

Abstract
The 18-kDa translocator protein (TSPO), also known as the peripheral benzodiazepine receptor, is a transmembrane protein in the outer mitochondrial membrane. TSPO has long been described as being indispensable for mitochondrial cholesterol import that is essential for steroid hormone production. In contrast to this initial proposition, recent experiments reexamining TSPO function have demonstrated that it is not involved in steroidogenesis. This fundamental change has forced a reexamination of the functional interpretations made for TSPO that broadly impacts both basic and clinical research across multiple fields. In this minireview, we recapitulate the key studies from 25 years of TSPO research and concurrently examine their limitations that perhaps led towards the incorrect association of TSPO and steroid hormone production. Although this shift in understanding raises new questions regarding the molecular function of TSPO, these recent developments are poised to have a significant positive impact for research progress in steroid endocrinology.
Translocator protein (TSPO), also known as the peripheral benzodiazepine receptor (PBR), was first identified in 1977 based on its distinct pharmacology with high affinity binding to benzodiazepines in peripheral tissues (1–4). The term “peripheral” was used to distinguish it from the plasma membrane “central” benzodiazepine receptor, a complex together with the γ-aminobutyric acid type A receptor that is important for inhibitory neurotransmission in the central nervous system (5, 6). In comparison, TSPO/PBR is present in mitochondria, and its functional properties have been a topic of active research for the past 25 years (reviewed in Refs. 7–9).
The pharmacology of benzodiazepines provided most of the early information about TSPO. Benzodiazepine Ro5-4864 [4′-chlorodiazepam; 7-Chloro-5-(4-chlorophenyl)-1,3-dihydro-1-methyl-2H-1,4-benzodiazepin-2-one] and a nonbenzodiazepine PK11195 [an isoquinoline carboxamide derivative, 1-(2-Chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide] were initially established as prototypical TSPO-binding chemicals, because they bind to TSPO but not to γ-aminobutyric acid type A receptor (10, 11). Based on thermodynamic studies (12), and their opposing effects on neuronal seizures (13), PK11195 was classified as an antagonist and Ro5-4864 as an agonist. This pharmacology has been extensively used in attempts to elucidate the physiological relevance of TSPO (7). Although these studies did not readily reveal TSPO function, the ability of these chemicals in detecting TSPO with reasonable accuracy, and the pathological TSPO up-regulation seen at sites of inflammation led to the development of TSPO as a diagnostic target (14). Radiolabeled forms of these chemicals that bind TSPO could be used to detect inflammatory lesions in vivo in a variety of human diseases using positron emission tomography (15, 16). Clinical trials for different TSPO-binding agents for this application are either in progress or have been recently completed (http://clinicaltrials.gov; identifiers: NCT02181582, NCT01547780, NCT01527695, NCT01428505, NCT02086240, NCT02062099, and NCT01613703) and, thus, remain an area of active research.
TSPO amino acid sequence shows conservation throughout evolution (17). TSPO in the photosynthetic bacteria Rhodobacter sphaeroides shows a 33.5% identity to human TSPO. Functional conservation was also highlighted in that mammalian TSPO could substitute for the bacterial version in negatively regulating photosynthesis genes in response to oxygen (18). Both human and mouse Tspo genes translate to a 169-amino acid protein with 81% sequence homology (19, 20). Expression of TSPO has been reported in different tissues including heart, brain, lung, spleen, testis, ovary, adrenal, kidney, bone marrow, salivary gland, adipose tissue, skin, and liver (21–23); and within these tissues, TSPO expression is regional and/or cell type specific (24).
In eukaryotic cells, subcellular fractionation and detection using TSPO-binding chemicals showed that TSPO was enriched in the outer mitochondrial membrane (OMM) (21, 25, 26). To a lesser extent, binding sites have also been observed in the plasma membrane of erythrocytes (27). Antibody-based labeling confirmed the predominant presence of TSPO in the mitochondria but also suggested localization in plasma membrane of adrenocortical cells (28), the nucleus in adult male germ cells (29), and some types of cancer cells (30, 31). However, when a highly specific rabbit monoclonal antibody against TSPO recently released in the market (catalog number ab109497; Abcam) was used, TSPO expression in adult male germ cells was not detected (24). This provided an indication that immunolocalization using polyclonal antisera against TSPO may detect nonspecific epitopes. In addition, use of TSPO-binding chemicals for precise localization may also lead to detection of alternate targets (32, 33). Therefore, it is prudent to consider that the existence of nonmitochondrial forms of mammalian TSPO remain to be definitively established. That being said, in lower eukaryotes like Arabidopsis thaliana, TSPO has been localized to the endoplasmic reticulum and Golgi stacks (34). Due to these distinctions, a homologous function for TSPO across kingdoms is viewed as unlikely and has certainly not been established.
The first evidence that mammalian TSPO could be associated with steroid hormone production was the observation that treatment of steroidogenic cells with TSPO-binding chemicals could induce progesterone production (35, 36).
Mitochondrial Cholesterol Transport and Steroidogenesis
Cholesterol is the common precursor for all steroid hormones. The cleavage of the side chain of cholesterol to form pregnenolone was determined to be the first step in the biosynthesis of steroids. This reaction occurred via the activity of the P450 side chain cleavage enzyme (P450scc/CYP11A1), which resided on the matrix side of the inner mitochondrial membrane (IMM) (37). Because the IMM is relatively cholesterol poor (38), it was determined that cholesterol would have to cross the OMM, the aqueous intermembrane space, and the IMM, to reach the P450scc. It was further determined that because cholesterol is hydrophobic, it could not traverse the aqueous space in a time frame rapid enough to produce steroids as quickly as was observed (39). Studies in the late 1950s in cultured adrenal tissues concluded that the nature of this transfer in all probability required the rapid de novo synthesis of a protein whose task was to aid in the transfer of cholesterol to the IMM (40). Studies in the early and mid 1960s confirmed that the putative regulator(s) of cholesterol transfer and hence, steroid hormone synthesis, was a protein moiety (41). At that point in time the search for the putative protein regulator of steroidogenesis became the focus of a number of laboratories and over the ensuing decades candidate proteins such as the sterol carrier protein 2 (SCP2) (42), the steroidogenesis-activator polypeptide (43, 44), the TSPO, and the steroidogenic acute regulatory protein (StAR) were placed in contention for being the protein responsible for the acute regulation of cholesterol transfer and, thus, steroidogenesis.
Each of the proteins mentioned appeared to have characteristics that made them credible candidates for this function. However, research progress on SCP2 based on studying gene-deleted mice ruled out an involvement for SCP2 in steroidogenesis (45). After detailed evaluation, steroidogenesis-activator polypeptide was identified as a peptide fragment of the glucose-regulated protein 78 (GRP78) (46), a master regulator of the unfolded protein response (47). As a result, GRP78 knockout mice died at an early embryonic stage (48), but studies examining GRP78 conditional knockouts showed a phenotype in oncogenic signaling (49). Examination of GRP78 structure showed a nucleotide-binding domain and a peptide substrate-binding domain (50), without any characteristics that indicate potential for lipid transport. Therefore, a function for GRP78 in acute regulation of cholesterol transport appeared unlikely. In contrast, TSPO and StAR have been topics of continued investigation in great detail.
The protein now known as StAR was first observed in cultured adrenal, Leydig and corpus luteum cells (51, 52), and was subsequently cloned (53). It was determined that expression of StAR in steroidogenic cells in the absence of hormone stimulation resulted in an increase in steroid biosynthesis (53). It was further demonstrated that expression of StAR in a nonsteroidogenic cell line rendered steroidogenic through transfection of the cholesterol side chain cleavage system, also resulted in a significant increase in steroid production (54). Perhaps the most compelling observation supporting the indispensable role of StAR in steroidogenesis was the finding that mutations in the StAR gene were responsible for the potentially fatal disease lipoid congenital adrenal hyperplasia (lipoid CAH), a disease in which severely afflicted individuals are unable to synthesize steroids (54). Examination of StAR-null mice showed a phenotype that was essentially identical to the human lipoid CAH phenotype, strongly supporting the indispensable role for StAR in steroidogenesis (55). StAR is translated as a preprotein containing an N-terminal cleavable mitochondrial targeting sequence. Crystal structure of the StAR-related lipid transfer (START) domain confirmed the potential of StAR to function as a cholesterol-shuttling protein (56).
In the case of TSPO, the first observation that chemicals that bind to this “receptor” could stimulate steroid biosynthesis was made in Y1 adrenal tumor cells in 1989 (35). Subsequent experiments in the MA-10 Leydig tumor cell line reinforced the hypothesis that synthetic small molecule drugs that bind TSPO could increase steroid hormone production (36). In support of these xenobiotic effects, a naturally occurring intracellular protein, the acyl-CoA-binding protein (ACBP/ACBD1), previously known as the diazepam-binding inhibitor that interacts with TSPO, increased steroid synthesis in steroidogenic cells (57–60). Subsequent experiments using knockdown technology to inhibit TSPO expression (61, 62) also indicated that the presence of TSPO was essential for steroid synthesis. Further, it was reported that the presence of a higher affinity ligand-binding site on TSPO was responsible for the constitutive biosynthesis of steroids observed in the R2C rat Leydig tumor cell line (63). Supporting this mechanism, targeted disruption of TSPO in these R2C cells was shown to obliterate their ability to produce steroid hormones (64). TSPO amino acid sequence also showed the existence of a cholesterol-binding amino acid consensus (CRAC) motif, suggesting a molecular mechanism for cholesterol binding and/or transport (65). Collectively, these points of evidence led to a model that TSPO was a cholesterol transport channel in the OMM, and the conclusion that it played an indispensable role in steroidogenesis (66). However, this did not entirely fit with initial mechanisms proposed for StAR action.
Mitochondrial cholesterol transport mediated by StAR was initially proposed as a shuttling event in that either StAR transfers cholesterol molecules, one at a time, to the IMM; or the START domain of StAR may alter the mitochondrial membrane to allow for the passage of cholesterol, before reaching its final resting place in the mitochondrial matrix (67). However, these models for StAR action required modifications, because deletion of 62 amino acids at the N-terminus of StAR, which disrupts mitochondrial StAR import but not the START domain structure, did not affect its cholesterol transfer and steroid production capabilities (68, 69). Moreover, a translocase of outer membrane 20 (Tom20)-StAR fusion protein affixed to the OMM could induce steroidogenesis, suggesting that StAR entry into the mitochondrion was not necessary and that the functional effects mediated by StAR occur at the level of the OMM (70). Using these same cells expressing OMM-affixed StAR-TOM20 fusion protein, it was demonstrated that TSPO knockdown using antisense oligonucleotides could reduce steroidogenic capacity (62). These findings ultimately led to a conclusion that there was a functional cooperation between StAR and TSPO necessary to mediate mitochondrial cholesterol transport required for steroid hormone biosynthesis. Therefore, a model in which StAR is considered the “initiator” of cholesterol transport and TSPO is considered the “gate” for cholesterol entry into mitochondria was established (71).
Translocator Protein Is not Involved in Steroidogenesis
Recent work on TSPO has challenged the previous literature, and refutes TSPO involvement in steroidogenesis. A conditional knockout mouse with TSPO deletion in Leydig cells (TspocΔ/Δ) demonstrated that TSPO was not essential for testosterone production in vivo (72). Neither TSPO transcripts nor protein was detectable in the testes of these TspocΔ/Δ mice, and yet testis development, sperm count, and fertility were unaffected. Subsequently, it was demonstrated that global TSPO deletion (Tspo−/−) in mice did not affect viability, fertility, and the ability to generate steroid hormones (23). Tspo−/− mice did not show any apparent abnormalities and produced both gonadal and adrenal steroid hormones at levels similar to control mice. This was a rather surprising discovery, because it was in direct contrast to a highly cited review article that described unpublished data indicating that Tspo−/− mice were early embryonic lethal (66). In an attempt to investigate why the above in vivo findings did not agree with previously published in vitro models, experiments using small interfering RNA-knockdown of TSPO expression were performed in different steroidogenic cell lines (MA-10, MLTC, Y1, and R2C). A decrease in TSPO expression of more than 80% in these cell lines showed no adverse effect on steroid hormone production (23). This was again in direct contrast to previous conclusions that TSPO knockdown inhibited steroidogenic capacity (62). As further evidence, it was demonstrated that complete disruption of TSPO in the MA-10 mouse Leydig cell line (MA-10:TspoΔ/Δ) did not have any effect on steroidogenesis (73). Again, this contradicted previous reports that targeted mono-allelic disruption of TSPO in the R2C rat Leydig cell line obliterated the steroidogenic potential of these cells (64) and that cells with more than 70% TSPO knockdown cannot survive (74, 75). In an attempt to investigate whether the steroidogenic response mediated by pharmacology of TSPO-binding chemicals was distinct from physiological TSPO function, effect of the prototypical TSPO-binding reagent PK11195 was investigated. Exposure of both Tspo intact and Tspo knockout MA-10 cells to PK11195 showed similar increases in steroid hormone production, demonstrating that the effect of PK11195 on steroidogenesis is not mediated through TSPO (73). This suggests that previous studies reporting a steroidogenic effect for TSPO-binding chemicals including PK11195 (35, 36) were observing off-target effects.
The global null Tspo−/− phenotype was recently reproduced in an unconnected study that independently generated another Tspo−/− mouse (76). This study also showed that isolated steroidogenic mitochondria devoid of TSPO did not have any deficits in cholesterol transport, suggesting that TSPO was not involved in this mitochondrial process (76).
Limitations of Studies Linking Translocator Protein to Steroidogenic Function
There were 2 key connections that initially made a function for TSPO in steroidogenesis plausible; they were its localization to the OMM, and its high level of expression noted in steroidogenic cells (25). However, although these provided interesting associations, they were not direct indications of its function. As another layer of support, often presented was the demonstration that hypophysectomy could decrease TSPO expression in the adrenal and testes (77). If this study is more closely examined, rats were analyzed 23 days after hypophysectomy, during which body weights had decreased significantly (to 55% of the control rats), and the adrenal was in a severe state of involution (only 28% the weight of control adrenals) (77). It should be noted that loss of adrenal weight after hypophysectomy is mainly due to shrinkage of the adrenal cortex (78); however, depletion of Ro5-4864 binding (used as a measure of TSPO) in this study was calculated based on whole adrenal weight (77). It should also be noted that depletion of mitochondrial CYP11A1 activity after hypophysectomy shows an almost linear drop to just 10% of control values in just 7 days, a time point when the adrenal weight was still 65% compared with control adrenals (79). A subsequent study by another group demonstrated that the decrease in Ro5-4864 binding in the adrenal after hypophysectomy in rats was associated with mitochondrial depletion and that changes to TSPO expression after ACTH restoration was not temporally related to the induction of steroidogenesis (80). Although this study by Cavallaro et al (80) provided an early indication that previous evidence for TSPO was clearly circumstantial, it was rarely referenced (14 citations to date), whereas mention of previous work by Anholt et al (77) was very common (115 citations to date).
TSPO pharmacology
Pharmacology of TSPO-binding chemicals was often presented as direct evidence for the role of TSPO in steroid hormone production (35, 36). Among the different TSPO-binding chemicals tested, PK11195 was the most potent inducer of steroidogenesis followed by Ro5-4864. However, the response itself was very modest (maximal levels were 80-fold lower than a response seen with human chorionic gonadotropin) and transient (progesterone production reached maximal levels within 40 minutes, with no progressive accumulation, and levels remained unchanged for the next 4 hours in such experiments). Although these observations suggested a physical response to treatment such as that resulting from membrane perturbation (81), rather than a sustained physiological response (as seen with human chorionic gonadotropin), this possibility was not investigated. Moreover, the effects of TSPO-binding chemicals were not always consistent. PK11195 and Ro5-4864 had distinct binding sites on TSPO (82, 83) and were shown to have different thermodynamic properties, and were predicted to be antagonistic and agonistic, respectively (12), even before studies linking TSPO and steroidogenesis. In agreement with this prediction and contrary to a stimulatory effect on steroidogenesis, it was demonstrated that treatment with PK11195 completely inhibited adrenal steroidogenesis stimulated by ACTH in hypophysectomized rats (84). In another study, PK11195 was shown to inhibit brain pregnenolone synthesis in adrenalectomized-castrated rats, whereas another TSPO-binding chemical FGIN-1-27 (N,N-Dihexyl-2-(4-fluorophenyl)indole-3-acetamide) was shown to increase pregnenolone production under similar conditions (85). Effects consistent with an inhibitory role for PK11195 in steroidogenesis and other effects were reported in several other studies (86–89). Also, when placental explants were exposed to Ro5-4864, a biphasic effect was observed; Ro5-4864 stimulated a 2.4-fold increase in progesterone levels at a low dose but caused a significant decrease compared with baseline at high doses (90). In another study, Ro5-4864 was found to suppress estradiol production in vivo, whereas PK11195 had no effect on ovarian steroidogenesis (91). In addition to these mixed effects, PK11195 has been reported to have other targets in the cell, including the constitutive androstane receptor (32) and the F1F0 ATP synthase (33). In essence, the fact that PK11195 effect on steroidogenesis happens in the absence of TSPO (73) indicates that previous assertions were incorrect.
ACBP/diazepam-binding inhibitor
Binding of ACBP and its processed peptides to TSPO was shown to stimulate steroid hormone production in isolated mitochondria (58). In hypophysectomized rats, although ACTH did not acutely increase the amount of adrenal ACBP or TSPO, it increased the rate of ACBP processing concurrently with hormone production (84). This ACTH-induced increase in ACBP processed peptides was interpreted as playing an important role in steroidogenesis. In support, knockdown of ACBP expression using a cholesterol-linked phosphorothioate oligodeoxynucleotide antisense sequence was shown to decrease steroid hormone production in cultured MA-10 Leydig tumor cells (92). ACBP is present in eukaryotes ranging from yeast to mammals (93) and was demonstrated to play a role in maintaining the intracellular acyl-CoA ester pool size (94) and synthesis of very long chain fatty acids and sphingolipids (95). A spontaneous mutant mouse nm1054 cataloged at The Jackson Laboratory (96, 97) was subsequently identified to also contain a mutation in the ACBP gene locus (98). Loss of ACBP in these nm1054 mice was linked to fatty acid metabolism abnormalities in skin and hair (98). The subsequent generation of Acbp knockout mice (Acbp−/−) resulted in a finding that they were early embryonic lethal at the blastocyst stage of development (99). However, a second independent Acbp−/− mouse generated showed that they were viable and fertile and developed normally but displayed delayed metabolic adaptation to weaning (100). Further studies on this latter mouse (101, 102) showed that metabolic abnormalities seen in skin and hair matched observations previously reported for the nm1054 mutation (98). There was no phenotypic evidence that indicated defects in steroid hormone production in these Acbp−/− mice. But why was the first Acbp−/− knockout mouse embryonic lethal? One explanation is that abnormal random insertion mutagenesis in embryonic stem cell clones could have resulted in lethality unrelated to ACBP deletion in the first Acbp−/− mouse line; validation to rule out this possibility was not presented (99). Nevertheless, more recent understanding of ACBP function does (101, 102), not indicate a link to steroidogenic function as previously concluded.
Cholesterol transport
Cholesterol is considered an endogenous ligand for TSPO. A CRAC motif that could bind to cholesterol was identified at the TSPO C-terminal region (103). The CRAC motif is a sequence pattern, L/V-X1–5-Y-X1–5-R/K, with a loose definition with only 1 unique residue and 2 large variable segments. It was pointed out that the genome of Streptococcus agalactiae that encodes 2094 known and hypothetical proteins, almost all of which will have no association with cholesterol, have 5737 CRAC motifs (2.7/protein and 1/122 amino acids) (104), an observation that is probably true for all organisms. Therefore, predicting a role of the CRAC motif as an indicator of cholesterol binding cannot be considered conclusive. For example, the CRAC motif in the nicotinic acetylcholine receptor was found in an energetically unfavorable location to bind cholesterol, and an alternative region of cholesterol binding was ultimately identified (105). For TSPO, there is evidence in the literature that cholesterol can interact with the CRAC motif (65, 103). Nevertheless, most functional CRAC motifs have been described in proteins that associate with cholesterol-rich membrane regions called raft microdomains, for example: myelin P0 (106) and caveolin-1 (107). Given the new findings that TSPO is not involved in steroid hormone production, it becomes necessary to ask the question whether there is enough functional evidence for TSPO in mitochondrial cholesterol transport? Direct experimental proof for mitochondrial cholesterol translocation by TSPO does not exist. As speculated previously (72), it is possible that the CRAC motif of TSPO could merely function in associating with cholesterol-rich raft microdomains. The voltage-dependent anion channel 1 on the OMM, which coprecipitates with TSPO, is a resident protein in raft microdomains (108). Therefore, TSPO membrane raft association via the CRAC motif could be the basis for its proximity to voltage-dependent anion channel 1.
Because StAR-mediated function is accomplished at the level of the OMM (70), it was believed that StAR interacts with TSPO to complete mitochondrial cholesterol transport (62). Using fluorescence resonance energy transfer (FRET), it was demonstrated that TSPO could interact with StAR in the OMM of Cos-7 cells (109). However, this same laboratory followed up on their FRET analysis using bioluminescence resonance energy transfer (BRET) and concluded that there was no evidence for StAR and TSPO interaction (110). The BRET analysis utilized cell populations rather than individual cells and 3 different cell types (CHO cells, MA-10 cells, and Cos-7 cells). Also in this report, bacterial and mammalian 2-hybrid assays failed to demonstrate a StAR-TSPO interaction (110). The authors concluded that because cells were selected for FRET analysis in their first report, it might have resulted in artifactual data (110). Nevertheless, this latter report did not fit the TSPO cholesterol transport model and was rarely referenced (15 citations to date), whereas mention of previous work to indicate that TSPO interacts with StAR was recurrent (111 citations to date).
TSPO structure
Structural features in TSPO were also presented as evidence for a role in cholesterol transport. But similar to other membrane proteins, building a reliable structural model for TSPO was a challenge. Initially, the murine TSPO was described as a 5 transmembrane α-helix (111). This was followed by a low-resolution structure of R. sphaeroides TSPO, constructed using electron cryo-microscopy (112). This formed the basis of early homology models, but these models were described to indicate that TSPO could form a hydrophobic channel-like interior core lined by the CRAC motif for cholesterol binding and translocation (8, 66, 113). Because it was not possible to assign amino acid sequences to this low-resolution TSPO structure as indicated by Korkhov et al (112), these early models were largely speculative based on presumed function at that time. A more recent high-resolution nuclear magnetic resonance (NMR) structure of TSPO showed that the side chains of the CRAC motif essential for cholesterol binding are located on the outside of the TSPO pointing towards the membrane environment (Figure 1) (83), suggesting that previous molecular dynamic simulations and homology models were not accurate.
Figure 1.
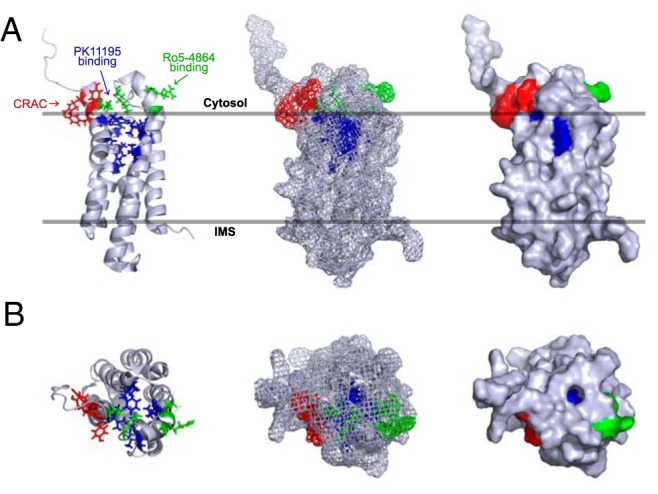
Structure of TSPO in OMM. A, Side view. TSPO has a 5 α-helix transmembrane structure. The CRAC motif (in red) is at the C- terminus (residues 147–159). Side chains of Tyr152, Tyr153, and Arg156, essential for cholesterol binding are located on the outside of the TSPO structure and point toward the membrane environment. PK11195-binding pocket (in purple) is formed by residues Ala23, Val26, Leu49, Ala50, Ile52, Trp107, Ala110, Leu114, Ala147, and Leu150, which do not involve side chains of CRAC motif amino acids. Binding site for Ro5-4864 (in green) is distinct from that of PK11195 and was identified to include residues Glu29, Arg32, Lys39, and Val154 (82). B, Top view. Cholesterol-binding side chains of the CRAC motif pointing outside of the TSPO structure and do not interfere with internal PK11195-binding site. From high-resolution NMR structure PDB ID, 2MGY (83).
The new structure for TSPO (83) supports our previous conjecture regarding position of the CRAC motif and that it could function in associating TSPO with raft microdomains in the membrane (72). Binding of PK11195 to TSPO appeared to stabilize its three dimensional structure (83). However, at the time this new structure was published, TSPO was still considered important for steroidogenesis, and accordingly, the authors attempted to relate its structural features to a cholesterol transport mechanism. Because there were no indications of a channel-like structure with a hydrophobic core as previously suggested (8), the authors speculated that the ability of cholesterol to dimerize could lead to the “known” oligomerization of TSPO, indicating that some transporters can function as dimers (83). But previous demonstration of TSPO oligomerization was not based on cholesterol dimerization. TSPO was demonstrated to oligomerize through covalent tyrosine cross-links under unique circumstances (114). In any case, the property that PK11195 binding increased cholesterol binding to TSPO polymers (114), which was used to explain a possibility for cholesterol transport (83), is not consistent. On the contrary, binding of PK11195 has been associated with the release of cholesterol from TSPO (103). This latter concept was recently used to explain why TSPO-binding chemicals could increase steroidogenesis even if TSPO is not a component of the natural steroidogenic machinery (115). Again, these arguments preceded the recent demonstration that PK11195 effect on steroidogenesis was not mediated by TSPO (73).
TSPO gene disruption
As genetic evidence, it was demonstrated that disruption of Tspo in the R2C rat Leydig cell line caused significant adverse morphological changes and obliterated steroidogenesis (64). Interestingly, disruption of one Tspo allele in this study resulted in a complete disappearance of TSPO protein. The study was based on a solitary anomalous clone of R2C cells. Subsequent studies have shown that mono allelic deletion of Tspo does not affect TSPO transcript/protein expression (23), and complete deletion of Tspo in the MA-10 mouse Leydig cell line did not affect morphology, proliferation, and/or steroidogenic function (73). Therefore, it is conceivable that other unrelated genetic aberrations associated with this R2C clone could have caused the 2–3 times slower cell proliferation, adverse morphological changes, and decrease in progesterone production as reported earlier (64). It was also shown that hormone production in these mono-allelic Tspo-disrupted R2C cells could be rescued after stable ectopic TSPO expression. However, there were clear indications that problems associated with Tspo-disrupted R2C cells could have spontaneously recovered; it was mentioned that proliferation rates rebounded after 3 months in culture and hormone production resumed after 2 years in culture without any intervention (64). Therefore, there are serious limitations to interpretations made using this mono-allelic Tspo-disrupted R2C cell model.
Another piece of genetic evidence was the report that global Tspo−/− mice were early embryonic lethal (66). The “evidence” appeared in a review article without experimental details or phenotypic characterization, but has been repeatedly referenced (297 citations to date) to indicate an important role for TSPO in basic cellular functions that included steroidogenesis (66). Two independent reports now provide solid data on phenotyping the Tspo−/− mouse and show that TSPO is not involved in viability, fertility, and steroidogenesis (23, 76). Therefore, we can only speculate that experimental problems hindered the first Tspo knockout attempt.
Human TSPO polymorphism
Mutations/polymorphisms for TSPO were previously sought and excluded in congenital lipoid adrenal hyperplasia patients (116). However, a common human polymorphism in TSPO (rs6971, leading to amino acid change Ala147Thr) has been demonstrated to cause differences in affinity of TSPO-binding chemicals used for diagnostic imaging (117, 118). Ala147 is part of the TSPO PK11195-binding pocket (83), suggesting that a change to Thr147 could indeed affect binding properties of PK11195 and other chemicals that bind to this region. Functionally, this rs6971 polymorphism was linked to adult separation anxiety in patients with depression (119). This same polymorphism was subsequently associated with decreased pregnenolone production by immune cells in both Thr147 homozygous and heterozygous individuals (120). Pregnenolone production by activated lymphocytes, specifically T helper 2 cells has been linked to functional immunosuppression (121). TSPO up-regulation has long been associated with immune activation and cellular responses (122). Therefore, this polymorphism may indicate an immune function for TSPO associated with its overexpression in inflammatory pathologies and cannot be considered as evidence for steroidogenesis as previously suggested (123).
Concluding Remarks
Functional redundancy is what immediately comes to mind, especially after a protein like TSPO whose function that has been extensively studied over the past 25 years is found inaccurate. In fact, redundancy was indeed offered as an explanation in a recent publication (115). As highlighted in this review, there were core problems with reproducibility of previous studies on TSPO (Table 1). Key findings with regard to gene deletion in vivo (66), knockdown in vitro (62), and gene disruption in vitro (64) were not reproducible in the same homologous systems (23, 72, 73, 76). Moreover, recent evidence also challenges core interpretations made based on the pharmacological targeting of TSPO (73). If there is indeed a redundant protein or mechanism, it is not clear why these did not become apparent in earlier studies that asserted an indispensable role for TSPO in the steroidogenic machinery. Nevertheless, possibilities for a redundant function by the TSPO paralog TSPO2 were considered and ruled out in recent studies (23, 72, 76).
Table 1.
Reevaluating Evidence for TSPO Function in Steroidogenesis
| TSPO and Steroidogenesis | Evidence to the Contrary |
|---|---|
| 1) Trophic maintenance of TSPO expression by the pituitary. Hypophysectomy in rats resulted in TSPO depletion in the adrenal parallel with their atrophy (77). | Adrenocorticotropic hormone-induced TSPO expression in adrenals of hypophysectomized rats is not cause-effect related to its steroidogenic action (80). |
| 2) TSPO-binding chemicals (including PK11195) can induce steroid hormone production in Y1 adrenocortical cells (35), and MA-10 Leydig cells (36). | Effect of PK11195 in Tspo knockout MA-10 Leydig cell clones showed that the steroidogenic response is not mediated through TSPO (73). |
| 3) Mono-allelic disruption of Tspo in the R2C Leydig cell line resulted in loss of steroidogenic capacity (64). TSPO knockdown in MA-10 Leydig cells using antisense oligonucleotides inhibited their ability to synthesize steroid hormone (62). | TSPO knockdown did not affect steroid hormone production in Y1 adrenocortical cells, MA-10 Leydig cells, and MLTC Leydig cells (23). Conditional deletion of Tspo in Leydig cells in vivo had no effect on testosterone production (72). Steroidogenic capacity was unaffected in CRISPR/Cas9-mediated Tspo knockout MA-10 Leydig cell clones (73). |
| 4) ACBP knockdown using a cholesterol-linked phosphorothioate oligodeoxynucleotide antisense sequence was shown to inhibit steroid hormone production in MA-10 Leydig tumor cells (92). | Phenotype of a spontaneous Acbp mutant (nm1054) (98), and Acbp knockout mice (100), link ACBP function to fatty acid metabolism and not steroidogenesis (101, 102). |
| 5) Tspo knockout mice are early embryonic lethal, suggesting involvement in basic functions necessary for embryonic development (66). Cells with >70% TSPO knockdown cannot survive (74, 75). | Two independent reports phenotype Tspo knockout mice showing that TSPO is not involved in viability, fertility or steroidogenesis (23, 76). |
| 6) TSPO and StAR function in a coordinated manner to transfer cholesterol into mitochondria (62). FRET detected a complex of TSPO and StAR in mitochondrial membranes (109).a | Data from Tspo knockout cells (73), and mice (23, 76), indicate that TSPO is dispensable for mitochondrial cholesterol transport and steroidogenesis. BRET detected no evidence for TSPO and StAR interaction (110).a |
| 7) Structural predictions for TSPO indicated a hydrophobic channel-like structure formed by the 5 transmembrane α-helices with the interior core lined by the CRAC motif capable of accommodating a cholesterol molecule (8, 66, 113). | High-resolution NMR structure of TSPO indicates a tight bundle of the 5 transmembrane α-helices with the CRAC motif located on the outside of TSPO pointing towards the membrane environment (83). |
Testing for TSPO-StAR interaction by FRET and BRET were follow-up studies from the same lab published in 2001 and 2007, respectively; in their BRET publication, they suggested that selection of individual cells for FRET analysis may have resulted in artifacts.
The mechanism of StAR action in mitochondrial cholesterol import remains to be completely defined at this point in time (Figure 2). However, now that we know TSPO is not a “cholesterol channel,” nor that it is a requisite requirement in cholesterol transport, creates a void and generates some key questions regarding the process by which cholesterol manages to enter the matrix and access the P450scc for steroid production. There are certainly some indications that StAR could cooperate with a support system, but this system is yet to be completely characterized (124). Recent developments regarding TSPO have cleared the pathway for future research progress on cholesterol transport and have also made it clear that TSPO is not a player in this process. Future progress will be made keeping these facts in consideration.
Figure 2.
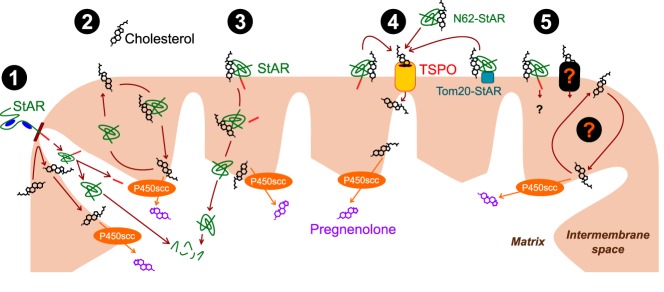
Evolution of mitochondrial cholesterol import models. Cholesterol transport to the IMM and its conversion to pregnenolone by P450scc are essential steps for all steroid hormone production. The first generation of models indicated that StAR could independently accomplish cholesterol import to the IMM: 1) StAR was carried by chaperones and entered mitochondria creating “contact sites” between the outer and IMMs. Cholesterol is able to cross from the outer to the IMM at these contact sites. On entering the matrix, StAR is assembled, cleaved, and subsequently degraded. 2) StAR worked at the level of the mitochondrial intermembrane space shuttling cholesterol from the outer to the IMM. 3) StAR could bind and carry cholesterol in one direction across to the IMM, then enters the matrix to be cleaved and subsequently degraded. The second generation of models appeared after TSPO was introduced as a functional partner for StAR action: 4) StAR resided in the OMM and delivered cholesterol to TSPO that acted as a cholesterol channel to directly transport it to the IMM through contact sites between the 2 membranes. This could explain why N-terminal StAR truncation (N62-StAR) that disrupts its mitochondrial targeting and the Tom20-StAR fusion protein that resides in the OMM could still mediate cholesterol import. Given the new information on TSPO and its high-resolution structure, these elements need to be modified. In addition, recent information suggests that movement of StAR and cleavage/degradation in the mitochondrial matrix might be a terminal event that occurs after its function is completed. Therefore, existing questions are: 5) Is there a channel at the level of the OMM that is essential for cholesterol import? Is there a mechanism that shuttles cholesterol across the intermembrane space? Do we completely understand the mechanism of StAR action? These are topics that will require investigation.
At this point in time, a functional designation for TSPO is still actively being sought (reviewed in Ref. 125). As an example of this, a previous model describing TSPO involvement in cell death processes affecting mitochondrial permeability transition was also challenged (126). Despite not being part of the steroidogenic machinery, TSPO expression in inflammatory pathologies, and use of TSPO-binding chemicals as a diagnostic tool will continue to make it an attractive target of study. But the new understanding of what TSPO does not do with respect to steroidogenesis (23, 72, 73, 126) has forced reevaluation/reinterpretation of numerous studies on TSPO function both within and outside the field of steroidogenesis.
Acknowledgments
We apologize to those whose work could not be cited directly due to space constraints.
This work was supported by the Cornell College of Agriculture and Life Sciences (startup) (V.S.), the National Institutes of Health Grant HD-17481 and Robert A. Welch Foundation (D.M.S.), and a fellowship from the Vietnam Education Foundation (L.N.T.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACBP
- acyl-CoA-binding protein
- BRET
- bioluminescence resonance energy transfer
- CRAC
- cholesterol-binding amino acid consensus
- FRET
- fluorescence resonance energy transfer
- GRP78
- glucose-regulated protein 78
- IMM
- inner mitochondrial membrane
- NMR
- nuclear magnetic resonance
- OMM
- outer mitochondrial membrane
- PBR
- benzodiazepine receptor
- PK11195
- 1-(2-Chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide
- P450scc
- P450 side chain cleavage enzyme
- Ro5-4864
- 7-Chloro-5-(4-chlorophenyl)-1,3-dihydro-1-methyl-2H-1,4-benzodiazepin-2-one
- SCP2
- sterol carrier protein 2
- StAR
- steroidogenic acute regulatory protein
- START
- StAR-related lipid transfer
- TSPO
- translocator protein
- TOM20
- translocase of outer membrane 20.
References
- 1. Braestrup C, Albrechtsen R, Squires RF. High densities of benzodiazepine receptors in human cortical areas. Nature. 1977;269:702–704. [DOI] [PubMed] [Google Scholar]
- 2. Braestrup C, Squires RF. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc Natl Acad Sci USA. 1977;74:3805–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Regan JW, Yamamura HI, Yamada S, Roeske WR. High affinity [3H]flunitrazepam binding: characterization, localization, and alteration in hypertension. Life Sci. 1981;28:991–998. [DOI] [PubMed] [Google Scholar]
- 4. Davies LP, Huston V. Peripheral benzodiazepine binding sites in heart and their interaction with dipyridamole. Eur J Pharmacol. 1981;73:209–211. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi H, Nagashima A, Koshino C. Effect of γ-aminobutyryl-choline upon the electrical activity of the cerebral cortex. Nature. 1958;182:1443–1444. [DOI] [PubMed] [Google Scholar]
- 6. Macdonald R, Barker JL. Benzodiazepines specifically modulate GABA-mediated postsynaptic inhibition in cultured mammalian neurones. Nature. 1978;271:563–564. [DOI] [PubMed] [Google Scholar]
- 7. Gavish M, Bachman I, Shoukrun R, et al. Enigma of the peripheral benzodiazepine receptor. Pharmacol Rev. 1999;51:629–650. [PubMed] [Google Scholar]
- 8. Rupprecht R, Papadopoulos V, Rammes G, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–988. [DOI] [PubMed] [Google Scholar]
- 9. Gatliff J, Campanella M. The 18 kDa translocator protein (TSPO): a new perspective in mitochondrial biology. Curr Mol Med. 2012;12:356–368. [DOI] [PubMed] [Google Scholar]
- 10. Le Fur G, Perrier ML, Vaucher N, et al. Peripheral benzodiazepine binding sites: effect of PK 11195, 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide. I. In vitro studies. Life Sci. 1983;32:1839–1847. [DOI] [PubMed] [Google Scholar]
- 11. Benavides J, Malgouris C, Imbault F, et al. “Peripheral type” benzodiazepine binding sites in rat adrenals: binding studies with [3H]PK 11195 and autoradiographic localization. Arch Int Pharmacodyn Ther. 1983;266:38–49. [PubMed] [Google Scholar]
- 12. Le Fur G, Vaucher N, Perrier ML, et al. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci. 1983;33:449–457. [DOI] [PubMed] [Google Scholar]
- 13. Bénavidès J, Guilloux F, Allam DE, et al. Opposite effects of an agonist, RO5-4864, and an antagonist, PK 11195, of the peripheral type benzodiazepine binding sites on audiogenic seizures in DBA/2J mice. Life Sci. 1984;34:2613–2620. [DOI] [PubMed] [Google Scholar]
- 14. Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Banati RB, Newcombe J, Gunn RN, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123(pt 11):2321–2337. [DOI] [PubMed] [Google Scholar]
- 16. Chauveau F, Boutin H, Van Camp N, Dollé F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35:2304–2319. [DOI] [PubMed] [Google Scholar]
- 17. Yeliseev AA, Kaplan S. A sensory transducer homologous to the mammalian peripheral-type benzodiazepine receptor regulates photosynthetic membrane complex formation in Rhodobacter sphaeroides 2.4.1. J Biol Chem. 1995;270:21167–21175. [DOI] [PubMed] [Google Scholar]
- 18. Yeliseev AA, Krueger KE, Kaplan S. A mammalian mitochondrial drug receptor functions as a bacterial “oxygen” sensor. Proc Natl Acad Sci USA. 1997;94:5101–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Riond J, Mattei MG, Kaghad M, et al. Molecular cloning and chromosomal localization of a human peripheral-type benzodiazepine receptor. Eur J Biochem. 1991;195:305–311. [DOI] [PubMed] [Google Scholar]
- 20. Bucan M, Gatalica B, Nolan P, et al. Comparative mapping of 9 human chromosome 22q loci in the laboratory mouse. Hum Mol Genet. 1993;2:1245–1252. [DOI] [PubMed] [Google Scholar]
- 21. Anholt RR, De Souza EB, Oster-Granite ML, Snyder SH. Peripheral-type benzodiazepine receptors: autoradiographic localization in whole-body sections of neonatal rats. J Pharmacol Exp Ther. 1985;233:517–526. [PubMed] [Google Scholar]
- 22. Wang HJ, Fan J, Papadopoulos V. Translocator protein (Tspo) gene promoter-driven green fluorescent protein synthesis in transgenic mice: an in vivo model to study Tspo transcription. Cell Tissue Res. 2012;350:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tu LN, Morohaku K, Manna PR, et al. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem. 2014;289:27444–27454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morohaku K, Phuong NS, Selvaraj V. Developmental expression of translocator protein/peripheral benzodiazepine receptor in reproductive tissues. PLoS One. 2013;8:e74509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anholt RR, Pedersen PL, De Souza EB, Snyder SH. The peripheral-type benzodiazepine receptor. Localization to the mitochondrial outer membrane. J Biol Chem. 1986;261:576–583. [PubMed] [Google Scholar]
- 26. Basile AS, Skolnick P. Subcellular localization of “peripheral-type” binding sites for benzodiazepines in rat brain. J Neurochem. 1986;46:305–308. [DOI] [PubMed] [Google Scholar]
- 27. Olson JM, Ciliax BJ, Mancini WR, Young AB. Presence of peripheral-type benzodiazepine binding sites on human erythrocyte membranes. Eur J Pharmacol. 1988;152:47–53. [DOI] [PubMed] [Google Scholar]
- 28. Oke BO, Suarez-Quian CA, Riond J, Ferrara P, Papadopoulos V. Cell surface localization of the peripheral-type benzodiazepine receptor (PBR) in adrenal cortex. Mol Cell Endocrinol. 1992;87:R1–R6. [DOI] [PubMed] [Google Scholar]
- 29. Manku G, Wang Y, Thuillier R, Rhodes C, Culty M. Developmental expression of the translocator protein 18 kDa (TSPO) in testicular germ cells. Curr Mol Med. 2012;12:467–475. [PubMed] [Google Scholar]
- 30. Venturini I, Zeneroli ML, Corsi L, et al. Up-regulation of peripheral benzodiazepine receptor system in hepatocellular carcinoma. Life Sci. 1998;63:1269–1280. [DOI] [PubMed] [Google Scholar]
- 31. Hardwick M, Fertikh D, Culty M, Li H, Vidic B, Papadopoulos V. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999;59:831–842. [PubMed] [Google Scholar]
- 32. Li L, Chen T, Stanton JD, Sueyoshi T, Negishi M, Wang H. The peripheral benzodiazepine receptor ligand 1-(2-chlorophenyl-methylpropyl)-3-isoquinoline-carboxamide is a novel antagonist of human constitutive androstane receptor. Mol Pharmacol. 2008;74:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seneviratne MS, Faccenda D, De Biase V, Campanella M. PK11195 inhibits mitophagy targeting the F1Fo-ATPsynthase in Bcl-2 knock-down cells. Curr Mol Med. 2012;12:476–482. [DOI] [PubMed] [Google Scholar]
- 34. Guillaumot D, Guillon S, Déplanque T, et al. The Arabidopsis TSPO-related protein is a stress and abscisic acid-regulated, endoplasmic reticulum-Golgi-localized membrane protein. Plant J. 2009;60:242–256. [DOI] [PubMed] [Google Scholar]
- 35. Mukhin AG, Papadopoulos V, Costa E, Krueger KE. Mitochondrial benzodiazepine receptors regulate steroid biosynthesis. Proc Natl Acad Sci USA. 1989;86:9813–9816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Papadopoulos V, Mukhin AG, Costa E, Krueger KE. The peripheral-type benzodiazepine receptor is functionally linked to Leydig cell steroidogenesis. J Biol Chem. 1990;265:3772–3779. [PubMed] [Google Scholar]
- 37. Churchill PF, Kimura T. Topological studies of cytochromes P-450scc and P-45011 β in bovine adrenocortical inner mitochondrial membranes. Effects of controlled tryptic digestion. J Biol Chem. 1979;254:10443–10448. [PubMed] [Google Scholar]
- 38. Cheng B, Hsu DK, Kimura T. Utilization of intramitochondrial membrane cholesterol by cytochrome P-450-dependent cholesterol side-chain cleavage reaction in bovine adrenocortical mitochondria: steroidogenic and non-steroidogenic pools of cholesterol in the mitochondrial inner membranes. Mol Cell Endocrinol. 1985;40:233–243. [DOI] [PubMed] [Google Scholar]
- 39. Rennert H, Cheng YJ, Strauss JF., 3rd Intracellular cholesterol dynamics in steroidogenic cells: a contemporary view. In: Adashi EY, LP, ed. The Ovary. New York, NY: Raven Press;1993:147–164. [Google Scholar]
- 40. Ferguson JJ., Jr Protein synthesis and adrenocorticotropin responsiveness. J Biol Chem. 1963;238:2754–2759. [PubMed] [Google Scholar]
- 41. Garren LD, Ney RL, Davis WW. Studies on the role of protein synthesis in the regulation of corticosterone production by adrenocorticotropic hormone in vivo. Proc Natl Acad Sci USA. 1965;53:1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vahouny GV, Chanderbhan R, Kharroubi A, Noland BJ, Pastuszyn A, Scallen TJ. Sterol carrier and lipid transfer proteins. Adv Lipid Res. 1987;22:83–113. [DOI] [PubMed] [Google Scholar]
- 43. Pedersen RC, Brownie AC. Cholesterol side-chain cleavage in the rat adrenal cortex: isolation of a cycloheximide-sensitive activator peptide. Proc Natl Acad Sci USA. 1983;80:1882–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pedersen RC, Brownie AC. Steroidogenesis-activator polypeptide isolated from a rat Leydig cell tumor. Science. 1987;236:188–190. [DOI] [PubMed] [Google Scholar]
- 45. Seedorf U, Raabe M, Ellinghaus P, et al. Defective peroxisomal catabolism of branched fatty acyl coenzyme A in mice lacking the sterol carrier protein-2/sterol carrier protein-x gene function. Genes Dev. 1998;12:1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li XA, Warren DW, Gregoire J, Pedersen RC, Lee AS. The rat 78,000 dalton glucose-regulated protein (GRP78) as a precursor for the rat steroidogenesis-activator polypeptide (SAP): the SAP coding sequence is homologous with the terminal end of GRP78. Mol Endocrinol. 1989;3:1944–1952. [DOI] [PubMed] [Google Scholar]
- 47. Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11:2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wey S, Luo B, Tseng CC, et al. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2012;119:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wisniewska M, Karlberg T, Lehtio L, et al. Crystal structures of the ATPase domains of four human Hsp70 isoforms: HSPA1L/Hsp70-hom, HSPA2/Hsp70–2, HSPA6/Hsp70B', and HSPA5/BiP/GRP78. PLoS One. 2010;5:e8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Krueger RJ, Orme-Johnson NR. Acute adrenocorticotropic hormone stimulation of adrenal corticosteroidogenesis. Discovery of a rapidly induced protein. J Biol Chem. 1983;258:10159–10167. [PubMed] [Google Scholar]
- 52. Pon LA, Hartigan JA, Orme-Johnson NR. Acute ACTH regulation of adrenal corticosteroid biosynthesis. Rapid accumulation of a phosphoprotein. J Biol Chem. 1986;261:13309–13316. [PubMed] [Google Scholar]
- 53. Clark BJ, Wells J, King SR, Stocco DM. The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J Biol Chem. 1994;269:28314–28322. [PubMed] [Google Scholar]
- 54. Lin D, Sugawara T, Strauss JF, 3rd, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995;267:1828–1831. [DOI] [PubMed] [Google Scholar]
- 55. Caron KM, Soo SC, Wetsel WC, Stocco DM, Clark BJ, Parker KL. Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc Natl Acad Sci USA. 1997;94:11540–11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tsujishita Y, Hurley JH. Structure and lipid transport mechanism of a StAR-related domain. Nat Struct Biol. 2000;7:408–414. [DOI] [PubMed] [Google Scholar]
- 57. Papadopoulos V, Berkovich A, Krueger KE. The role of diazepam binding inhibitor and its processing products at mitochondrial benzodiazepine receptors: regulation of steroid biosynthesis. Neuropharmacology. 1991;30:1417–1423. [DOI] [PubMed] [Google Scholar]
- 58. Papadopoulos V, Berkovich A, Krueger KE, Costa E, Guidotti A. Diazepam binding inhibitor and its processing products stimulate mitochondrial steroid biosynthesis via an interaction with mitochondrial benzodiazepine receptors. Endocrinology. 1991;129:1481–1488. [DOI] [PubMed] [Google Scholar]
- 59. Papadopoulos V, Guarneri P, Kreuger KE, Guidotti A, Costa E. Pregnenolone biosynthesis in C6-2B glioma cell mitochondria: regulation by a mitochondrial diazepam binding inhibitor receptor. Proc Natl Acad Sci USA. 1992;89:5113–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garnier M, Boujrad N, Oke BO, et al. Diazepam binding inhibitor is a paracrine/autocrine regulator of Leydig cell proliferation and steroidogenesis: action via peripheral-type benzodiazepine receptor and independent mechanisms. Endocrinology. 1993;132:444–458. [DOI] [PubMed] [Google Scholar]
- 61. Kelly-Hershkovitz E, Weizman R, Spanier I, et al. Effects of peripheral-type benzodiazepine receptor antisense knockout on MA-10 Leydig cell proliferation and steroidogenesis. J Biol Chem. 1998;273:5478–5483. [DOI] [PubMed] [Google Scholar]
- 62. Hauet T, Yao ZX, Bose HS, et al. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol Endocrinol. 2005;19:540–554. [DOI] [PubMed] [Google Scholar]
- 63. Garnier M, Boujrad N, Ogwuegbu SO, Hudson JR, Jr, Papadopoulos V. The polypeptide diazepam-binding inhibitor and a higher affinity mitochondrial peripheral-type benzodiazepine receptor sustain constitutive steroidogenesis in the R2C Leydig tumor cell line. J Biol Chem. 1994;269:22105–22112. [PubMed] [Google Scholar]
- 64. Papadopoulos V, Amri H, Li H, Boujrad N, Vidic B, Garnier M. Targeted disruption of the peripheral-type benzodiazepine receptor gene inhibits steroidogenesis in the R2C Leydig tumor cell line. J Biol Chem. 1997;272:32129–32135. [DOI] [PubMed] [Google Scholar]
- 65. Li H, Yao Z, Degenhardt B, Teper G, Papadopoulos V. Cholesterol binding at the cholesterol recognition/interaction amino acid consensus (CRAC) of the peripheral-type benzodiazepine receptor and inhibition of steroidogenesis by an HIV TAT-CRAC peptide. Proc Natl Acad Sci USA. 2001;98:1267–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Papadopoulos V, Amri H, Boujrad N, et al. Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids. 1997;62:21–28. [DOI] [PubMed] [Google Scholar]
- 67. Stocco DM. StAR protein and the regulation of steroid hormone biosynthesis. Annu Rev Physiol. 2001;63:193–213. [DOI] [PubMed] [Google Scholar]
- 68. Arakane F, Sugawara T, Nishino H, et al. Steroidogenic acute regulatory protein (StAR) retains activity in the absence of its mitochondrial import sequence: implications for the mechanism of StAR action. Proc Natl Acad Sci USA. 1996;93:13731–13736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang X, Liu Z, Eimerl S, et al. Effect of truncated forms of the steroidogenic acute regulatory protein on intramitochondrial cholesterol transfer. Endocrinology. 1998;139:3903–3912. [DOI] [PubMed] [Google Scholar]
- 70. Bose HS, Lingappa VR, Miller WL. Rapid regulation of steroidogenesis by mitochondrial protein import. Nature. 2002;417:87–91. [DOI] [PubMed] [Google Scholar]
- 71. Papadopoulos V, Miller WL. Role of mitochondria in steroidogenesis. Best Pract Res Clin Endocrinol Metab. 2012;26:771–790. [DOI] [PubMed] [Google Scholar]
- 72. Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology. 2014;155:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tu LN, Zhao AH, Stocco DM, Selvaraj V. PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology 2014:en20141707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Veenman L, Papadopoulos V, Gavish M. Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des. 2007;13:2385–2405. [DOI] [PubMed] [Google Scholar]
- 75. Amri H, Culty M, Gaillard JL, Teper G, Papadopoulos V. The peripheral-type benzodiazepine receptor and adrenal steroidogenesis. Curr Opin Endocrinol Diabetes. 1999;6:179–184. [Google Scholar]
- 76. Banati RB, Middleton RJ, Chan R, et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat Commun. 2014;5:5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Anholt RR, De Souza EB, Kuhar MJ, Snyder SH. Depletion of peripheral-type benzodiazepine receptors after hypophysectomy in rat adrenal gland and testis. Eur J Pharmacol. 1985;110:41–46. [DOI] [PubMed] [Google Scholar]
- 78. Deane HW, Greep RO. A morphological and histochemical study of the rat's adrenal cortex after hypoph ysectomy, with comments on the liver. Am J Anat. 1946;79:117–145. [DOI] [PubMed] [Google Scholar]
- 79. Kimura T. Effects of hypophysectomy and ACTH administration on the level of adrenal cholesterol side-chain desmolase. Endocrinology. 1969;85:492–499. [DOI] [PubMed] [Google Scholar]
- 80. Cavallaro S, Pani L, Guidotti A, Costa E. ACTH-induced mitochondrial DBI receptor (MDR) and diazepam binding inhibitor (DBI) expression in adrenals of hypophysectomized rats is not cause-effect related to its immediate steroidogenic action. Life Sci. 1993;53:1137–1147. [DOI] [PubMed] [Google Scholar]
- 81. Hatty CR, Le Brun AP, Lake V, et al. Investigating the interactions of the 18kDa translocator protein and its ligand PK11195 in planar lipid bilayers. Biochim Biophys Acta. 2014;1838:1019–1030. [DOI] [PubMed] [Google Scholar]
- 82. Farges R, Joseph-Liauzun E, Shire D, Caput D, Le Fur G, Ferrara P. Site-directed mutagenesis of the peripheral benzodiazepine receptor: identification of amino acids implicated in the binding site of Ro5-4864. Mol Pharmacol. 1994;46:1160–1167. [PubMed] [Google Scholar]
- 83. Jaremko L, Jaremko M, Giller K, Becker S, Zweckstetter M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science. 2014;343:1363–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cavallaro S, Korneyev A, Guidotti A, Costa E. Diazepam-binding inhibitor (DBI)-processing products, acting at the mitochondrial DBI receptor, mediate adrenocorticotropic hormone-induced steroidogenesis in rat adrenal gland. Proc Natl Acad Sci USA. 1992;89:10598–10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Romeo E, Cavallaro S, Korneyev A, et al. Stimulation of brain steroidogenesis by 2-aryl-indole-3-acetamide derivatives acting at the mitochondrial diazepam-binding inhibitor receptor complex. J Pharmacol Exp Ther. 1993;267:462–471. [PubMed] [Google Scholar]
- 86. Korneyev A, Pan BS, Polo A, Romeo E, Guidotti A, Costa E. Stimulation of brain pregnenolone synthesis by mitochondrial diazepam binding inhibitor receptor ligands in vivo. J Neurochem. 1993;61:1515–1524. [DOI] [PubMed] [Google Scholar]
- 87. Auta J, Romeo E, Kozikowski A, Ma D, Costa E, Guidotti A. Participation of mitochondrial diazepam binding inhibitor receptors in the anticonflict, antineophobic and anticonvulsant action of 2-aryl-3-indoleacetamide and imidazopyridine derivatives. J Pharmacol Exp Ther. 1993;265:649–656. [PubMed] [Google Scholar]
- 88. Tsuda M, Suzuki T, Misawa M. Subsensitivity to mitochondrial diazepam binding inhibitor receptor agonist FGIN-1-27-induced antiseizure effect in diazepam-withdrawn mice. Life Sci. 1998;62:PL213–217. [DOI] [PubMed] [Google Scholar]
- 89. Frye CA, Paris JJ, Rhodes ME. Increasing 3α,5α-THP following inhibition of neurosteroid biosynthesis in the ventral tegmental area reinstates anti-anxiety, social, and sexual behavior of naturally receptive rats. Reproduction. 2009;137:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barnea ER, Fares F, Gavish M. Modulatory action of benzodiazepines on human term placental steroidogenesis in vitro. Mol Cell Endocrinol. 1989;64:155–159. [DOI] [PubMed] [Google Scholar]
- 91. Weizman R, Leschiner S, Schlegel W, Gavish M. Peripheral-type benzodiazepine receptor ligands and serum steroid hormones. Brain Res. 1997;772:203–208. [DOI] [PubMed] [Google Scholar]
- 92. Boujrad N, Hudson JR, Jr, Papadopoulos V. Inhibition of hormone-stimulated steroidogenesis in cultured Leydig tumor cells by a cholesterol-linked phosphorothioate oligodeoxynucleotide antisense to diazepam-binding inhibitor. Proc Natl Acad Sci USA. 1993;90:5728–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Knudsen J, Jensen MV, Hansen JK, Faergeman NJ, Neergaard TB, Gaigg B. Role of acylCoA binding protein in acylCoA transport, metabolism and cell signaling. Mol Cell Biochem. 1999;192:95–103. [DOI] [PubMed] [Google Scholar]
- 94. Mandrup S, Jepsen R, Skott H, et al. Effect of heterologous expression of acyl-CoA-binding protein on acyl-CoA level and composition in yeast. Biochem J. 1993;290(pt 2):369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gaigg B, Neergaard TB, Schneiter R, et al. Depletion of acyl-coenzyme A-binding protein affects sphingolipid synthesis and causes vesicle accumulation and membrane defects in Saccharomyces cerevisiae. Mol Biol Cell. 2001;12:1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ohgami RS, Campagna DR, Greer EL, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005;37:1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ohgami RS, Campagna DR, Antiochos B, et al. nm1054: a spontaneous, recessive, hypochromic, microcytic anemia mutation in the mouse. Blood. 2005;106:3625–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lee L, DeBono CA, Campagna DR, Young DC, Moody DB, Fleming MD. Loss of the acyl-CoA binding protein (Acbp) results in fatty acid metabolism abnormalities in mouse hair and skin. J Invest Dermatol. 2007;127:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Landrock D, Atshaves BP, McIntosh AL, Landrock KK, Schroeder F, Kier AB. Acyl-CoA binding protein gene ablation induces pre-implantation embryonic lethality in mice. Lipids. 2010;45:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Neess D, Bloksgaard M, Bek S, et al. Disruption of the acyl-CoA-binding protein gene delays hepatic adaptation to metabolic changes at weaning. J Biol Chem. 2011;286:3460–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bloksgaard M, Bek S, Marcher AB, et al. The acyl-CoA binding protein is required for normal epidermal barrier function in mice. J Lipid Res. 2012;53:2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Neess D, Bek S, Bloksgaard M, Marcher AB, Færgeman NJ, Mandrup S. Delayed hepatic adaptation to weaning in ACBP-/- mice is caused by disruption of the epidermal barrier. Cell Rep. 2013;5:1403–1412. [DOI] [PubMed] [Google Scholar]
- 103. Li H, Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139:4991–4997. [DOI] [PubMed] [Google Scholar]
- 104. Palmer M. Cholesterol and the activity of bacterial toxins. FEMS Microbiol Lett. 2004;238:281–289. [DOI] [PubMed] [Google Scholar]
- 105. Baier CJ, Fantini J, Barrantes FJ. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci Rep. 2011;1:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Luo X, Sharma D, Inouye H, et al. Cytoplasmic domain of human myelin protein zero likely folded as β-structure in compact myelin. Biophys J. 2007;92:1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang G, Xu H, Li Z, Li F. Interactions of caveolin-1 scaffolding and intramembrane regions containing a CRAC motif with cholesterol in lipid bilayers. Biochim Biophys Acta. 2014;1838:2588–2599. [DOI] [PubMed] [Google Scholar]
- 108. Herrera JL, Diaz M, Hernández-Fernaud JR, et al. Voltage-dependent anion channel as a resident protein of lipid rafts: post-transductional regulation by estrogens and involvement in neuronal preservation against Alzheimer's disease. J Neurochem. 2011;116:820–827. [DOI] [PubMed] [Google Scholar]
- 109. West LA, Horvat RD, Roess DA, Barisas BG, Juengel JL, Niswender GD. Steroidogenic acute regulatory protein and peripheral-type benzodiazepine receptor associate at the mitochondrial membrane. Endocrinology. 2001;142:502–505. [DOI] [PubMed] [Google Scholar]
- 110. Bogan RL, Davis TL, Niswender GD. Peripheral-type benzodiazepine receptor (PBR) aggregation and absence of steroidogenic acute regulatory protein (StAR)/PBR association in the mitochondrial membrane as determined by bioluminescence resonance energy transfer (BRET). J Steroid Biochem Mol Biol. 2007;104:61–67. [DOI] [PubMed] [Google Scholar]
- 111. Murail S, Robert JC, Coïc YM, et al. Secondary and tertiary structures of the transmembrane domains of the translocator protein TSPO determined by NMR. Stabilization of the TSPO tertiary fold upon ligand binding. Biochim Biophys Acta. 2008;1778:1375–1381. [DOI] [PubMed] [Google Scholar]
- 112. Korkhov VM, Sachse C, Short JM, Tate CG. Three-dimensional structure of TspO by electron cryomicroscopy of helical crystals. Structure. 2010;18:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bernassau JM, Reversat JL, Ferrara P, Caput D, Lefur G. A 3D model of the peripheral benzodiazepine receptor and its implication in intra mitochondrial cholesterol transport. J Mol Graph. 1993;11:236–244, 235. [DOI] [PubMed] [Google Scholar]
- 114. Delavoie F, Li H, Hardwick M, Robert JC, et al. In vivo and in vitro peripheral-type benzodiazepine receptor polymerization: functional significance in drug ligand and cholesterol binding. Biochemistry. 2003;42:4506–4519. [DOI] [PubMed] [Google Scholar]
- 115. Midzak A, Papadopoulos V. Binding domain-driven intracellular trafficking of sterols for synthesis of steroid hormones, bile acids and oxysterols. Traffic. 2014;15:895–914. [DOI] [PubMed] [Google Scholar]
- 116. Lin D, Chang YJ, Strauss JF, 3rd, Miller WL. The human peripheral benzodiazepine receptor gene: cloning and characterization of alternative splicing in normal tissues and in a patient with congenital lipoid adrenal hyperplasia. Genomics. 1993;18:643–650. [DOI] [PubMed] [Google Scholar]
- 117. Owen DR, Yeo AJ, Gunn RN, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mizrahi R, Rusjan PM, Kennedy J, et al. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [(18)F]-FEPPA. J Cereb Blood Flow Metab. 2012;32:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Costa B, Pini S, Martini C, et al. Ala147Thr substitution in translocator protein is associated with adult separation anxiety in patients with depression. Psychiatr Genet. 2009;19:110–111. [DOI] [PubMed] [Google Scholar]
- 120. Costa B, Pini S, Gabelloni P, et al. The spontaneous Ala147Thr amino acid substitution within the translocator protein influences pregnenolone production in lymphomonocytes of healthy individuals. Endocrinology. 2009;150:5438–5445. [DOI] [PubMed] [Google Scholar]
- 121. Mahata B, Zhang X, Kolodziejczyk AA, et al. Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep. 2014;7:1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu GJ, Middleton RJ, Hatty CR, et al. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. 2014;24:631–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Papadopoulos V. On the role of the translocator protein (18-kDa) TSPO in steroid hormone biosynthesis. Endocrinology. 2014;155:15–20. [DOI] [PubMed] [Google Scholar]
- 124. Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS. Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J Biol Chem. 2015;290:2604–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Selvaraj V, Stocco DM. The changing landscape in translocator protein (TSPO) function [published online 2015]. Trends Endocrinol Metab. doi:10.1016/j.tem.2015.02.007. Published Online March 20, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Šileikyte J, Blachly-Dyson E, Sewell R, et al. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (TSPO). J Biol Chem. 2014;289:13769–13781. [DOI] [PMC free article] [PubMed] [Google Scholar]