

Abstract
In vitro slice studies have revealed that there are significant differences in the spontaneous firing activity between anteroventral periventricular/periventricular preoptic nucleus (AVPV/PeN) and arcuate nucleus (ARC) kisspeptin (Kiss1) neurons in females. Although both populations express similar endogenous conductances, we have discovered that AVPV/PeN Kiss1 neurons express a subthreshold, persistent sodium current (INaP) that dramatically alters their firing activity. Based on whole-cell recording of Kiss1-Cre-green fluorescent protein (GFP) neurons, INaP was 4-fold greater in AVPV/PeN vs ARC Kiss1 neurons. An LH surge-producing dose of 17β-estradiol (E2) that increased Kiss1 mRNA expression in the AVPV/PeN, also augmented INaP in AVPV/PeN neurons by 2-fold. Because the activation threshold for INaP was close to the resting membrane potential (RMP) of AVPV/PeN Kiss1 neurons (−54 mV), it rendered them much more excitable and spontaneously active vs ARC Kiss1 neurons (RMP = −66 mV). Single-cell RT-PCR revealed that AVPV/PeN Kiss1 neurons expressed the requisite sodium channel α-subunit transcripts, NaV1.1, NaV1.2, and NaV1.6 and β subunits, β2 and β4. Importantly, NaV1.1α and -β2 transcripts in AVPV/PeN, but not ARC, were up-regulated 2- to 3-fold by a surge-producing dose of E2, similar to the transient calcium current channel subunit Cav3.1. The transient calcium current collaborates with INaP to generate burst firing, and selective blockade of INaP by riluzole significantly attenuated rebound burst firing and spontaneous activity. Therefore, INaP appears to play a prominent role in AVPV/PeN Kiss1 neurons to generate spontaneous, repetitive burst firing, which is required for the high-frequency-stimulated release of kisspeptin for exciting GnRH neurons and potentially generating the GnRH surge.
Kisspeptin (Kiss1) neurons in the anteroventral and more caudal periventricular preoptic nucleus (AVPV/PeN) are thought to be vital presynaptic neurons that drive GnRH neuronal excitability (1, 2), and it has been shown that high-frequency stimulation of these neurons (>5 Hz) is required to induce kisspeptin release and hence excitation of GnRH neurons (3). Both a low threshold, transient calcium current (IT) and a hyperpolarization-activated, cyclic nucleotide-gated current (Ih) are prominently expressed in both AVPV/PeN and arcuate nucleus Kiss1 neurons (4, 5). Kiss1 neurons also express the corresponding critical channel transcripts, Cav3 and HCN, which underlie these currents (5, 6). In addition, AVPV/PeN Kiss1 neurons express a prominent persistent Na+ current (INaP) that is activated approximately 10 mV negative to the threshold for the transient, rapidly inactivating, sodium current (INaT), which underlies the action potential generation (5). Although 17β-estradiol (E2) treatment increases both Ih and IT and the excitability of AVPV/PeN Kiss1 neurons (5, 7), nothing is known about the physiological regulation of INaP in Kiss1 neurons during the female reproductive cycle.
Persistent sodium currents help sculpt firing patterns of central nervous system (CNS) neurons (8–10). Na+ channel proteins that underlie the persistent sodium current are complexes of a 260-kDa α-subunit in association with one or two auxiliary β-subunits (β1–4) of 33–34 kDa (11). Three different α-subunits are expressed in the adult mammalian brain, NaV1.1, NaV1.2, and NaV1.6, all of which can contribute to INaP. The α-subunits associate with one or more β-subunits, which regulate not only the trafficking of α-subunits but also their gating properties. Although it is cellular environmental dependent, β1-, β2-, and β3-subunits all promote persistent sodium currents in CNS neurons (8–10). Therefore, NaV1.1-, NaV1.2-, and NaV1.6-α subunits not only generate INaT but also associate with β-subunits to produce a regenerative depolarizing current in the subthreshold voltage range, ie, the persistent sodium current. Although the magnitude of INaP is only about 2%–5% of the transient sodium current, it is activated approximately 10 mV negative to INaT, in which few voltage-gated channels (eg, potassium channels) are active and the input resistance is high (10), so the depolarizing effects of this current are greatly amplified. Therefore, INaP shapes repetitive firing, generates rhythmicity, and amplifies excitatory postsynaptic potentials (12). INaP modulates pacemaking activity in numerous CNS neurons including cerebellar Purkinje neurons (13, 14), CA1 pyramidal neurons (15), ventral tegmental dopamine neurons (16), hypothalamic suprachiasmatic neurons (17), tuberomammilary histamine neurons (18), and GnRH neurons (19), all of which depend on the interplay between INaP, IT, and Ih (20). Also, neurotransmitters that affect any of these conductances greatly facilitate burst firing activity. Therefore, we hypothesized that similar to its effects on IT and Ih, E2 increases INaP in AVPV/PeN Kiss1 neurons and consequently the burst firing capability of these vital reproductive neurons.
Materials and Methods
Animals
All procedures performed with animals were in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by our local committee on animal care and use. Kiss1-Cre-green fluorescent protein (Kiss1-Cre-GFP; C57BL6/J and S129 background) mice were produced by Dr Robert Steiner and his colleagues at the University of Washington (4). Offspring that carried the transgene were identified by PCR on the genomic DNA extracted from tail biopsies. Transgenic animals were maintained as heterozygous by breeding with wild-type C57BL6/J mice. Animals were housed under constant temperature and light in a 12-hour light, 12-hour dark cycle, with lights on at 6:00 am [zeitgeber time (ZT) 0] and lights off at 6:00 pm (ZT12). Food and water were provided ad libitum. Heterozygous female animals between 8 and 16 weeks of age were used for all experiments. Bilateral ovariectomies (OVX) were performed under inhalant isofluorane anesthesia. Carprofen (Rimadyl; Pfizer) was given immediately after a surgery at a dose of 4 mg/kg for analgesia. After OVX, animals were treated with E2, which has been shown to induce an LH surge in Kiss1-Cre-GFP mice (5). Five days (d 5) after ovariectomy, animals were treated with a sc injection of 0.25 μg 17β-estradiol benzoate in sesame oil (50 μl) at ZT4. On the following day (d 6), animals were given a sc injection of 1.5–2.0 μg 17β-estradiol benzoate in sesame oil. On the day of the induced surge (d 7), E2-treated animals (high-E2 group; uterine weights 90–130 mg) were killed at approximately ZT4.
An additional group of animals was treated as above but received less E2 as a model for the diestrous stage of the estrous cycle. These animals received injections of 0.0625 μg 17β-estradiol benzoate in sesame oil on day 5 and/or day 6 after OVX and were used for experiments on day 7 (low-E2 group; uterine weights 30–44 mg). Other animals used for cell harvesting of arcuate nucleus (ARC) Kiss1 neurons were OVX and received oil vehicle treatment (uterine weights 13–18 mg).
Slice preparation
On the day of each experiment, the animal was killed by decapitation and the brain was removed from the skull. The brainstem was removed, and the resulting block was mounted on a cutting vibratome and submerged in ice-cold oxygenated (95% O2-5% CO2) high-sucrose artificial cerebral spinal fluid (aCSF) (in millimoles: 208 sucrose, 2 KCl, 1 MgCl2, 1.25 NaH2PO4, 10 HEPES, 26 NaHCO3, 10 dextrose, 2 MgSO4, and 1 CaCl2). Slices of 220–250 μm thickness were cut through the AVPV/PeN area and the ARC. Slices were transferred to an auxiliary chamber containing oxygenated aCSF at room temperature (in millimoles: 124 NaCl, 5 KCl, 1.44 NaH2PO4, 5 HEPES, 10 dextrose, 26 NaHCO3, 2 MgCl2, and 2 CaCl2) and allowed to recover for at least 1 hour. Also, the uterus was removed, trimmed of fat, blotted dry, and weighed. Uterine weights served as an indicator of circulating E2 levels (5, 21).
Cell harvesting and RT-PCR
Tissue slices (250 μm) containing the AVPV/PeN were microdissected and used for neuronal dispersion and harvesting as described previously (5, 22). Briefly, the tissue was treated with protease (from Streptomyces griseus; Sigma) at 37°C for 15 minutes and then washed three times with low Ca2+ aCSF (1 mM Ca2+) and two times in aCSF. Using flame-polished glass Pasteur pipettes of decreasing sizes, tissues were titrated and dispersed neurons were plated onto a 60-mm cell culture dish with a glass bottom. Oxygenated aCSF at room temperature was constantly perfused into the dish at a rate of 2 ml/min. Individual GFP-positive and several GFP-negative neurons were harvested into a standard glass pipette (1.5 mm outer diameter/0.83 mm inner diameter; World Precision Instruments) using the XenoWorks microinjector system (Sutter Instruments). Importantly, GFP-negative cells were always negative to Kiss1 mRNA, whereas GFP-expressing cells were positive to Kiss1.
Only fully intact, healthy neurons that showed uniform GFP fluorescence (or no fluorescence) and were anchored to the glass plate were harvested.
Neurons were ejected into a siliconized 0.5-ml tube containing a solution of 1× Invitrogen Superscript III buffer, 15 U RNasin (Promega), 10 mM of dithiothreitol, and diethylpyrocarbonate-treated water in a total of 5 μl (single cells) or 8 μl (pools of cells). Cells were harvested individually or as pools (five cells/pool) and were frozen on dry ice and stored at −80°C until further processing.
Harvested cells were reverse transcribed according to established protocols (22). The cDNA was stored at −20°C. The positive and negative controls included preoptic or arcuate hypothalamic RNA samples with reverse transcriptase added (+RT) or without reverse transcriptase (−RT), single cells without reverse transcriptase, aCSF from the surrounding area, and water blank. Each cell or pool of cells was then evaluated by using single-cell PCR or quantitative real-time PCR (qPCR), respectively.
Primer design
PCR primers were designed using Clone Manager Software (Sci Ed Software). Primers were designed to cross at least one intron/exon boundary. Only primers that demonstrated a single-peak melting curve and had efficiency between 95% and 100% were used. Primer sequences for single-cell PCR were as follows: NaV1.1α (accession number NM_018733), forward primer 3393–3414 nt, reverse primer 3587–3608 nt, 216 bp; NaV1.2α (accession number NM_001099298), forward primer 894–911 nt, reverse primer 1066–1083 nt, 190 bp; NaV1.6α (accession number NM_001077499), forward primer 3448–3468 nt, reverse primer 3561–3581 nt, 134 bp; NaVβ2 (accession number NM_001014761), forward primer 85–104 nt; reverse primer 313–332 nt, 248 bp; NaVβ4 (accession number NM_001013390), forward primer 216–237 nt, reverse primer 351–372 nt, 157 bp. Primer sequences for qPCR were as follows: NaV1.1α (118 bp product), forward primer 3501–3519 nt, reverse primer 3598–3618 nt; NaV1.2α (130 bp product), forward primer 3925–3946 nt, reverse primer 4033–4054 nt; NaV1.6α (82 bp product), forward primer 4290–4309 nt, reverse primer 4352–4371 nt; NaVβ2 (105 bp product), forward primer 187–206 nt, reverse primer 272–291 nt; NaVβ4 (117 bp product), forward primer 24–41 nt, reverse primer 123–140 nt; glyceraldehyde-3-phosphate dehydrogenase (GAPDH; accession number NM_008084), forward primer 689–706 nt, reverse primer 764–781 nt, 93 bp. Primers for CaV3.1, Kiss1, and β-actin were as described previously (5, 22).
Single-cell PCR and qPCR
Analysis of single cells was performed on 3 μl of cDNA as described previously (22). Conditions for the PCR were as follows: initial denaturation for 2 minutes at 95°C; 50 cycles of amplification at 63°C for NaV1.1α, NaV1.2α, and NaV1.6α; 50 cycles of amplification at 61°C for NaVβ2 and NaVβ4, followed by a final extension at 72°C for 5 minutes. PCR products were visualized with ethidium bromide on a 2% agarose gel. Analysis by qPCR was performed using the Power SybrGreen master mix method as described previously (22, 23) on a Life Technologies Quant Studio 7 Flex System (Life Technologies). cDNA samples of AVPV/PeN neurons were run in 3- to 4-μl duplicates for the target genes (Kiss1, NaV1.1α, NaV1.2α, NaV1.6α, NaVβ2, NaVβ4, CaV3.1) and 2 μl duplicates for the reference genes (β-actin, GAPDH), and the mean Δcycle threshold of the low E2-treated pool values was used as a calibrator when comparing the mRNA quantities with the high E2-treated samples. The mRNA expression of Kiss1, NaV1.1α, NaV1.2α, NaV1.6α, and NaVβ2 (NaVβ4 was not detected in ARC Kiss1 neurons) was also quantified in harvested arcuate kisspeptin neurons from OVX high E2, as compared with OVX oil-treated animals as described above with the exception that GAPDH was used as the reference gene (see below). Data were analyzed following the comparative ΔΔcycle threshold method described previously (22, 23) and are reported as relative amounts of mRNA expression to the calibrator.
Electrophysiological recording
Whole-cell patch clamp recordings were conducted from GFP-labeled Kiss1 neurons with the use of an Olympus BX51 (Olympus) or a Zeiss Axioskop FS upright microscope (Carl Zeiss Microscopy) equipped with fluorescence and infrared differential interference contrast imaging devices (1, 4, 24, 25). Patch pipettes (1.5 mm outer diameter borosilicate glass; A-M Systems) were pulled on a Flaming/Brown puller (model P-97; Sutter Instrument Co) and filled with the following internal solution (in millimoles): 128 potassium gluconate, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 2 or 4 ATP, and 0.25 GTP; adjusted to pH 7.3 with KOH; 290 mOsm. Pipettes filled with the above internal solution had a resistance of 2–3 MΩ. In whole-cell configuration, and access resistance was less than 25 MΩ and was 80% compensated for the INaT. Generally, INaP is measured from the current activated by a slow voltage ramp (<100 mV/sec) from −80 to −20 mV (17, 18).
In preliminary experiments we examined the INaP with voltage ramps from 20 mV/sec to 100 mV/sec and found that the amplitude of INaP decreased with the decrease in the ramp speed and reached a state at 20 mV/sec when the INaP was relatively stable and action currents (from the unclamped axon) were eliminated (Figure 1A). Therefore, we measured the INaP at a ramp speed of 20 mV/sec in the absence and presence of tetrodotoxin (TTX; 1 μM), which blocks the persistent (and transient) sodium current. To record INaT, a step voltage command of 100 milliseconds from −60 mV to −20 mV with a step size of 2.5 mV was applied to cells. Postinhibitory rebound burst firing was examined at resting membrane potentials after a series of hyperpolarizing steps of 1 second duration from −70 mV to −120 mV to recruit active T-type calcium channels (23). The spontaneous firing of AVPV/PeN and arcuate Kiss1 neurons were examined in the presence of ionotropic glutamate and γ-aminobutyric acid (GABA)A receptor blockers [10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 50 μM DL-2-amino-5-phosphonopentanoic acid (AP-5), and 100 μM picrotoxin]. The liquid junction potential of −10 mV was corrected for in all analyses. Consecutive current traces were filtered at 2–10 kHz and acquired at a sampling rate of 0.5–5 kHz. Electrophysiological signals were amplified with an Axopatch 200B amplifier and digitized with a Digidata 1440A (Molecular Devices), or with an Axopatch 1D amplifier and digitized with a Digidata 1322 (Axon Instruments). Data were analyzed with Clampfit software (version 9.0 or 10.0; Molecular Devices).
Figure 1.

INaP in AVPV/PeN and ARC Kiss1 neurons. A, Representative recordings showing the relationship between INaP and voltage ramps (speeds from 20 mV/sec to 100 mV/sec) from an AVPV/PeN Kiss1 neuron. B and C, Voltage clamp recordings showing INaP generated by a voltage ramp from −80 to −20 mV at 20 mV/sec before (black trace; control) and after (red trace) application of TTX (1 μM) in representative AVPV/PeN (B) and ARC (C) Kiss1 neurons. The black straight line indicates the linear part of the whole-cell current in the presence of TTX. The horizontal stippled line indicates the zero current level. The vertical stippled line marks the resting membrane potential, and the arrow points to activation threshold for the INaP. D, Comparison of INaP density measured at −45 mV in AVPV/PeN Kiss1 neurons and ARC Kiss1 neurons from high-E2 animals. **, P < .01, n = 9–14; Student's t test.
Drugs or chemicals
Riluzole, CNQX, AP-5, and picrotoxin were purchased from Tocris Bioscience and were made up as a stock of 10, 10, 50, and 100 mM, respectively, in DMSO. TTX was purchased from Alomone Laboratories and was made up as a 1-mM stock in Milli-Q water.
Data analysis
PCR/qPCR
For quantification of mRNA expression differences between low-E2 (n = 5) and high-E2 (n = 5–6) animals, Kiss1 neuronal pools were analyzed using real-time qPCR as described above. Three pools of five cells each were analyzed from each animal, and the average mRNA expression relative to β-actin was determined for each animal and used for further analysis of mean ± SEM, and differences in gene expression were analyzed by a two-tailed Student's t test or one-way ANOVA. Note that β-actin was used as a reference gene for quantification of mRNA expression in AVPV/PeN Kiss1 neurons because its mRNA was not regulated by E2 in these neurons. For quantification of mRNA expression in arcuate Kiss1 neurons we used GAPDH rather than β-actin because β-actin, but not GAPDH, was regulated by E2 in these neurons.
Electrophysiology
The spontaneous firing frequencies of cells were measured at their resting membrane potential. The INaP activation threshold was defined as the voltage in which 5% of the maximum INaP was activated (15). Data were analyzed with Mini Analysis (Synaptosoft Inc) and Clampfit 9.2 (Molecular Devices) software. Graphs were plotted using GraphPad Prism 4, Sigma Plot 8.0, and Macromedia Freehand 10 software. Comparisons between different treatments were performed using a paired or unpaired Student's t test. Differences were considered significant if the probability of error was less than 5%. All data are presented as mean ± SEM.
Results
INaP in AVPV/PeN and ARC Kiss1 neurons
Kiss1 neurons in the AVPV/PeN and ARC of females exhibit dramatically different firing activities (4, 5, 7, 26, 27). Because both AVPV/PeN and ARC Kiss1 neurons express IT and Ih but exhibit these differences in firing activity (4, 5), we hypothesized that the expression of INaP in these two distinct populations of Kiss1 neurons would account for these differences. First, we isolated INaP in AVPV/PeN and ARC Kiss1 neurons using a ramp voltage protocol in the presence or absence of sodium channel blocker TTX (see Materials and Methods). Similar to other hypothalamic neurons (17, 18), INaP in Kiss1 neurons was evoked by slowly ramping the membrane voltage from −80 to −20 mV at 20 mV/sec. In this way we were able to isolate INaP without contamination of INaT. Figure 1A illustrates that INaP decreased with the ramp speed from 100 mV/s to 40 mV/s, but stabilized between 40 mV/sec and 20 mV/sec. Also, we observed that at 20 mV/sec the INaT was eliminated in most cells. Therefore, a ramp speed of 20 mV/sec was chosen for later experiments, and cells that displayed INaT in the ramp were excluded from the analysis (17, 18). In AVPV/PeN and ARC Kiss1 neurons, the INaT was not activated until −43.1 ± 2.4 mV and −41.4 ± 1.8 mV (n = 14) mV, respectively, and was inactivated with the slow voltage ramp speed of 20 mV/sec (Figure 1A). Therefore, we measured INaP at −45 mV by subtracting the linear background current obtained in the presence of TTX. INaP was approximately 4-fold greater in AVPV/PeN Kiss1 neurons vs ARC Kiss1 neurons (1.16 ± 0.18 pA/pF, n = 14 vs 0.34 ± 0.11 pA/pF, n = 9, respectively; Figure 1).
Also, the activation threshold for INaP was significantly different between the two population of neurons (AVPV/PeN: −54.2 ± 1.3 mV, n = 14 vs ARC: −50.1 ± 1.5 mV, n = 9) (Figure 1, B and C). Furthermore, the whole-cell current between the resting membrane potential and the INaP threshold was inward (depolarizing) in the majority (88%) of AVPV/PeN Kiss1 neurons (Figure 1B) but outward (hyperpolarizing) in the majority (70%) of ARC Kiss1 neurons (Figure 1C), which is consistent with the fact that the resting membrane potential of AVPV/PeN Kiss1 neurons was depolarized (−54.6 ± 0.8 mV, n = 15) relative to the resting membrane potential of ARC Kiss1 neurons (−65.9 ± 1.2 mV, n = 13). However, the activation threshold for INaT was not significantly different between the two groups of Kiss1 neurons (AVPV/PeN neurons: 43.1 ± 2.4 mV, n = 14 vs ARC neurons: −41.4 ± 1.8 mV, n = 9).
E2 increases persistent sodium current in AVPV/PeN Kiss1 neurons
Previous studies showed that the excitability of Kiss1 neurons was regulated by circulating levels of E2 (4, 5, 7, 26, 27). Therefore, we examined whether E2 regulates the expression of INaP in AVPV/PeN and ARC Kiss1 neurons by comparing the whole-cell current density from both low-E2 and high-E2 animals. GFP neurons in the AVPV/PeN from low-E2 animals harvested in the slice during whole-cell recording (or after dispersion) were found consistently to be positive to Kiss1 mRNA (Figure 2, A and B) as described previously in high-E2 animals (5). Importantly, as shown in Figure 2, A and C, E2 increased the current density of INaP by greater than 2-fold in AVPV/PeN Kiss1 (1.25 ± 0.18 pA/pF for high-E2 vs 0.55 ± 0.14 pA/pF for low-E2, P < .01) but not in ARC Kiss1 neurons (0.38 ± 0.11 pA/pF for high-E2 vs 0.49 ± 0.23 pA/pF for low-E2).
Figure 2.

High estradiol treatment increased INaP in AVPV/PeN Kiss1 neurons but not in ARC Kiss1 neurons. A, INaP current in AVPV/PeN Kiss1 neurons is increased in high-E2 as compared with low-E2 animals. **, P < .01, Student's t test. B, qPCR measurements of NaV1.1α and NaVβ2 mRNAs in AVPV/PeN Kiss1 neuronal pools (three pools per animal) from low-E2 and high-E2 OVX mice (n = 5 animals in each group). **, P < .01, n = 10–14; Student's t test. The gel figures in panels A and B illustrate that AVPV/PeN GFP neurons from low-E2 Kiss1 animals harvested during recording in the slice (A) or after dispersion (B) are Kiss1 positive. C, There was no difference in the magnitude of INaP in ARC Kiss1 neurons between high-E2 and low-E2 animals. D, In ARC Kiss1 neurons, Kiss1 mRNA expression was decreased with high-E2 treatment vs vehicle treatment; however, NaV1.1α and NaVβ2 mRNA expression from high-E2 groups were not different from vehicle-treated OVX females. ***, P < .001, n = 5; Student's t test.
To see whether the increase in the amplitude of INaP in AVPV/PeN Kiss1 neurons was correlated to an increased mRNA expression of NaVα and/or the NaVβ subunits, we first determined which α- and β-subunits were expressed in AVPV/PeN Kiss1 neurons, and then we compared the expression levels in high-E2 vs low-E2 animals using quantitative PCR on pooled cells. Based on sc RT-PCR, NaV1.1α, NaV1.2α, NaV1.6α, NaVβ2 and NaVβ4 were all expressed in AVPV/PeN Kiss1 neurons (n = 50 cells from four animals). Based on qPCR, the mRNA expression of NaVα and NaVβ subunits in AVPV/PeN Kiss1 neurons were as follows: NaV1.2α was similar to or greater than NaV1.6α, which was greater than NaV1.1α (P < .05; ANOVA, n = 3 animals), whereas NaVβ2 was the predominant β-subunit expressed in these cells, and NaVβ4 mRNA was barely detectable. Moreover, our qPCR results showed that NaV1.1α mRNA expression in AVPV/PeN Kiss1 neurons was increased by 2- to 3-fold, and the β2-subunit was increased by 2-fold in high-E2 compared with low-E2 females (Figure 2B). Also, as found previously in tissue, Kiss1 mRNA was significantly increased in Kiss1 neuronal pools in high-E2 vs low-E2 groups (Figure 2B). In addition, NaV1.2α (high-E2, 1.61 ± 0.22; low-E2, 1.03 ± 0.04) and NaV1.6α (high-E2, 2.40 ± 0.43; low-E2, 1.22 ± 0.19) were significantly increased in AVPV/PeN Kiss1 neuronal pools from high-E2 females (P < .05; n = 5 animals in each group). In ARC Kiss1 neurons, NaV1.2α was greater than NaV1.6α which was greater than NaV1.1α (P < .05–0.01; ANOVA, n = 3 animals), and NaVβ2, but not β4, was also expressed. Comparing Kiss1 neurons from OVX vehicle-treated vs high-E2 animals revealed that NaV1.1α and Navβ2 mRNA levels were not altered by E2 treatment, whereas Kiss1 mRNA levels were greatly reduced (Figure 2D). Also, similar to NaV1.1α, the mRNA levels of NaV1.2α and NaV1.6α were also not regulated by E2 (data not shown).
Persistent sodium current and firing properties of AVPV/PeN Kiss1 neurons
Given that persistent sodium currents are critical for generating repetitive firing in the face of depolarizing synaptic input, we used the relatively selective INaP blocker riluzole (28) to abrogate INaP to examine the contribution of INaP to the overall excitability of AVPV/PeN Kiss1 neurons. In voltage clamp, riluzole (10 μM) robustly inhibited the INaP (74.1% ± 3.3%, n = 6; Figure 3A) but had negligible effects (<10% inhibition) on INaT (Figure 3A, inset). In current clamp, riluzole decreased the baseline firing activity of Kiss1 neurons by 88% ± 3.5% (n = 3) (Figure 3B). Moreover, riluzole attenuated the sustained firing induced by a depolarizing current (20 pA) injection by greater than 2-fold (Figure 3, C and D).
Figure 3.
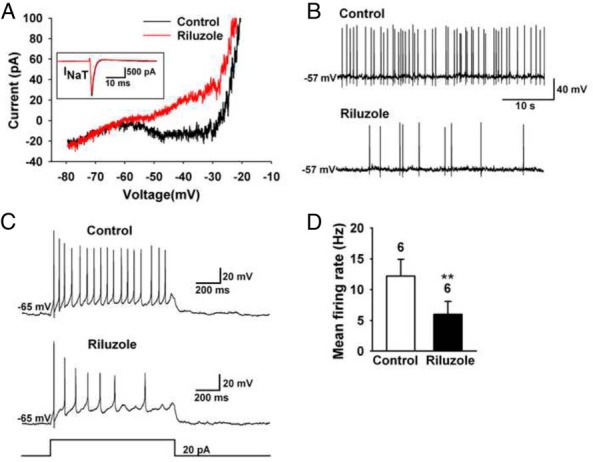
Riluzole blocked INaP and inhibited firing in AVPV/PeN Kiss1 neurons. A, Representative recordings showing that treatment with riluzole (10 μM) for 10 minutes inhibited the INaP in an AVPV/PeN neuron. The inset shows that the INaT was only slightly inhibited (<10%) by riluzole. B, Representative recording showing that riluzole inhibited the spontaneous firing of an AVPV/PeN neuron by 90%. C, The current-induced firing before and after exposure to riluzole. The current injection protocol (20 pA, 1 sec duration) is illustrated at the bottom. D, Summary of the inhibitory effects of riluzole (illustrated in panel C) on the mean firing rate. **, P < .01, n = 6; Student's t test.
Because INaP collaborates with IT to generate repetitive burst firing and we found that IT in AVPV/PeN Kiss1 neurons was augmented by 6-fold in high-E2 females (5), we measured CaV3.1 mRNA levels in these neurons. Indeed, high-E2 compared with low-E2 increased the mRNA expression of CaV3.1 by 2.5-fold (Figure 4A). In high-E2 animals, we also ascertained whether INaP could modulate the transient calcium channel-mediated rebound burst firing (see Materials and Methods). AVPV/PeN Kiss1 neurons typically generated two to four action potentials during rebound burst firing under control conditions (Figure 4B). However in the presence of the INaP blocker riluzole (10 μM), the rebound burst firing was reduced to one to two spikes, and the interspike interval between the first and second spikes increased from 12 to 18 milliseconds. Riluzole had no effect on the amplitude of T-type calcium current (Figure 4B, inset). The summarized results in Figure 4, C and D, show that inhibiting INaP by riluzole significantly reduced the mean number of action potentials (ie, fast sodium spikes) generated during the rebound burst from 3.0 ± 0.37 to 1.5 ± 0.22 spikes and increased the interspike interval from 13.3 ± 1.3 to 19.1 ± 2.0 milliseconds (n = 6). Finally, because both IT and INaP are critical for generating spontaneous firing and both are augmented in AVPV/PeN Kiss1 neurons by E2, we examined the spontaneous firing of these neurons in the presence of ionotropic glutamate and GABAA receptor blockers (10 μM CNQX, 50 μM AP-5, and 100 μM picrotoxin). As one would predict, the mean firing rate of AVPV/PeN Kiss1 neurons from high-E2 vs low-E2 females was significantly increased (3.7 ± 0.6 Hz in high-E2 vs 1.5 ± 0.2 Hz in low-E2, P < .05; Figure 5). However, ARC Kiss1 neurons from low-E2 and high-E2 animals were not significantly different in the mean firing rate (0.04 ± 0.02 Hz for low-E2 vs 0.06 ± 0.03 Hz for high-E2).
Figure 4.

INaP modulates T-type calcium channel-mediated rebound burst firing. A, qPCR measurements of CaV3.1 mRNA in AVPV/PeN Kiss1 neuronal pools (three pools per animal) from low-E2 and high-E2 mice (n = 5 animals in each group). **, P < .01, Student's t test. B, Representative recordings showing that in control conditions hyperpolarizing a cell to −95 mV for 1 second from −60 mV induced rebound burst firing with four action potentials (upper trace); however, in the presence of riluzole (10 μM), the same hyperpolarization from −60 mV induced rebound burst firing with only two action potentials, and the interspike interval from the first to the second action potential increased from 12 milliseconds to 18 milliseconds. The inset (middle trace) shows that riluzole had no effect on the T-type calcium current amplitude. C and D, Summary of the effects of riluzole on the number of action potentials (spikes) in the rebound burst and interspike intervals. **, P < .01, n = 6; Student's t test.
Figure 5.
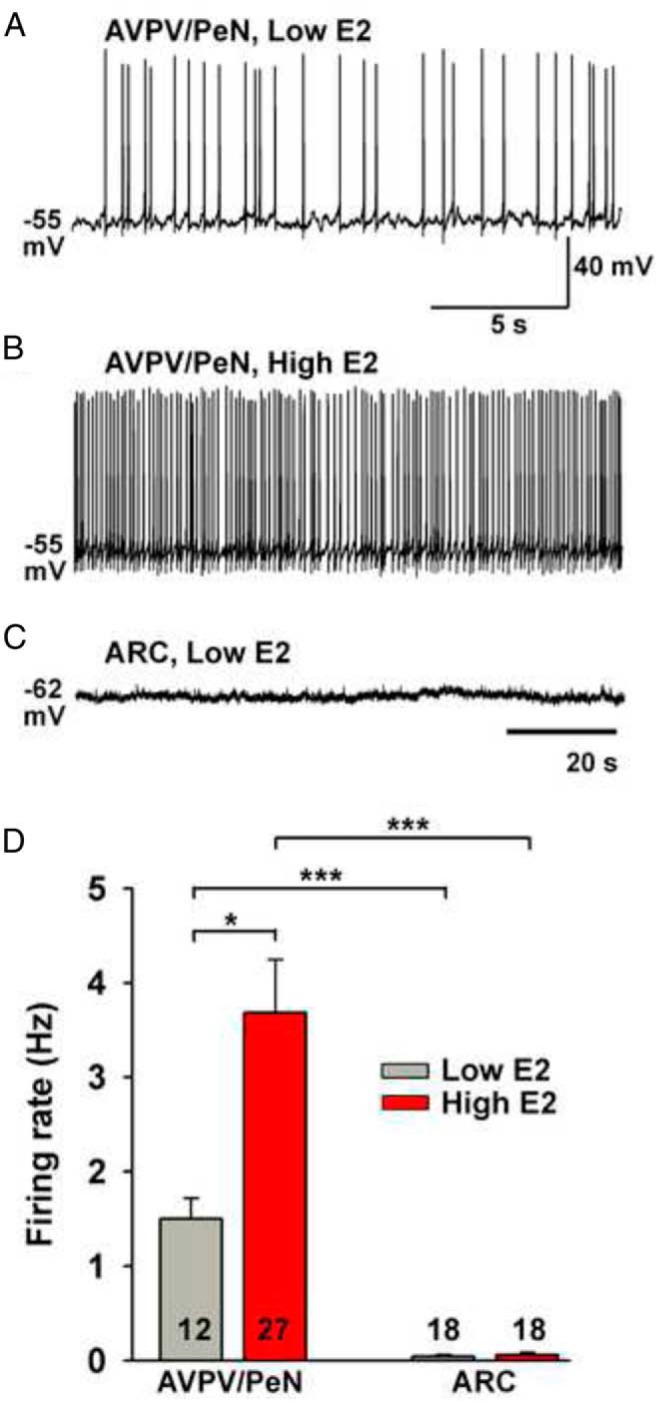
E2 increases the excitability of AVPV/PeN Kiss1 neurons. A and B, The spontaneous firing rate of Kiss1 neurons was increased in high-E2 vs low-E2 animals in the presence of ionic glutamatergic and GABAergic receptor blockers (10 μM CNQX, 50 μM AP-5, and 100 μM picrotoxin). C, A representative recording showing that E2-treated ARC Kiss1 neurons are silent in the presence of ionic glutamatergic and GABAergic receptor blockers. D, Summary of the effects E2 treatment on the spontaneous firing of AVPV/PeN and ARC Kiss1 neurons. *, P < .05; ***, P < .001, Student's t test.
Discussion
We have discovered a fundamental difference between AVPV/PeN and ARC Kiss1 neurons that may account for the dramatic differences in their spontaneous firing activity: AVPV/PeN Kiss1 neurons express a persistent sodium current that is approximately 4-fold greater than that of ARC Kiss1 neurons. The activation threshold and peak voltage were more negative in AVPV/PeN Kiss1 neurons than ARC Kiss1 neurons. Moreover, the resting membrane potential of AVPV/PeN Kiss1 neurons (−54 mV) was close to the threshold potential for activation of INaP, whereas the resting membrane potentials of ARC Kiss1 neurons (−64 mV) was significantly hyperpolarized relative to the threshold potential of INaP (4, 5). As shown previously, AVPV/PeN Kiss1 neurons tend to fire spontaneously (5, 26, 29), whereas ARC neurons are mostly silent except when they are depolarized to the INaP threshold (4, 26). Therefore, the more depolarized resting membrane potential in combination with the expression of the INaP may account for the fundamental differences in the spontaneous activity and burst firing capability of AVPV/PeN vs ARC Kiss1 neurons.
Although all of the adult NaVα subunits were detected and regulated by E2 in AVPV/PeN Kiss1 neurons, we found that NaV1.1 was the most highly up-regulated by a surge-inducing dose of E2. The β2 subunit, which was also expressed and highly up-regulated by E2 in AVPV/PeN Kiss1 neurons, in combination with the pore-forming α-subunit will generate the persistent sodium current (8–10). As predicted from the regulation of the subunit mRNA expression, INaP was augmented by 2-fold in AVPV/PeN Kiss1 neurons from high-E2 females. Neither NaV1α nor NaVβ mRNA expression was regulated by E2 in ARC Kiss1 neurons, which corresponded to the lack of E2 effects on INaP in these neurons. Therefore, it appears that there is a close correlation between ion channel mRNA expression and function in Kiss1 neurons as shown previously in other hypothalamic neurons (23, 30). Moreover, INaP in combination with the other endogenous conductances in Kiss1 neurons promoted the burst firing that is required for kisspeptin release (see below). Indeed, selective blockade of INaP with riluzole significantly diminished the ability of Kiss1 neurons to fire spontaneously and to burst fire after a hyperpolarizing stimulus.
Persistent sodium currents have been reported to drive spontaneous firing and rhythmic (pacemaking) burst firing in many CNS neurons (15, 17–19, 31, 32). Likewise, INaP appears to play a critical role in the control of spontaneous repetitive firing and T-type calcium channel-mediated burst firing of AVPV/PeN Kiss1 neurons. The function of INaP in different cells is determined by its interaction with other subthreshold conductances including IT, Ih, and inwardly rectifying potassium conductances (20, 32, 33). For example, hippocampal CA1 pyramidal neurons are normally quiescent, but they express a robust INaP and fire spontaneously when depolarized by muscarinic agonists that bring the membrane potential into the activation voltage range of INaP (15). Therefore, to drive spontaneous firing, the resting membrane potential of the cell should be close to or above the activation threshold of INaP. For AVPV/PeN Kiss1 neurons, the mean resting membrane potential is −54 mV, which is right at the activation threshold of INaP; thus, they can fire spontaneously (5, 7, 27). In contrast, the resting membrane potential of ARC Kiss1 neurons is −64 mV, which is farther from the activation threshold of INaP (−50 mV); therefore, they are mostly quiescent except when they are depolarized by glutamate, neurokinin B (4, 34), and other metabotropic receptors (eg, leptin receptor) that are coupled to activation of canonical transient receptor potential channels (TRPCs) (25). With depolarizing stimuli, which can be induced with current injection, INaP increases exponentially, and AVPV/PeN neurons fire in a repetitive high frequency manner. Indeed, we found that blockade of INaP by riluzole attenuated spontaneous repetitive firing (ie, Figure 3B). Furthermore, Amarillo et al (20) predicted that INaP in combination with IT will enhance burst firing in thalamocortical neurons, due to the fact that the activation of IT depolarizes the membrane potential into the activation range of INaP, which boosts the generation of action potentials during a burst firing event. As predicted, riluzole significantly diminished the number of spikes during rebound burst firing of AVPV/PeN Kiss1 neurons (ie, Figure 4).
Generally, high-frequency burst firing is required for peptide release from neurons (35–37). Studies from Liu et al (3) confirmed that kisspeptin release from AVPV/PeN Kiss1 neurons is frequency dependent, and a minimum of 5 Hz stimulation is required to induce kisspeptin-mediated excitation of GnRH neurons. It is noteworthy that T-type calcium channel-mediated burst firing reaches frequencies of 15–200 Hz in AVPV/PeN Kiss1 neurons, which is an ideal range for triggering the release of kisspeptin (3, 5). On the other hand, low-frequency spontaneous firing would most probably not evoke kisspeptin release but could induce GABA release from AVPV/PeN Kiss1 neuronal terminals (3). Activation of GABAA receptors causes short-lived excitation of GnRH neurons (38, 39), whereas activation of the Gq-coupled kisspeptin receptor (also known as GPR54) causes a sustained depolarization that lasts tens of minutes in an in vitro slice preparation (1, 40, 41). One caveat is that high-frequency stimulation may also release more GABA, which could spill over and activate GABAB receptors postsynaptically in GnRH neurons (42). Although activation of GABAB receptors hyperpolarizes GnRH neurons via activation of G protein-coupled K+ channels (43–45), this hyperpolarization could facilitate the subsequent actions of kisspeptin by recruiting more T-type calcium channels through deinactivation of IT and activation of Ih at more hyperpolarized potentials (5, 23). This scenario needs to be elucidated using optogenetic tools to differentially evoke the release of GABA or kisspeptin from these AVPV/PeN neurons and study the complexity of the postsynaptic responses using select pharmacological ligands. However, previous and present findings clearly show that IT, Ih, and INaP are all up-regulated by surge-inducing doses of E2, and the interaction of INaP and IT enhances the spontaneous activity and high-frequency burst firing that is necessary for the release of kisspeptin from AVPV/PeN neurons.
Acknowledgments
We thank Ms Uyen-Vy Navarro for excellent technical assistance and Dr Casey C Nestor for editorial comments on earlier versions of the manuscript.
The content of this work is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.
This work was supported by National Institutes Health R01 Grants NS 038809 (to M.J.K.), NS 043330 (to O.K.R.), and DK 068098 (to M.J.K. and O.K.R.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- aCSF
- artificial cerebral spinal fluid
- AP-5
- DL-2-amino-5-phosphonopentanoic acid
- ARC
- arcuate nucleus
- AVPV/PeN
- anteroventral periventricular/periventricular preoptic nucleus
- CNQX
- 6-cyano-7-nitroquinoxaline-2,3-dione
- CNS
- central nervous system
- E2
- 17β-estradiol
- GABA
- γ-aminobutyric acid
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GFP
- green fluorescent protein
- Ih
- hyperpolarization-activated, cyclic nucleotide-gated current
- INaP
- persistent sodium current
- INaT
- transient, rapidly inactivating, sodium current
- IT
- transient calcium current
- Kiss1
- kisspeptin
- OVX
- ovariectomy
- qPCR
- quantitative real-time PCR
- TRPC
- transient receptor potential channel
- TTX
- tetrodotoxin
- ZT
- zeitgeber time.
References
- 1. Zhang C, Roepke TA, Kelly MJ, Rønnekleiv OK. Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28:4423–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang C, Bosch MA, Rønnekleiv OK, Kelly MJ. Kisspeptin activation of TRPC4 channels in female GnRH neurons requires PIP2 depletion and cSrc kinase activation. Endocrinology. 2013;154:2772–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu X, Porteous R, d'Anglemont de Tassigny X, et al. Frequency-dependent recruitment of fast amino acid and slow neuropeptide neurotransmitter release controls gonadotropin-releasing hormone neuron excitability. J Neurosci. 2011;31:2421–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gottsch ML, Popa SM, Lawhorn JK, et al. Molecular properties of Kiss1 neurons in the arcuate nucleus of the mouse. Endocrinology. 2011;152:4298–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang C, Tonsfeldt KJ, Qiu J, et al. Molecular mechanisms that drive estradiol-dependent burst firing of Kiss1 neurons in the rostral periventricular preoptic area. Am J Physiol Endocrinol Metab. 2013;305:E1384–E1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Navarro VM, Gottsch ML, Wu M, et al. Regulation of NKB pathways and their roles in the control of Kiss1 neurons in the arcuate nucleus of the male mouse. Endocrinology. 2011;152:4265–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piet R, Boehm U, Herbison AE. Estrous cycle plasticity in the hyperpolarization-activated current Ih is mediated by circulating 17β-estradiol in preoptic area kisspeptin neurons. J Neurosci. 2013;33:10828–10839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Isom LL, Ragsdale DS, De Jongh KS, et al. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–442. [DOI] [PubMed] [Google Scholar]
- 9. Qu Y, Curtis R, Lawson D, et al. Differential modulation of sodium channel gating and persistent sodium currents by the β1, β2, and β3 subunits. Mol Cell Neurosci. 2001;18:570–580. [DOI] [PubMed] [Google Scholar]
- 10. Aman TK, Grieco-Calub TM, Chen C, et al. Regulation of persistent Na current by interactions between β subunits of voltage-gated Na channels. J Neurosci. 2009;29:2027–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol. 2012;590:2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol. 1996;58:349–362. [DOI] [PubMed] [Google Scholar]
- 13. Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar purkinje neurons. J Neurosci. 1997;17:4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carter BC, Giessel AJ, Sabatini BL, Bean BP. Transient sodium current at subthreshold voltages: activation by EPSP waveforms. Neuron. 2012;75:1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamada-Hanff J, Bean BP. Persistent sodium current drives conditional pacemaking in CA1 pyramidal neurons under muscarinic stimulation. J Neurosci. 2013;33:15011–15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci. 2010;30:7401–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jackson AC, Yao GL, Bean BP. Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J Neurosci. 2004;24:7985–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. [DOI] [PubMed] [Google Scholar]
- 19. Oka Y. Characterization of TTX-resistant persistent Na+ current underlying pacemaker potentials of fish gonadotropin-releasing hormone (GnRH) neurons. J Neurophysiol. 1996;75:2397–2404. [DOI] [PubMed] [Google Scholar]
- 20. Amarillo Y, Zagha E, Mato G, Rudy B, Nadal MS. The interplay of seven subthreshold conductances controls the resting membrane potential and the oscillatory behavior of thalamocortical neurons. J Neurophysiol. 2014;112:393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bronson FH, Vom Saal FS. Control of the preovulatory release of luteinizing hormone by steroids in the mouse. Endocrinology. 1979;104:1247–1255. [DOI] [PubMed] [Google Scholar]
- 22. Bosch MA, Tonsfeldt KJ, Rønnekleiv OK. mRNA expression of ion channels in GnRH neurons: subtype-specific regulation by 17β-estradiol. J Mol Cell Endocrinol. 2013;367:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang C, Bosch MA, Rick EA, Kelly MJ, Rønnekleiv OK. 17β-Estradiol regulation of T-type calcium channels in gonadotropin-releasing hormone neurons. J Neurosci. 2009;29:10552–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang C, Bosch MA, Levine JE, Rønnekleiv OK, Kelly MJ. Gonadotropin-releasing hormone neurons express K(ATP) channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. J Neurosci. 2007;27:10153–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qiu J, Fang Y, Bosch MA, Rønnekleiv OK, Kelly MJ. Guinea pig kisspeptin neurons are depolarized by leptin via activation of TRPC channels. Endocrinology. 2011;152:1503–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Croft S, Piet R, Mayer C, Mai O, Boehm U, Herbison AE. Spontaneous kisspeptin neuron firing in the adult mouse reveals marked sex and brain region differences but no support for a direct role in negative feedback. Endocrinology. 2012;153:1–10. [DOI] [PubMed] [Google Scholar]
- 27. Frazão R, Cravo RM, Donato J, Jr, et al. Shift in Kiss1 cell activity requires estrogen receptor α. J Neurosci. 2013;33:2807–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lamas JA, Romero M, Reboreda A, Sánchez E, Ribeiro SJ. A riluzole- and valproate-sensitive persistent sodium current contributes to the resting membrane potential and increases the excitability of sympathetic neurones. Pflugers Arch. 2009;458:589–599. [DOI] [PubMed] [Google Scholar]
- 29. Ducret E, Gaidamaka G, Herbison AE. Electrical and morphological characteristics of anteroventral periventricular nucleus kisspeptin and other neurons in the female mouse. Endocrinology. 2010;151:2223–2232. [DOI] [PubMed] [Google Scholar]
- 30. Roepke TA, Qiu J, Smith AW, Rønnekleiv OK, Kelly MJ. Fasting and 17β-estradiol differentially modulate the M-current in neuropeptide Y neurons. J Neurosci. 2011;17:11825–11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu N, Enomoto A, Tanaka S, et al. Persistent sodium currents in mesencephalic v neurons participate in burst generation and control of membrane excitability. J Neurophysiol. 2005;93:2710–2722. [DOI] [PubMed] [Google Scholar]
- 32. Li J, Baccei ML. Pacemaker neurons within newborn spinal pain circuits. J Neurosci. 2011;31:9010–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Surmeier DJ, Mercer JN, Chan CS. Autonomous pacemakers in the basal ganglia: who needs excitatory synapses anyway? Curr Opin Neurobiol. 2005;15:312–318. [DOI] [PubMed] [Google Scholar]
- 34. de Croft S, Boehm U, Herbison AE. Neurokinin B activates arcuate kisspeptin neurons through multiple tachykinin receptors in the male mouse. Endocrinology. 2013;154:2750–2760. [DOI] [PubMed] [Google Scholar]
- 35. Bicknell RJ. Optimizing release from peptide hormone secretory nerve terminals. J Exp Biol. 1988;139:51–65. [DOI] [PubMed] [Google Scholar]
- 36. Shakiryanova D, Tully A, Hewes RS, Deitcher DL, Levitan ES. Activity-dependent liberation of synaptic neuropeptide vesicles. Nat Neurosci. 2005;8:173–178. [DOI] [PubMed] [Google Scholar]
- 37. Masterson SP, Li J, Bickford ME. Frequency-dependent release of substance P mediates heterosynaptic potentiation of glutamatergic synaptic responses in the rat visual thalamus. J Neurophysiol. 2010;104:1758–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DeFazio RA, Heger S, Ojeda SR, Moenter SM. Activation of A-type γ-aminobutyric receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16:2872–2891. [DOI] [PubMed] [Google Scholar]
- 39. Herbison AE, Moenter SM. Depolarising and hyperpolarising actions of GABAA receptor activation on gonadotrophin-releasing hormone neurons: towards an emerging consensus. J Neuroendocrinol. 2011;23:557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu X, Lee K, Herbison AE. Kisspeptin excites gonadotropin-releasing hormone (GnRH) neurons through a phospholipase C/calcium-dependent pathway regulating multiple ion channels. Endocrinology. 2008;149:4605–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology. 2008;149:1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scanziani M. GABA spillover activates postsynaptic GABAB receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. [DOI] [PubMed] [Google Scholar]
- 43. Lagrange AH, Rønnekleiv OK, Kelly MJ. Estradiol-17β and μ-opioid peptides rapidly hyperpolarize GnRH neurons: a cellular mechanism of negative feedback? Endocrinology. 1995;136:2341–2344. [DOI] [PubMed] [Google Scholar]
- 44. Zhang C, Bosch MA, Rønnekleiv OK, Kelly MJ. GABAB receptor mediated inhibition of GnRH neurons is suppressed by kisspeptin-GPR54 signaling. Endocrinology. 2009;150:2388–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu X, Herbison AE. Estrous cycle- and sex-dependent changes in pre- and postsynaptic GABAB control of GnRH neuron excitability. Endocrinology. 2011;152:1–9. [DOI] [PubMed] [Google Scholar]