenesis: pituitary primordium formation and cell specification. Development. 2002;129:329–337. [DOI] [PubMed] [Google Scholar]
- 32. Lange C, Calegari F. Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle. 2010;9:1893–1900. [DOI] [PubMed] [Google Scholar]
- 33. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. [DOI] [PubMed] [Google Scholar]
- 34. Kozar K, Ciemerych MA, Rebel VI, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491. [DOI] [PubMed] [Google Scholar]
- 35. Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012;22:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zindy F, van Deursen J, Grosveld G, Sherr CJ, Roussel MF. INK4d-deficient mice are fertile despite testicular atrophy. Mol Cell Biol. 2000;20:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Depamphilis ML, de Renty CM, Ullah Z, Lee CY. “The Octet”: eight protein kinases that control mammalian DNA replication. Front Physiol. 2012;3:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol. 2013;5:a008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bilodeau S, Roussel-Gervais A, Drouin J. Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol Cell Biol. 2009;29:1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monahan P, Rybak S, Raetzman LT. The notch target gene HES1 regulates cell cycle inhibitor expression in the developing pituitary. Endocrinology. 2009;150:4386–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin DM, Skidmore JM, Fox SE, Gage PJ, Camper SA. Pitx2 distinguishes subtypes of terminally differentiated neurons in the developing mouse neuroepithelium. Dev Biol. 2002;252:84–99. [DOI] [PubMed] [Google Scholar]
- 42. Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(Web Server issue):W273–279 http://www.ncbi.nlm.nih.gov/pubmed/15215394 Accessed July 1, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sloop KW, Dwyer CJ, Rhodes SJ. An isoform-specific inhibitory domain regulates the LHX3 LIM homeodomain factor holoprotein and the production of a functional alternate translation form. J Biol Chem. 2001;276:36311–36319. [DOI] [PubMed] [Google Scholar]
- 44. Windle JJ, Weiner RI, Mellon PL. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol. 1990;4:597–603. [DOI] [PubMed] [Google Scholar]
- 45. Brinkmeier ML, Gordon DF, Dowding JM, et al. Cell-specific expression of the mouse glycoprotein hormone α-subunit gene requires multiple interacting DNA elements in transgenic mice and cultured cells. Mol Endocrinol. 1998;12:622–633. [DOI] [PubMed] [Google Scholar]
- 46. Schagat T, Paguio A, Kopish K. Normalizing genetic reporter assays: approaches and considerations for increasing consistency and statistical significance. Cell Notes. 2007;(17):9–12. [Google Scholar]
- 47. Monahan P, Himes AD, Parfieniuk A, Raetzman LT. p21, an important mediator of quiescence during pituitary tumor formation, is dispensable for normal pituitary development during embryogenesis. Mech Dev. 2012;128:640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carthon BC, Neumann CA, Das M, et al. Genetic replacement of cyclin D1 function in mouse development by cyclin D2. Mol Cell Biol. 2005;25:1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang P, Liegeois NJ, Wong C, et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. [DOI] [PubMed] [Google Scholar]
- 50. Geng Y, Yu Q, Sicinska E, et al. Cyclin E ablation in the mouse. Cell. 2003;114:431–443. [DOI] [PubMed] [Google Scholar]
- 51. Lawless C, Wang C, Jurk D, Merz A, Zglinicki T, Passos JF. Quantitative assessment of markers for cell senescence. Exp Gerontol. 2010;45:772–778. [DOI] [PubMed] [Google Scholar]
- 52. Japon MA, Rubinstein M, Low MJ. In situ hybridization analysis of anterior pituitary hormone gene expression during fetal mouse development. J Histochem Cytochem. 1994;42:1117–1125. [DOI] [PubMed] [Google Scholar]
- 53. Pope C, McNeilly JR, Coutts S, Millar M, Anderson RA, McNeilly AS. Gonadotrope and thyrotrope development in the human and mouse anterior pituitary gland. Dev Biol. 2006;297:172–181. [DOI] [PubMed] [Google Scholar]
- 54. Zheng Q, Zhao Y. The diverse biofunctions of LIM domain proteins: determined by subcellular localization and protein-protein interaction. Biol Cell. 2007;99:489–502. [DOI] [PubMed] [Google Scholar]
- 55. Ericson J, Norlin S, Jessell TM, Edlund T. Integrated FGF and BMP signaling controls the progression of progenitor cell differentiation and the emergence of pattern in the embryonic anterior pituitary. Development. 1998;125:1005–1015. [DOI] [PubMed] [Google Scholar]
- 56. Takuma N, Sheng HZ, Furuta Y, Ward JM, et al. Formation of Rathke's pouch requires dual induction from the diencephalon. Development. 1998;125:4835–4840. [DOI] [PubMed] [Google Scholar]
- 57. Castinetti F, Saveanu A, Reynaud R, et al. A novel dysfunctional LHX4 mutation with high phenotypical variability in patients with hypopituitarism. J Clin Endocrinol Metab. 2008;93:2790–2799. [DOI] [PubMed] [Google Scholar]
- 58. Baek SH, Kioussi C, Briata P, et al. Regulated subset of G1 growth-control genes in response to derepression by the Wnt pathway. Proc Natl Acad Sci USA. 2003;100:3245–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ward RD, Raetzman LT, Suh H, Stone BM, Nasonkin IO, Camper SA. Role of PROP1 in pituitary gland growth. Mol Endocrinol. 2005;19:698–710. [DOI] [PubMed] [Google Scholar]
- 60. Quereda V, Malumbres M. Cell cycle control of pituitary development and disease. J Mol Endocrinol. 2009;42:75–86. [DOI] [PubMed] [Google Scholar]
- 61. Salehi F, Agur A, Scheithauer BW, Kovacs K, Lloyd RV, Cusimano M. Biomarkers of pituitary neoplasms: a review (part II). Neurosurgery. 2010;67:1790–1798; discussion 1798. [DOI] [PubMed] [Google Scholar]
- 62. Ciemerych MA, Sicinski P. Cell cycle in mouse development. Oncogene. 2005;24:2877–2898. [DOI] [PubMed] [Google Scholar]
- 63. Geng Y, Whoriskey W, Park MY, et al. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–777. [DOI] [PubMed] [Google Scholar]
- 64. Geng Y, Yu Q, Sicinska E, Das M, Bronson RT, Sicinski P. Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proc Natl Acad Sci USA. 2001;98:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fero ML, Rivkin M, Tasch M, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. [DOI] [PubMed] [Google Scholar]
- 66. Nakayama K, Ishida N, Shirane M, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. [DOI] [PubMed] [Google Scholar]
- 67. Franklin DS, Godfrey VL, Lee H, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 δ 20/+ mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9:1021–1027. [PubMed] [Google Scholar]
- 69. Chesnokova V, Kovacs K, Castro AV, Zonis S, Melmed S. Pituitary hypoplasia in Pttg−/− mice is protective for Rb+/− pituitary tumorigenesis. Mol Endocrinol. 2005;19:2371–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chesnokova V, Zonis S, Kovacs K, et al. p21(Cip1) restrains pituitary tumor growth. Proc Natl Acad Sci USA. 2008;105:17498–17503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moons DS, Jirawatnotai S, Parlow AF, Gibori G, Kineman RD, Kiyokawa H. Pituitary hypoplasia and lactotroph dysfunction in mice deficient for cyclin-dependent kinase-4. Endocrinology. 2002;143:3001–3008. [DOI] [PubMed] [Google Scholar]
- 72. Jirawatnotai S, Aziyu A, Osmundson EC, et al. Cdk4 is indispensable for postnatal proliferation of the anterior pituitary. J Biol Chem. 2004;279:51100–51106. [DOI] [PubMed] [Google Scholar]
- 73. Malumbres M, Sotillo R, Santamaria D, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. [DOI] [PubMed] [Google Scholar]
- 74. Martin A, Odajima J, Hunt SL, et al. Cdk2 is dispensable for cell cycle inhibition and tumor suppression mediated by p27(Kip1) and p21(Cip1). Cancer Cell. 2005;7:591–598. [DOI] [PubMed] [Google Scholar]
- 75. Santamaria D, Barriere C, Cerqueira A, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. [DOI] [PubMed] [Google Scholar]
- 76. van der Meer T, Chan WY, Palazon LS, et al. Cyclin A1 protein shows haplo-insufficiency for normal fertility in male mice. Reproduction. 2004;127:503–511. [DOI] [PubMed] [Google Scholar]
- 77. Brandeis M, Rosewell I, Carrington M, et al. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc Natl Acad Sci USA. 1998;95:4344–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 1995;9:2364–2372. [DOI] [PubMed] [Google Scholar]
- 79. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. [DOI] [PubMed] [Google Scholar]
- 80. Latres E, Malumbres M, Sotillo R, et al. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. [DOI] [PubMed] [Google Scholar]
- 82. Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. [DOI] [PubMed] [Google Scholar]
- 83. Jackson RJ, Engelman RW, Coppola D, Cantor AB, Wharton W, Pledger WJ. p21Cip1 nullizygosity increases tumor metastasis in irradiated mice. Cancer Res. 2003;63:3021–3025. [PubMed] [Google Scholar]
- 84. Salvador JM, Hollander MC, Nguyen AT, et al. Mice lacking the p53-effector gene Gadd45a develop a lupus-like syndrome. Immunity. 2002;16:499–508. [DOI] [PubMed] [Google Scholar]
- 85. Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. [DOI] [PubMed] [Google Scholar]
- 86. Pfaffle R, Klammt J. Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab. 2011;25:43–60. [DOI] [PubMed] [Google Scholar]
- 87. Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–174. [DOI] [PubMed] [Google Scholar]
- 88. Zhao Y, Morales DC, Hermesz E, Lee WK, Pfaff SL, Westphal H. Reduced expression of the LIM-homeobox gene Lhx3 impairs growth and differentiation of Rathke's pouch and increases cell apoptosis during mouse pituitary development. Mech Dev. 2006;123:605–613. [DOI] [PubMed] [Google Scholar]
- 89. Pfaff SL, Mendelsohn M, Stewart CL, Edlund T, Jessell TM. Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell. 1996;84:309–320. [DOI] [PubMed] [Google Scholar]
- 90. Sharma K, Sheng HZ, Lettieri K, et al. LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell. 1998;95:817–828. [DOI] [PubMed] [Google Scholar]
- 91. Elshatory Y, Everhart D, Deng M, Xie X, Barlow RB, Gan L. Islet-1 controls the differentiation of retinal bipolar and cholinergic amacrine cells. J Neurosci. 2007;27:12707–12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Thaler JP, Koo SJ, Kania A, et al. A postmitotic role for Isl-class LIM homeodomain proteins in the assignment of visceral spinal motor neuron identity. Neuron. 2004;41:337–350. [DOI] [PubMed] [Google Scholar]
- 93. Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–1374. [DOI] [PubMed] [Google Scholar]
- 94. el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. [DOI] [PubMed] [Google Scholar]
- 95. Heldring N, Joseph B, Hermanson O, Kioussi C. Pitx2 expression promotes p21 expression and cell cycle exit in neural stem cells. CNS Neurol Disord Drug Targets. 2012;11:884–892. [DOI] [PubMed] [Google Scholar]
- 96. Cao H, Florez S, Amen M, et al. Tbx1 regulates progenitor cell proliferation in the dental epithelium by modulating Pitx2 activation of p21. Dev Biol. 2010;347:289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. [DOI] [PubMed] [Google Scholar]
- 98. el-Deiry WS, Tokino T, Waldman T, et al. Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res. 1995;55:2910–2919. [PubMed] [Google Scholar]
- 99. Xiao H, Hasegawa T, Miyaishi O, Ohkusu K, Isobe K. Sodium butyrate induces NIH3T3 cells to senescence-like state and enhances promoter activity of p21WAF/CIP1 in p53-independent manner. Biochem Biophys Res Commun. 1997;237:457–460. [DOI] [PubMed] [Google Scholar]
- 100. Tajima T, Hattori T, Nakajima T, Okuhara K, Tsubaki J, Fujieda K. A novel missense mutation (P366T) of the LHX4 gene causes severe combined pituitary hormone deficiency with pituitary hypoplasia, ectopic posterior lobe and a poorly developed sella turcica. Endocr J. 2007;54:637–641. [DOI] [PubMed] [Google Scholar]
- 101. Reynaud R, Albarel F, Saveanu A, et al. Pituitary stalk interruption syndrome in 83 patients: novel HESX1 mutation and severe hormonal prognosis in malformative forms. Eur J Endocrinol. 2011;164:457–465. [DOI] [PubMed] [Google Scholar]
- 102. Castinetti F, Reynaud R, Saveanu A, et al. Clinical and genetic aspects of combined pituitary hormone deficiencies. Ann Endocrinol (Paris). 2008;69:7–17. [DOI] [PubMed] [Google Scholar]
- 103. Dateki S, Fukami M, Uematsu A, et al. Mutation and gene copy number analyses of six pituitary transcription factor genes in 71 patients with combined pituitary hormone deficiency: identification of a single patient with LHX4 deletion. J Clin Endocrinol Metab. 2010;95:4043–4047. [DOI] [PubMed] [Google Scholar]
- 104. Pfaeffle RW, Hunter CS, Savage JJ, et al. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J Clin Endocrinol Metab. 2008;93:1062–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hung TM, Hu RH, Ho CM, et al. Downregulation of α-fetoprotein expression by LHX4: a critical role in hepatocarcinogenesis. Carcinogenesis. 2011;32:1815–1823. [DOI] [PubMed] [Google Scholar]
- 106. Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–7947. [DOI] [PubMed] [Google Scholar]
- 107. Varela C, Denis JA, Polentes J, et al. Recurrent genomic instability of chromosome 1q in neural derivatives of human embryonic stem cells. J Clin Invest. 2012;122:569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Faria C, Miguens J, Antunes JL, et al. Pediatric brain tumors: genetics and clinical outcome. J Neurosurg Pediatr. 2010;5:263–270. [DOI] [PubMed] [Google Scholar]
- 109. Tomlinson GE. Cytogenetics of hepatoblastoma. Front Biosci. 2012;4:1287–1292. [DOI] [PubMed] [Google Scholar]
- 110. Hoglund M, Gisselsson D, Hansen GB, Mitelman F. Statistical dissection of cytogenetic patterns in lung cancer reveals multiple modes of karyotypic evolution independent of histological classification. Cancer Genet Cytogenet. 2004;154:99–109. [DOI] [PubMed] [Google Scholar]
- 111. Davis SW, Castinetti F, Carvalho LR, et al. Molecular mechanisms of pituitary organogenesis: In search of novel regulatory genes. Mol Cell Endocrinol. 2010;323:4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]