

Abstract
Context and Objective:
Oncocytic thyroid carcinoma, also known as Hürthle cell thyroid carcinoma, accounts for only a small percentage of all thyroid cancers. However, this malignancy often presents at an advanced stage and poses unique challenges to patients and clinicians. Surgical resection of the tumor accompanied in some cases by radioactive iodine treatment, radiation, and chemotherapy are the established modes of therapy. Knowledge of the perturbed oncogenic pathways can provide better understanding of the mechanism of disease and thus opportunities for more effective clinical management.
Design and Patients:
Initially, two oncocytic thyroid carcinomas and their matched normal tissues were profiled using whole genome sequencing. Subsequently, 72 oncocytic thyroid carcinomas, one cell line, and five Hürthle cell adenomas were examined by targeted sequencing for the presence of mutations in the multiple endocrine neoplasia I (MEN1) gene.
Results:
Here we report the identification of MEN1 loss-of-function mutations in 4% of patients diagnosed with oncocytic thyroid carcinoma. Whole genome sequence data also revealed large regions of copy number variation encompassing nearly the entire genomes of these tumors.
Conclusion:
Menin, a ubiquitously expressed nuclear protein, is a well-characterized tumor suppressor whose loss is the cause of MEN1 syndrome. Menin is involved in several major cellular pathways such as regulation of transcription, control of cell cycle, apoptosis, and DNA damage repair pathways. Mutations of this gene in a subset of Hürthle cell tumors point to a potential role for this protein and its associated pathways in thyroid tumorigenesis.
Hürthle or oncocytic cells of the thyroid are follicular-derived cells with a large nucleus, prominent nucleolus, and an abundant accumulation of mitochondria resulting in a distinct granular appearance on histology sections (1). Nodules consisting of 75% or greater oncocytic cells are categorized as Hürthle cell neoplasms; those demonstrating capsular or vascular invasion or presence of distant metastasis are diagnosed as malignant tumors, rendering fine-needle aspiration cytology as an inadequate technique for diagnosis of Hürthle cell malignancies (1). Hürthle cell thyroid carcinoma is considered an oxyphilic variant of follicular thyroid cancers by some (2) whereas others regard it as a separate subtype of differentiated thyroid cancers (3). Nonetheless, it is treated according to the same established guidelines for papillary and follicular neoplasms, namely surgical removal of all or part of the gland, radioactive iodine treatment, and to a lesser extent, with chemotherapy and radiation treatment (3). Oncocytic carcinoma is a rare entity accounting for only 3–7% of differentiated thyroid cancers, yet it demonstrates more aggressive behavior with 5-year survival rates ranging from 50–60% (1, 4). Demographic comparison of 3311 oncocytic carcinoma patients with 59 585 individuals diagnosed with papillary or follicular carcinomas from The Surveillance, Epidemiology, and End Results (SEER) database between 1988 and 2009 showed higher prevalence of oncocytic carcinomas among older men who generally present with larger tumors, more advanced disease, and demonstrate lower disease-specific survival (4).
The uncommon occurrence of this subtype of thyroid cancer has hindered the complete characterization of this malignancy on the molecular level and hence the genetic changes associated with this cancer and their roles in tumorigenesis are not entirely understood. Here we report the profile of two tumors on the whole genome scale in addition to identification of recurrent inactivating mutations in the multiple endocrine neoplasia I (MEN1) tumor suppressor gene in five of 74 oncocytic thyroid carcinomas.
Materials and Methods
Study samples
Whole genome sequencing experiments were performed for two flash-frozen sporadic oncocytic thyroid carcinomas and matched peripheral blood specimens; one tumor was a primary disease whereas the second specimen was a metastatic lesion obtained from a different patient presenting with lymph node involvement. Neither patient had a family history of cancer or multiple endocrine neoplasia (MEN) syndrome. Both patients provided written informed consent for genomic profiling. The specimens were collected as part of a research project approved by the University of British Columbia Cancer Agency Research Ethics Board and Mayo Clinic Institutional Review Board and are in accordance with the Declaration of Helsinki. Seventy-two formalin-fixed paraffin-embedded (FFPE) oncocytic carcinoma specimens (six accompanied by matched adjacent normal tissue), five FFPE Hürthle cell adenomas, and one oncocytic cell line, XTC.UC1, were included in the targeted sequencing validation experiment.
DNA sequencing and bioinformatic analysis
DNA extracted from the frozen tumor tissues and blood were subjected to high-throughput whole genome sequencing (Supplemental Methods). Sanger sequencing was subsequently used as an orthogonal technique for the verification of somatic MEN1 mutations in both patients. Following this verification experiment, 17 pairs of primers were designed to amplify nine protein-coding exons of MEN1 in a larger cohort of oncocytic tumors (Supplemental Methods and Supplemental Table 1). All datasets have been deposited at the European Genome-Phenome Archive (http://www.ebi.ac.uk/ega/, accession number EGAS00001000940). Somatic alterations including single nucleotide mutations, small insertions and deletions, large structural variants, and gene fusions as well as regions of copy number variation and loss of heterozygosity were identified from the whole genome datasets; sequence reads from the targeted validation experiment were also examined for the presence of any alterations (Supplemental Methods).
Results
Copy number analysis revealed large regions of somatic copy number alteration in both tumors. The metastatic tumor had a change in every chromosome with one copy loss of chromosomes 1, 2, 3, 4, 6, 8, 9, 11, 14, 15, 16, 21, and X while the remaining chromosomes had gained extra copies. The primary tumor had a striking profile, showing copy-neutral loss of heterozygosity of chromosomes 1, 2, 3, 4, 6, 8, 9, 11, 13, 14, 15, 17, and 22 (Figure 1). Loss of heterozygosity while maintaining two chromosomal copies is likely the result of chromosomal amplification of a mostly haploid genome during the evolution of the tumor (5). Copy number data also points to the high amplification of mitochondrial genome confirming the increase in mitochondrial numbers in both tumors. De novo assembly of sequence reads found no gene fusions in these tumors.
Figure 1.

Copy number variation (CNV) and loss of heterozygosity (LOH) regions in two separate oncocytic thyroid tumors. From the outer ring in: chromosomes, primary tumor CNV, (unrelated) metastatic tumor CNV, primary tumor LOH, and metastatic tumor LOH.
We identified 51 and 157 somatic single nucleotide variants and indels in the primary and metastatic genomes, respectively (Supplemental Tables 2 and 3). Of particular interest was a splice site mutation in EWSR1 in the primary tumor, a hemizygous frame-shift deletion in the DNA mismatch repair gene MSH2 and a heterozygous missense mutation in BRCA1 in the metastatic tumor. Mutational analysis revealed somatic mutations in multiple endocrine neoplasia 1 gene in both tumors; MEN1 was the only shared mutated gene in these specimens and it harbored two distinct somatic single nucleotide deletions. The primary tumor showed a homozygous deletion of a single base, leading to a shift in the reading frame and as a result the deletion of amino acids 592 to the end of MEN1. The metastatic tumor showed a hemizygous deletion of a single base, causing a frame-shift in MEN1 starting from amino acid 521 in exon 10. This particular deletion has previously been identified in the germline of patients diagnosed with MEN type I disorder (6) and also in three large intestine carcinomas and one liver carcinoma (COSM1355794) (7). Both deletions were present at close to 100% allele frequency after correction for normal tissue contamination. Their presence and somatic nature were verified in patients using Sanger sequencing.
Targeted sequencing of the validation cohort provided a high depth of sequence coverage of the MEN1 gene (Supplemental Figure 1). Most tumor specimens did not have matched adjacent normal tissues to distinguish somatic and germline mutations; however, given the mutational profiles of the original two tumors and known tumor suppressive role of MEN1 (8), we examined the samples for the presence of likely loss-of-function mutations such as nonsense single nucleotide variants and indels. We found small deletions in three patients diagnosed with oncocytic carcinoma in the validation cohort with mutational allele frequencies of 17.8%, 10.7%, and 3.2%. Adjacent normal tissue was available for one of these three and it did not harbor the MEN1 mutation. Two of these mutations led to frameshift alterations starting at amino acids 178 and 252. Although, these are likely loss-of-function mutations, it is unknown what effect the third identified alteration, an in-frame deletion, would have on protein function. Furthermore, we found small deletions in an additional four patients with allele frequency levels (1.5%, 1.4%, 1.3%, and 1%) close to the sequencing technology's inherent error rate, and hence we have not included these in the population's mutational rate estimate despite a previous publication reporting one of these as a somatic mutation in sporadic parathyroid adenomas (9) (Figure 2). All the above-mentioned deletions, including those found at low allele frequencies, were identified through both alignment-based and de novo assembly-based variant calling methods. No loss-of-function mutations were observed in the XTC.UC1 cell line, benign Hürthle cell tumors, or the normal specimens.
Figure 2.
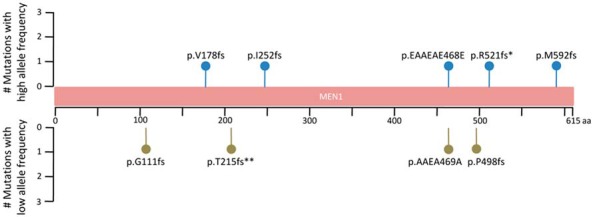
The identified mutations throughout MEN1. Although five mutations were found at high allele frequencies and thus are high-confident calls, extra four mutations were found to have low allele frequencies between 1 and 2% (Supplemental Table 4). *, This mutation has previously been described in patients diagnosed with MEN1 disorder (6). It has also been detected in three large intestine carcinoma specimens and one liver carcinoma (COSM1355794). **, This mutation has previously been found in sporadic parathyroid adenomas (9) (COSM255131).
Discussion
Oncocytic thyroid carcinoma, a rare entity accounting for only 2–3% of all thyroid cancers, often presents in a metastatic setting and hence has a poor prognosis (10). Genomic studies of oncocytic carcinomas are limited and the molecular aberrations driving this malignancy are not entirely understood. Activating NRAS mutations, frequently found in follicular thyroid cancers (10), were identified in three of 27 oncocytic carcinomas (11) leading perhaps to the conclusion that a subset of these malignancies might be derived from follicular thyroid cancers. It has been suggested that in contrast with these oxyphilic variants of papillary or follicular carcinomas, in “true” or “primary” Hürthle cell tumors the aberrations leading to oncocytic phenotype occur prior, rather than subsequent, to neoplastic change(s) such as NRAS mutations (12). It is conceivable that these different disease subtypes are subjected to distinct oncogenesis mechanisms. In this study, we aimed to provide molecular profiles of those oncocytic malignancies that lack the most commonly mutated thyroid cancer genes. We found MEN1 mutations in three of 72 (4.2%) patients diagnosed with oncocytic thyroid carcinoma. In the initial two tumors that underwent whole genome sequencing, complete loss of MEN1 through the loss of the remaining wild-type allele was observed. In agreement with previous reports on tumor-suppressive role of MEN1 (8), the mutational profile of these tumors, namely homozygous and hemizygous frameshift deletions, has lent itself to a strong argument for a causative role of this tumor suppressor gene in oncocytic thyroid malignancies.
Germline loss-of-function mutations of MEN1 are recognized as the single predisposing event in both familial and sporadic cases of MEN type 1 disorder. Affected individuals develop tumors of two or more endocrine organs with the majority found in the parathyroid, pancreatic islets, duodenal endocrine cells, and anterior pituitary (8). A wide array of mutations including frame-shift, nonsense, missense, and in-frame deletions throughout MEN1, with no distinct hotspot mutations, along with loss of the wild-type allele have been identified in association with this syndrome (8, 13, 14). Moreover, somatic inactivating mutations of both MEN1 alleles are found in sporadic endocrine tumors unrelated to MEN type 1 disorder; these include benign parathyroid tumors (15), carcinoids tumors of the lung (16), gastrinomas, and insulinomas (17). In contrast with its prominent role in benign endocrine tumorigenesis, MEN1 has not been implicated as the main driver of malignancy in any cancer types. A query of the cBioPortal database (18), which contains mutational data from several large scale studies, revealed a low mutation rate in MEN1 (Supplemental Figure 2). This query also included 401 cases of papillary thyroid carcinoma from The Cancer Genome Atlas project, where only one patient was found to have a MEN1 mutation.
The current study is perhaps underestimating the prevalence of MEN1 mutations in the oncocytic thyroid carcinoma population. Intratumor heterogeneity, frequently observed in most cancers but specifically in oncocytic carcinomas (5) and low tumor content, a common characteristic of tissue cores obtained from FFPE specimens, might have resulted in underestimating MEN1 mutation rate. In addition, we cannot rule out the presence of untranslated, intronic, or promoter alterations or epigenomic silencing of the gene. Nonetheless, this study implicates MEN1 in the pathogenesis of a subset of oncocytic thyroid carcinomas. Further mutational analyses in this rare cancer type, preferably using microdissected regions of flash-frozen tissues, are warranted and promise to aid in unraveling the mechanism of disease initiation and progression.
Acknowledgments
We are greatly indebted to the patients for their participation in this study. We would also like to acknowledge the contribution of the BC Cancer Agency Genome Sciences Centre biospecimen, library construction, and sequencing cores to this work.
This work was supported by the Canadian Cancer Society Research Institute (2010–700329 to S.J.M.J) and the National Cancer Institute at the National Institutes of Health (R01CA136665 to J.A.C and R.C.S). Financial support also comes from a generous gift from Alfred D. and Audrey M. Petersen and National Cancer Institute (P30-CA015083–40 to R.C.S.) (B.A.P. Shared Resource) and in part from the 26.2 with Donna Foundation (to E.A.T.). K.K. is a recipient of the doctoral fellowship from the Canadian Institutes of Health Research. A.M.C. is a recipient of Crawford Research Fellowship at Mayo Clinic Jacksonville. M.A.M. is UBC Canada Research Chair in Genome Science.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- FFPE
- formalin-fixed paraffin-embedded
- MEN
- multiple endocrine neoplasia
- MEN1
- multiple endocrine neoplasia I gene
- SEER
- Surveillance, Epidemiology, and End Results database.
References
- 1. Montone KT, Baloch ZW, LiVolsi VA. The thyroid Hürthle (oncocytic) cell and its associated pathologic conditions: a surgical pathology and cytopathology review. Arch Pathol Lab Med. 2008;132(8):1241–1250. [DOI] [PubMed] [Google Scholar]
- 2. Hedinger C, Williams ED, Sobin LH. The WHO histological classification of thyroid tumors: A commentary on the second edition. Cancer. 1989;63(5):908–911. [DOI] [PubMed] [Google Scholar]
- 3. Cooper DS, Doherty GM, Haugen BR, et al. Revised American thyroid association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167–1214. [DOI] [PubMed] [Google Scholar]
- 4. Goffredo P, Roman SA, Sosa JA. Hurthle cell carcinoma: A population-level analysis of 3311 patients. Cancer. 2013;119(3):504–511. [DOI] [PubMed] [Google Scholar]
- 5. Corver WE, Ruano D, Weijers K, et al. Genome haploidisation with chromosome 7 retention in oncocytic follicular thyroid carcinoma. PLoS One. 2012;7(6):e38287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bassett JH, Forbes SA, Pannett AA, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet. 1998;62(2):232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Forbes SA, Bindal N, Bamford S, et al. COSMIC: Mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276(5311):404–407. [DOI] [PubMed] [Google Scholar]
- 9. Cromer MK, Starker LF, Choi M, et al. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab. 2012;97(9):E1774–E1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13(3):184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ganly I, Ricarte Filho J, Eng S, et al. Genomic dissection of hurthle cell carcinoma reveals a unique class of thyroid malignancy. J Clin Endocrinol Metab. 2013;98(5):E962–E972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Máximo V, Sobrinho-Simões M. Hürthle cell tumours of the thyroid. A review with emphasis on mitochondrial abnormalities with clinical relevance. Virchows Arch. 2000;437(2):107–115. [DOI] [PubMed] [Google Scholar]
- 13. Wautot V, Vercherat C, Lespinasse J, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: Search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat. 2002;20(1):35–47. [DOI] [PubMed] [Google Scholar]
- 14. Agarwal SK, Kester MB, Debelenko LV, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet. 1997;6(7):1169–1175. [DOI] [PubMed] [Google Scholar]
- 15. Heppner C, Kester MB, Agarwal SK, et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet. 1997;16(4):375–378. [DOI] [PubMed] [Google Scholar]
- 16. Debelenko LV, Brambilla E, Agarwal SK, et al. Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum Mol Genet. 1997;6(13):2285–2290. [DOI] [PubMed] [Google Scholar]
- 17. Zhuang Z, Vortmeyer AO, Pack S, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 1997;57(21):4682–4686. [PubMed] [Google Scholar]
- 18. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]