

Abstract
Context:
Aberrant cellular oxygen sensing is a leading theory for development of pheochromocytoma (PHEO) and paraganglioma (PGL).
Objective:
The objective of the study was to test the hypothesis that chronic hypoxia in patients with cyanotic congenital heart disease (CCHD) increases the risk for PHEO-PGL.
Design/Setting/Participants:
We investigated the association between CCHD and PHEO-PGL with two complementary studies: study 1) an international consortium was established to identify congenital heart disease (CHD) patients with a PHEO-PGL diagnosis confirmed by pathology or biochemistry and imaging; study 2) the 2000–2009 Nationwide Inpatient Survey, a nationally representative discharge database, was used to determine population-based cross-sectional PHEO-PGL frequency in hospitalized CCHD patients compared with noncyanotic CHD and those without CHD using multivariable logistic regression adjusted for age, sex, and genetic PHEO-PGL syndromes.
Results:
In study 1, we identified 20 PHEO-PGL cases, of which 18 had CCHD. Most presented with cardiovascular or psychiatric symptoms. Median cyanosis duration for the CCHD PHEO-PGL cases was 20 years (range 1–57 y). Cases were young at diagnosis (median 31.5 y, range 15–57 y) and 7 of 18 had multiple tumors (two bilateral PHEO; six multifocal or recurrent PGL), whereas 11 had single tumors (seven PHEO; four PGL). PGLs were abdominal (13 of 17) or head/neck (4 of 17). Cases displayed a noradrenergic biochemical phenotype similar to reported hypoxia-related PHEO-PGL genetic syndromes but without clinical signs of such syndromes. In study 2, hospitalized CCHD patients had an increased likelihood of PHEO-PGL (adjusted odds ratio 6.0, 95% confidence interval 2.6–13.7, P < .0001) compared with those without CHD; patients with noncyanotic CHD had no increased risk (odds ratio 0.9, P = .48).
Conclusions:
There is a strong link between CCHD and PHEO-PGL. Whether these rare diseases coassociate due to hypoxic stress, common genetic or developmental factors, or some combination requires further investigation.
Cyanotic congenital heart disease (CCHD) refers to a subset of congenital heart disease (CHD) diagnoses that often present soon after birth with systemic hypoxemia and hypoxia related to impaired pulmonary flow and mixing of pulmonary and systemic venous blood. CCHD comprises approximately 10% of all CHD, or about 0.1% of all live births (1). A subset of patients with acyanotic defects later develop cyanosis, most commonly due to progressive pulmonary vascular disease and Eisenmenger syndrome. CCHD, inclusive of all types of CHD with resulting hypoxemia and hypoxia, has diverse multisystem effects. Secondary phenomena associated with chronic cyanosis in the setting of pulmonary vascular disease include erythrocytosis, hyperviscosity, cholelithiasis, cerebral abscess, vascular dysfunction, and hemoptysis (2). Most, but not all, patients with CCHD born in developed countries undergo surgical repair in childhood, resulting in either an elimination or reduction in the degree of hypoxemia.
Pheochromocytoma (PHEO) and paraganglioma (PGL) are neuroendocrine tumors arising from neural crest-derived cells or organs either in the adrenal gland (PHEO) or along the central sympathetic and parasympathetic chains (PGL), including the carotid body. These are relatively rare tumors in the general population, with a prevalence of 0.2–0.6% among hypertensive adults, 1.7% among hypertensive children, approximately 5% among incidentally discovered adrenal masses, and 0.05–0.1% in autopsy series (3). In the last decade, a convincing body of evidence linking hypoxia, hypoxia pathways, and pheochromocytoma-paraganglioma genetic syndromes has been generated (4–9). To date, more than a dozen inheritable genetic alterations have been implicated in pheochromocytoma-paraganglioma syndromes (9–11). A majority of these genes cluster in a common cellular pathway that activates hypoxia-inducible factors including succinate dehydrogenase (SDHx) genes, von Hippel-Lindau (VHL), and hypoxia induced factor 2A (HIF2A) (9, 10) These discoveries have led investigators to propose the pseudohypoxia hypothesis of PHEO-PGL development, in which genetic susceptibilities resulting in aberrant activation of hypoxia pathways in chromaffin cells are responsible for the pathogenesis of PHEO-PGL (9, 12). This hypothesis is particularly attractive, given prior observations linking hypoxia at an altitude with a higher prevalence of PHEO-PGL (13–15).
We hypothesized that exposure to chronic hypoxia in CCHD would increase the risk for developing PHEO-PGL. We tested this hypothesis using two complementary approaches: 1) development of a multicenter international consortium to identify all known cases of PHEO-PGL in patients with CHD; and 2) a systematic analysis of a nationally representative population-based hospitalization data set to determine whether patients with CCHD are at higher risk for PHEO-PGL when compared with those with noncyanotic CHD.
Materials and Methods
Multicenter case series
We developed an international consortium of nine adult congenital heart disease referral centers with the unified aim of identifying all known PHEO-PGL cases within our collective CHD patient populations to develop a centralized case series. Inclusion into the database required the following: age 18 years or older at the time of clinical presentation to one of our centers during or after the year 2000, a diagnosis of CHD, and a diagnosis of PHEO-PGL. CCHD was defined as the presence of CHD with a history of at least 1 year of chronic hypoxemia (arterial saturation ≤ 92%). The diagnosis of PHEO-PGL was defined by either pathologic confirmation of a PHEO or PGL, and/or a combination of suggestive biochemical markers drawn to evaluate hyperadrenergic symptoms (at least 2-fold elevations in plasma metanephrines or urinary catecholamines or metanephrines) in combination with supportive imaging findings (radiographic findings suggestive of PHEO or PGL). Ethics board approval was obtained at the coordinating centers (Brigham and Women's Hospital and Boston Children's Hospital) and the contributing sites (Hospital of the University of Pennsylvania and The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania; Columbia University Medical Center, New York City, New York; University Hospital Zurich, Zurich, Switzerland; University of California at Los Angeles Medical Center, Los Angeles, California; St Paul's Hospital, University of British Columbia, Vancouver, British Columbia, Canada; Nationwide Children's Hospital and The Ohio State University Wexner Medical Center, Columbus, Ohio; Department of Medicine, University Health Network and University of Toronto, Toronto, Ontario, Canada; and Medical College of Wisconsin, Milwaukee, Wisconsin).
For each identified case, the following data were collected: underlying CHD, timing of prior cardiac procedures and duration of cyanosis, clinical presentation of PHEO-PGL diagnosis, and other clinical information about each PHEO or PGL (laboratory testing, pathology, management) and clinical follow-up. Data were also collected on any clinical evidence or signs of known PHEO-PGL genetic syndromes including the following: thyroid nodules, medullary thyroid cancer, hypercalcemia, or hyperparathyroidism, oral mucosal neuromas, marfanoid habitus (multiple endocrine neoplasia [MEN], type 2); neurofibromas, freckling, cafe au lait spots, Lisch nodules/optic nerve tumors, skeletal dysplasia (neurofibromatosis type I); retinal angiomas, central nervous system or pancreatic masses, kidney masses, or renal cell carcinoma, epididymal cystadenomas (von Hippel-Lindau syndrome); kidney masses or renal cell carcinoma, gastrointestinal stromal tumors, or pituitary adenomas (succinate dehydrogenase deficiency).
Of the 20 cases reported, 18 were in patients with a history of chronic cyanosis. We report data on all 20 cases but focus the analysis and discussion on the 18 cases in patients with CCHD except where specifically noted.
Population-based study
We performed a retrospective, cross-sectional, observational study using the 2000–2009 Nationwide Inpatient Sample (NIS) administrative data for adults 18–75 years old hospitalized with any nonmaternal/childbirth diagnosis; NIS is the largest publicly available all-payer inpatient care database in the United States (16) and includes data on approximately 7 million to 8 million discharges annually. NIS includes a stratified sample designed to approximate a 20% sample of US nonfederal, short-term, general, and specialty hospitals. Demographic covariates included age, sex, and year of admission. PHEO-PGL was defined as the presence of pheochromocytoma [International Classification of Diseases, ninth revision (ICD-9), codes 227.0, 255.6] carotid body tumor (codes 194.5, 227.5) or paraganglioma (codes 194.6, 227.6, 237.3). CHD was defined as described elsewhere (17). Given the absence of historical data on cyanosis, the definition of CCHD differed for the population-based study and the case series; for the population-based study, CCHD was defined as any CHD with coexisting codes for cyanosis, hypoxemia, or secondary erythrocytosis (codes 782.5, 289.0, 799.0). To assess whether any association was independent of demographic and genetic confounders, we performed a multivariable logistic regression adjusting for available demographic variables (age, sex), clinical features known to occur with PHEO-PGL (hypertension, renal cell carcinoma), and known pheochromocytoma-paraganglioma syndromes (MEN code 258.0, von Hippel-Lindau disease (VHL) code 759.6, neurofibromatosis code 237.7). Using more specific codes (ie, code 258.02 for MEN type 2; code 237.71 for neurofibromatosis type 1) did not change the conclusions. The statistical analyses were performed using SAS for Windows 9.3 (SAS Institute Inc). Analyses used provided sample weights to produce national estimates and account for complex sample design and clustering by hospital (16).
Results
Multicenter case detection
Cardiac anatomy and history
In total, we identified 20 cases of PHEO-PGL in patients with CHD; 90% of these cases (18 of 20) had CCHD (Table 1). Most patients had complex cyanotic congenital heart disease, including double-inlet left ventricle (LV), tetralogy of Fallot, double-outlet right ventricle (RV) with pulmonary atresia, and tricuspid and pulmonary atresia (n = 16), whereas a smaller subset had Eisenmenger syndrome in the setting of a simple defect (n = 2) (Table 1). There was no apparent association between a specific subgroup of cardiac malformations and PHEO-PGL. Interestingly, both patients with noncyanotic CHD had aortic coarctation.
Table 1.
Demographic and clinical features of patients with CHD and PHEO-PGL
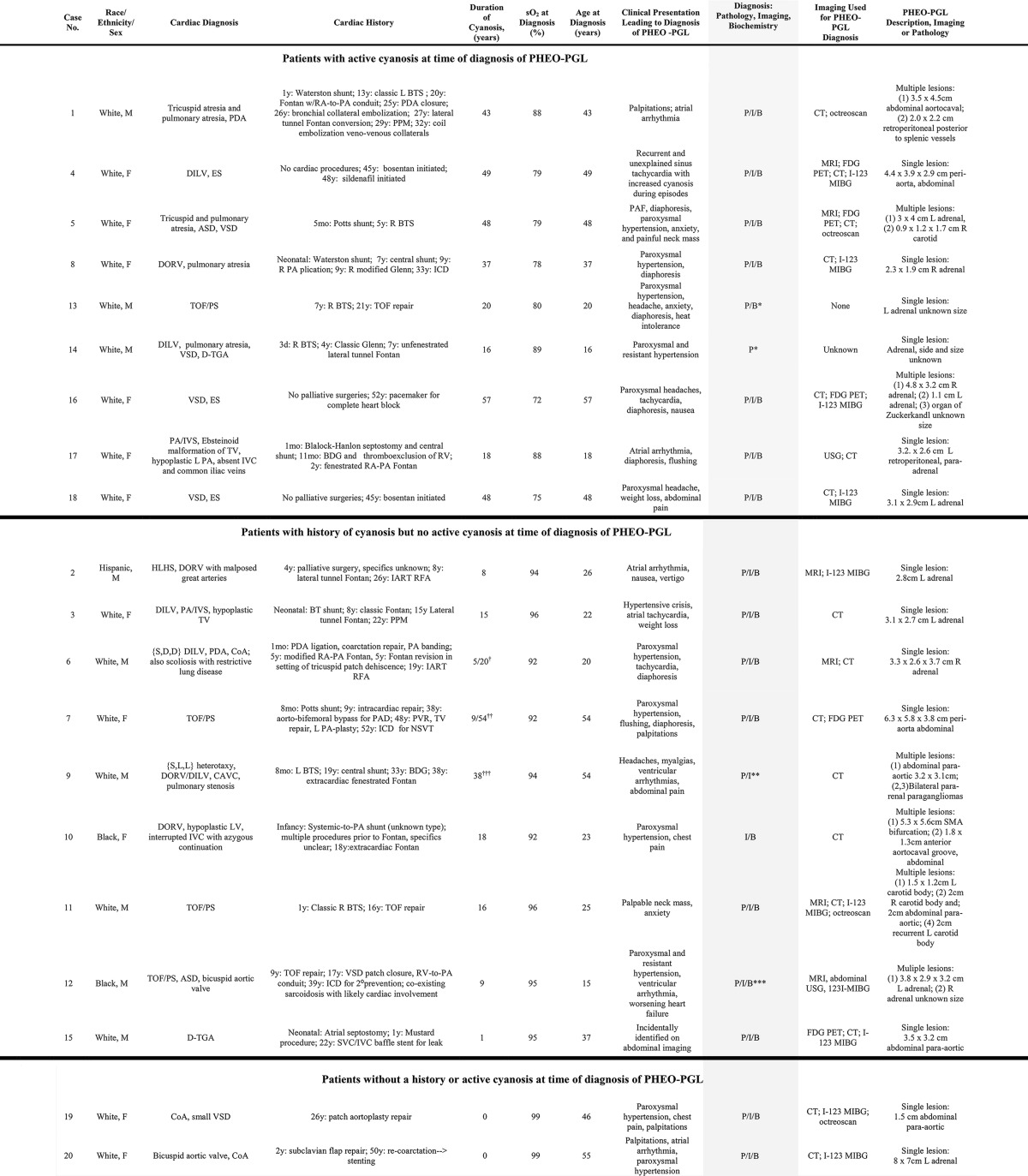
Abbreviations: ASD, atrial septal defect; BDG, bidirectional Glenn; BTS, Blalock-Taussig shunt; CAVC, complete atrioventricular canal defect; CoA, coarctation of the aorta; DILV, double-inlet left ventricle; DORV, double-outlet right ventricle; ES, Eisenmenger syndrome; FDG PET, 18-fluoro-deoxyglucose positron emission tomography; HLHS, hypoplastic left heart syndrome; IART, intraatrial reentrant tachycardia; ICD, implantable cardioverter defibrilator; I-123 MIBG, 123I-metaiodobenzylguanidine scintigraphy; IVC, inferior vena cava; L, left; PA, pulmonary artery; PAD, peripheral artery disease; PAF, paroxysmal atrial fibrillation; PA/IVS, pulmonary atresia, intact ventricular septum; PDA, patent ductus arteriosus; P/I/B, diagnosis by pathology, imaging, and biochemistry, respectively; PPM, permanent pacemaker; PVR, pulmonary valve replacement; R, right; RA, right atrium; RFA, ablation procedure; SVC, superior vena cava; TGA, transposition of the great arteries; TOF/PS, tetralogy of Fallot with pulmonary stenosis; TV, tricuspid valve; VSD, ventricular septal defect.
a Cases 13 and 14 underwent surgical resection of PHEO-PGL prior to presentation to the participating centers. Diagnosis of PHEO-PGL is based on available data that included primary information on biochemistry and pathology for case 13 but only historical documentation of consistency and confirmatory pathology for case 14.
b Severe cyanosis prior to Fontan. Documented resting saturations were 86%–92% between 10 and 20 years old.
c Severe cyanosis prior to TOF repair. Resting saturation was 92% in the decade prior to a PHEO-PGL diagnosis.
d Resting saturation after Fontan ranged from 91% to 94%, with exertional desaturation to a high percentage in the 80s.
e Diagnosis for case 9 was based on autopsy. An abdominal computed tomography scan performed shortly before the patient died suggested lymphadenopathy; PHEO-PGL was not suspected. The imaging findings suggestive of lymphadenopathy likely represented autopsy-documented paragangliomas.
f Diagnosis for case 12 differed for the two lesions. A left-sided pheochromocytoma was resected at age 15 years, with the diagnosis based on pathology. The right-sided lesion was diagnosed at age 31 years, and resection was pending at the time of data collection; the diagnosis for this lesion is based on imaging and biochemistry.
Patients with CCHD had a median age at PHEO-PGL diagnosis of 31.5 years (range 15–57 y), and the median cumulative duration of cyanosis was 20 years (range 1–57 y). Although the mean oxygen saturation at time of PHEO-PGL diagnosis was 87.4%, only half of the patients (9 of 18) were actively cyanotic at the time of diagnosis, whereas the other half (9 of 18) had a remote history of cyanosis. As expected, the nine patients diagnosed with PHEO-PGL while actively cyanotic, had a longer duration of cyanosis (37.3 ± 14.5 y vs 19.9 ± 15.5 y, Wilcoxon rank sums P = .02) and a lower oxygen saturation at diagnosis (80.9% ± 5.7% vs 94.0% ± 1.5%, Wilcoxon rank sums P < .001) when compared with the nine patients without active cyanosis at the time of diagnosis; however, there was no difference in the age at PHEO-PGL diagnosis between these groups (37.3 ± 14.5 y vs 30.7 ± 13.6 y, Wilcoxon rank sums P = .28).
Tumor and biochemical phenotype and genetic evaluation
Among patients with CCHD, 61% (11 of 18) presented with a single tumor (seven PHEOs and four PGLs), and 39% (seven of 18) presented with multiple tumors (two with bilateral PHEO and six with multifocal or recurrent PGL). In total, there were 17 individual PGL tumors identified among the 18 patients, of which 13 were intraabdominal and four were in the head and neck region. None of the tumors were malignant on histopathology, and no patients demonstrated imaging findings suggestive of metastatic disease. A dominant noradrenergic biochemical phenotype was observed among patients with CCHD and PHEO-PGL. Nearly all cases presented with greater than 3-fold elevations in either plasma normetanephrines or urinary norepinephrine or normetanephrine concentrations, with little or no elevation of epinephrine or dopamine metabolites (Table 2). Two patients did not have any available laboratory data and were confirmed using pathologic criteria. Case 14 was included based on clinical documentation of surgery for pathologically confirmed pheochromocytoma after development of hypertension, but primary records were not available.
Table 2.
Biochemical Phenotype of the 18 Cases of CCHD and PHEO-PGL
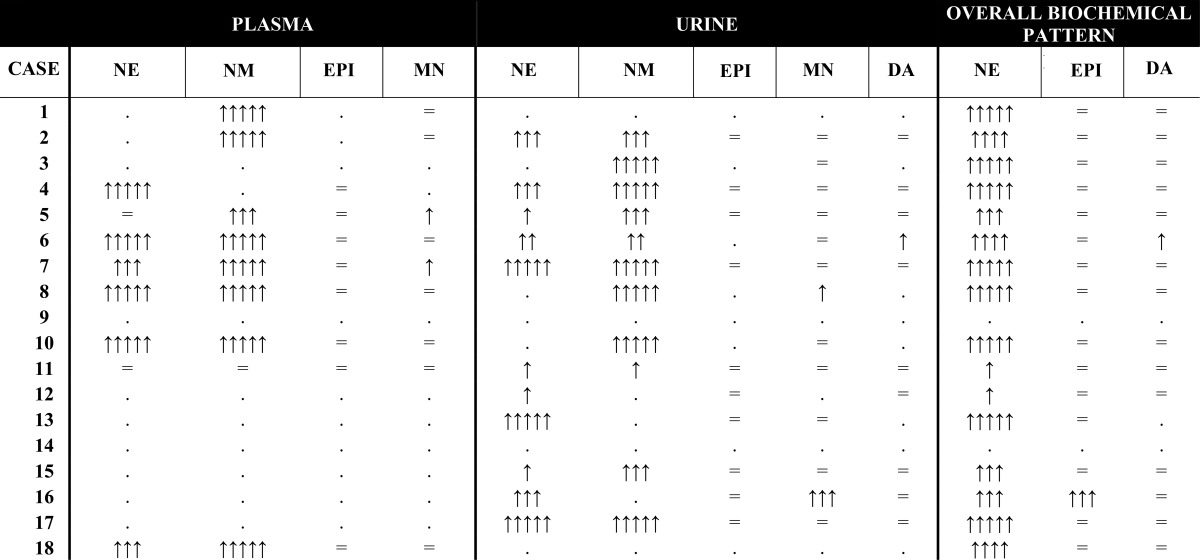
Arrows indicate the degree of elevation relative to the upper limit of normal in plasma, urine, and a composite summary of the overall biochemical pattern. Equal signs indicate the value in the reference range, and periods indicate that measurements were either not performed or not available to assess. NE, norepinephrine; NM, normetanephrine; EPI, epinephrine; MN, metanephrine; DA, dopamine.
↑↑↑↑↑ more than 8 × the upper limit of the reference range.
↑↑↑ 3–8 × the upper limit of the reference range.
↑↑ 2–3 × the upper limit of the reference range.
↑ more than 2 × the upper limit of the reference range.
Clinical genetic testing was incomplete. Genetic testing results were available for only five patients, and only three were tested for succinate dehydrogenase genes. One of the five patients was found to have a reported pathogenic missense SDHB mutation (G137A in exon 2), in the setting of nonmalignant, multifocal, recurrent PGL (case number 11, Table 1) and a strong family history (three affected siblings and one affected child). No genetic abnormalities were detected in the four other patients with partial testing (case numbers 3, 5, 7, and 17 in Table 1, including testing for RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, MAX, VHL). There were no other documented clinical or radiographic manifestations to suggest known genetic syndromes associated with pheochromocytoma-paraganglioma syndromes (no personal or family history of renal carcinoma, gastrintestinal stromal tumors, pituitary tumors, medullary thyroid carcinomas, vascular or ocular stigmata to suggest von Hippel-Lindau syndrome or signs or symptoms of neurofibromatosis).
Population-based analysis
We analyzed more than 40 million hospitalizations for adults younger than 75 years old for reasons other than pregnancy and childbirth between 2000 and 2009. There were 576 168 ± 20 596 admissions with CHD, of which 16 823 ± 691 (2.8% ± 0.1%) were for CCHD. We identified 108 413 ± 3205 admissions with a diagnosis code suggestive of PHEO-PGL, 50.8 ± 21.6 of which were in patients with CCHD. In the overall sample, those hospitalized with PHEO-PGL were older, more likely to be female, and more likely to have genetic pheochromocytoma-paraganglioma syndromes and associated features [eg, MEN, VHL, neurofibromatosis (NF), renal cell carcinoma, hypertension] (Table 3).
Table 3.
Population-Based Association Between CCHD and PHEO-PGL
| PHEO-PGL |
P Value | Univariate |
Multivariable |
||||
|---|---|---|---|---|---|---|---|
| Yes | No | OR | 95% CI | OR | 95% CI | ||
| Age, y | 55.9 ± 0.1 | 53.2 ± 0.1 | <.0001 | 1.013 | 1.012–1.015 | 1.013 | 1.012–1.014 |
| Female, % | 61.2 ± 0.3 | 51.8 ± 0.1 | <.0001 | 1.5 | 1.4–1.5 | 1.5 | 1.4–1.5 |
| Cyanotic CHD, per 105 | 4.7 ± 2.0 | 0.9 ± 0.0 | <.0001 | 5.5 | 2.4–12.5 | 6.0 | 2.6–13.7 |
| Noncyanotic CHD, per 105 | 19.6 ± 3.0 | 21.9 ± 0.7 | 0.48 | 0.9 | 0.7–1.2 | 0.9 | 0.7–1.5 |
| MEN, per 105 | 15.9 ± 3.1 | 0.3 ± 0.0 | <.0001 | 60.9 | 41.0–90.6 | 59.8 | 40.0–89.6 |
| VHL, per 105 | 26.8 ± 4.7 | 1.4 ± 3.1 | <.0001 | 19.7 | 14.3–27.3 | 15.9 | 11.5–22.1 |
| NF, per 105 | 34.8 ± 5.8 | 3.4 ± 0.1 | <.0001 | 10.3 | 7.4–14.2 | 10.8 | 7.8–15.0 |
| Renal cell cancer, per 105 | 207.3 ± 10.9 | 24.6 ± 0.5 | <.0001 | 8.6 | 7.8–9.4 | 8.2 | 7.5–9.1 |
| Hypertension, % | 62.6 ± 0.4 | 42.2 ± 0.0 | <.0001 | 2.3 | 2.2–2.4 | 2.2 | 2.2–2.3 |
| Hypothyroidism, % | 8.3 ± 0.2 | 6.8 ± 0.0 | <.0001 | 1.24 | 1.18–1.31 | 1.04 | 0.98–1.09 |
| Diabetes, % | 26.1 ± 0.4 | 22.7 ± 0.1 | <.0001 | 1.21 | 1.17–1.25 | 1.12 | 1.09–1.16 |
Results are shown as univariate and multivariable adjusted ORs. Data for nonpregnant patients 18–75 years old hospitalized between 2000 and 2009 were analyzed to determine the population-based frequency of hospitalization for cyanotic CHD and PHEO-PGL. PHEO-PGL was defined as PHEO (ICD-9 codes 194.0, 227.0, 255.6), PGL (codes 194.5/6, 237.3), or carotid body tumor (codes 194.5, 227.5). Cyanotic CHD was defined as any diagnostic code for CHD plus one of the following: cyanosis, hypoxemia, or secondary erythrocytosis (ICD-9 782.5, 289.0, 799.0). Multivariable logistic regression was performed, adjusting for age, gender, and established PHEO-PGL genetic syndromes and phenotypes associated with such syndromes, such as MEN (ICD-9 code 258.0), VHL (ICD-9 code 759.6), and NF (ICD-9 code 237.7), and hypertension diagnosis and renal cell cancer.
Of all admissions for patients with noncyanotic CHD, 0.05% ± 0.01% were associated with PHEO-PGL, in comparison with 0.3% ± 0.1% of CCHD admissions. CCHD was present in just less than 1 in 100 000 non-PHEO-PGL hospitalizations, compared with a greater than 5-fold higher proportion of hospitalizations for a patient with PHEO-PGL (0.9 vs 4.7 per 100 000 hospitalizations). The adjusted odds of hospitalization with PHEO-PGL among patients with CCHD was an odds ratio (OR) of 6.0 [95% confidence interval (CI) 2.6–13.7, P < .0001], whereas the odds of PHEO-PGL in noncyanotic CHD was equivalent to that seen in hospitalizations for patients without CHD (OR 0.9, P = .48) (Table 3). Although the absolute number of cases precludes a detailed reporting of diagnoses and associations within the group of patients with CCHD, the underlying diagnoses included ventricular septal defect (ICD-9 code 745.4), ostium secundum atrial septal defect (code 745.5), common atrium or atrioventricular canal defect (code 745.69), complete transposition of the great arteries (code 745.1), and common ventricle (code 745.3). None had Down syndrome (code 758.0).
Discussion
The co-occurrence of PHEO-PGL and CCHD has been reported anecdotally; however, based on the increasing number of candidate genes implicated in pheochromocytoma-paraganglioma syndromes that are in the hypoxia pathway (9–11) and observations linking hypoxia at altitude with PHEO-PGL prevalence (13–15), we speculated that these sporadic observations may reflect a deeper connection between chronic hypoxia exposure and an underlying susceptibility for PHEO-PGL. Herein we report an international experience of PHEO-PGL cases among patients with CCHD, the largest series to our knowledge. The data demonstrate a number of features suggestive of pseudohypoxic syndromic PHEO-PGL phenotypes in CCHD patients (young age of onset, multiple tumors, noradrenergic biochemical phenotype). Furthermore, we report a similar coassociation between these diseases in a large population-based sample. Patients hospitalized with a CCHD diagnosis are also much more likely to have a diagnosis of PHEO-PGL, independent of other known and reported risk factors for development of these tumors.
Although PHEO and PGL are rare tumors, they have the potential for malignant transformation and are often associated with significant adverse cardiovascular events such as arrhythmia, hypertension, and heart failure, particularly in patients with existing cardiovascular disease. Recent discoveries have revolutionized our insight into the pathogenesis of pheochromocytoma and paraganglioma by suggesting that up to 35–40% of these tumors are likely part of a greater pheochromocytoma-paraganglioma genetic syndrome (9–11). Notably, many of the identified susceptibility genes for PHEO-PGL have implicated dysregulated cellular responses to hypoxia (pseudohypoxia) as a leading explanation for the development of pheochromocytoma-paraganglioma syndromes.
This hypothesis of the genomic era, however, may reflect an extension of prior observations made in true hypoxic states. For example, carotid body glomus cells are O2-sensitive chemoreceptors, and animal models suggest that adrenal chromaffin cells are also sensitive to hypoxia in the postnatal period (18). Basic pathological studies have observed abnormal mitochondrial structure and appearance in PHEO and PGL tissue (19, 20). Hypoxemia is associated with carotid body hyperplasia, (21), and people living at high altitude are at increased risk for pheochromocytoma-paraganglioma syndromes (13–15, 22, 23). There is even one reported instance in which regression of a PGL was observed after relief of chronic hypoxemia (24).
A number of case reports have described the diagnosis of neuroendocrine tumors in patients with CCHD, mainly PHEO-PGL. The association between PHEO-PGL and CCHD was first suggested in a 1964 case series; almost a quarter of patients with histologically proven PHEO seen at Johns Hopkins Hospital (Baltimore, Maryland) between 1901 and 1962 (5 of 21) had CCHD (25). The co-occurrence of CCHD and PHEO-PGL has been repeatedly observed in case report form (26–47). Until the current investigation, however, there had been no systematic multicenter large series, limiting the ability to understand or confirm the clinical presentation and phenotype of PHEO-PGL in this group. Furthermore, there had been no epidemiological studies investigating the frequency of PHEO-PGL in patients with CCHD in comparison with that in patients with noncyanotic CHD or the general population, limiting inference on whether patients with CCHD truly have a higher risk for PHEO-PGL.
Several observations from the current report support a relationship between clinical hypoxemia and PHEO-PGL. First, all but two of the cases presented here had a history of an extended period of cyanosis prior to PHEO-PGL diagnosis. It is important to note that although the precise proportion is unknown, only a small minority of patients seen in adult congenital heart disease clinics have an extended history of cyanosis. The most common adult congenital heart disease diagnoses (eg, atrial and ventricular septal defects, bicuspid aortic valve, and coarctation of the aorta) are not associated with a high burden of hypoxemia.
Second, the age at the time of diagnosis of PHEO-PGL in this series was younger than seen in the overall population of patients with PHEO-PGL, and 40% of patients developed multiple tumors. Young age and multiple tumors are usually associated with high-risk or syndromic PHEO-PGL. The cumulative duration of cyanosis was also long, raising the possibility of an association between burden of hypoxemia and syndromic-type PHEO-PGL phenotypes. Further investigation is needed to confirm this hypothesis and to assess the relative contributions of timing, duration and severity of cyanosis.
Third, all of the cases displayed a biochemical phenotype suggestive of pseudohypoxic pheochromocytoma-paraganglioma syndromes (9–11). Biochemical characteristics of pheochromocytoma-paraganglioma syndromes have been consistently described: pheochromocytoma-paraganglioma syndromes resulting from mutations in the hypoxia pathway (cluster 1 genes: VHL, SDHx, HIF2A) present with a noradrenergic phenotype (markedly elevated norepinephrine and normetanephrines) with a notable absence of an adrenergic phenotype (essentially normal epinephrine and metanephrines) (48, 49). The CCHD patients with PHEO-PGL identified in our international cohort displayed striking noradrenergic biochemical profiles, with a distinct absence of any convincing adrenergic secretory patterns, providing tantalizing evidence that hypoxia or pseudohypoxia in CCHD may represent the causal risk factor for PHEO-PGL.
Whether the risk for PHEO-PGL is increased in CCHD due only to chronic hypoxia or the combination of chronic hypoxia with an underlying genetic susceptibility could not be completely assessed in our study. Although one of the cases had a known syndrome or genetic/pathological cause for PHEO-PGL (case number 11), only five patients underwent clinical genetic testing, and most of these did not have comprehensive assessment of all currently known pheochromocytoma-paraganglioma syndrome genes (11). The patient with an identified pathogenic SDHB mutation had the most severe phenotype with multiple PHEO-PGL recurrences, potentially reflecting an important gene-environment interaction. The incomplete genotyping in this series precludes us from concluding whether the prevalence of predisposing pheochromocytoma-paraganglioma syndrome genes is equivalent to the overall PHEO-PGL population or whether investigation for new forms of genetic predisposition may be warranted. Despite this limitation, our findings suggest that CCHD may represent a useful human model of increased PHEO-PGL risk in which focused investigations may be conducted to better understand the pathophysiology, genetic underpinnings, and gene-environment interactions of PHEO-PGL. Given that up to 35–40% of PHEO-PGL is now thought to be attributed to a reported germline gene alteration, and clinical genetic investigations are routinely recommended (3, 11), assessment for potential gene-hypoxia interaction represents an area for future investigation.
Prior case reports of PHEO-PGL and CHD focused entirely on patients with ongoing chronic cyanosis. This situation is increasingly infrequent in the contemporary era because most patients undergo intervention early in life. Although the vast majority of the patients in this series had a history of long-standing cyanosis, many had undergone biventricular repair or had been converted to a Fontan circulation long before the diagnosis of PHEO-PGL and were not hypoxemic at the time of diagnosis. It should be noted that a sizable subset of patients with Fontan circulation remain mildly hypoxemic (due to coronary sinus anatomy, patent fenestration, venovenous collaterals, pulmonary arteriovenous malformations, or parenchymal lung disease). Risk for PHEO-PGL in these patients could presumably be due to early severe cyanosis, chronic low-level hypoxemia, or both. The sample size precludes inferences regarding what variables predict risk (eg, severity, duration, and timing of cyanosis). The observation, however, does argue that the population at risk for PHEO-PGL includes those with repaired CHD who had historical exposure to hypoxemia, a growing group of patients.
The reported association has direct clinical implications. First, symptoms and complications associated with PHEO-PGL overlap strikingly with symptoms and complications of CCHD including palpitations, progressive arrhythmia, sudden cardiac arrest, fatigue, and malaise. PHEO-PGL represents a potentially curable cause of deterioration. PHEO-PGL should be considered as a cause of change in/development of new symptoms in these patients. Increased systemic adrenergic and noradrenergic tone may also provoke cardiovascular decline via neurohormonal effects in tenuous patients (50, 51). Although it would be reasonable to propose the need for greater clinical suspicion for PHEO-PGL and a lower threshold for serum and urine testing, there are no normative data to guide interpretation in this population, which would be expected to have higher baseline catecholamine levels (52). This would presumably be associated with a high burden of false-positive results, requiring further evaluation. Thus, although we believe the current observation should be integrated into care, the appropriate approach to doing so remains to be defined.
Limitations
Each study must be interpreted in the context of well-described limits of its particular design (case series and cross-sectional analysis of a large administrative data set), and this will not be further detailed. The two approaches tend to have complementary strengths and weaknesses; the consistency of conclusion is notable and supports the qualitative validity of each as presented. Specific limitations to the case ascertainment include variable and incomplete assessment of PHEO-PGL phenotype and genotype as well as a lack of detailed tumor tissue data. We cannot confidently conclude that our observation of concurrent cyanotic congenital heart disease and PHEO-PGL is independent of known genetic predispositions for PHEO-PGL because only five of the 20 patients were tested. However, our findings from the case-series and population-based study do support the hypoxia hypothesis of PHEO-PGL pathogenesis and may serve as a foundation upon which future studies can extend these results. Furthermore, we are unable to determine the exact incidence of PHEO-PGL in study 1 because the assessment of the true denominator of patients with CHD cannot be accurately determined for all of the participating centers. For the population-based study, we focused on patients with codes indicating both CHD and chronic ongoing cyanosis rather than identify patients by underlying diagnosis. We opted for this definition because many patients with cyanotic CHD diagnoses are now repaired very early in life, and we had no way to identify how long a given person had been cyanotic. All observational studies have the potential for bias and confounding; we attempted to minimize both of these with our analytical approach. The use of diagnosis codes for hypoxemia could result in ascertainment bias because we are unable to differentiate those individuals with chronic hypoxemia due to CCHD from those with CHD hospitalized with acute hypoxemia due to another cause. Although it highlights the possible pitfalls of this sort of analysis, this specific potential bias, however, is unlikely to explain the significantly higher association of PHEO-PGL diagnosis in those with diagnosis codes for CHD and hypoxemia or cyanosis or secondary erythrocytosis.
Conclusions
Patients with congenital heart disease and current or historical cyanosis are at increased risk for developing PHEO-PGL. Many symptoms often attributed to progressive CCHD overlap with those seen due to catecholamine secretion by these tumors. Clinicians should consider PHEO-PGL as a possible cause of otherwise unexplained clinical decline; early identification and treatment would presumably be associated with better outcomes. Further investigation is needed to identify the cause of this association, whether hypoxic stress, common genetic or developmental factors, or some combination.
Acknowledgments
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This was an Alliance for Adult Research in Congenital Cardiology study.
A.V. was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award K23HL111771. A.V. is also supported by a William Randolph Hearst Foundation Young Investigator Award. A.R.O., M.J.L., F.W., and M.N.S. were supported by the Dunlevie Family Fund.
Disclosure Summary: The authors have nothing to declare.
Footnotes
- ASD
- atrial septal defect
- CCHD
- cyanotic congenital heart disease
- CHD
- congenital heart disease
- CI
- confidence interval
- ICD-9
- International Classification of Diseases, ninth revision
- LV
- left ventricle
- MEN
- multiple endocrine neoplasia
- NF
- neurofibromatosis
- NIS
- Nationwide Inpatient Sample
- OR
- odds ratio
- PGL
- paraganglioma
- PHEO
- pheochromocytoma
- RV
- right ventricle
- VHL
- von Hippel-Lindau disease
- VSD
- ventricular septal defect.
References
- 1. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–1900. [DOI] [PubMed] [Google Scholar]
- 2. Opotowsky AR, Landzberg MJ, Beghetti M. The exceptional and far-flung manifestations of heart failure in Eisenmenger syndrome. Heart Fail Clin. 2014;10(1):91–104. [DOI] [PubMed] [Google Scholar]
- 3. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. [DOI] [PubMed] [Google Scholar]
- 4. Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287(5454):848–851. [DOI] [PubMed] [Google Scholar]
- 5. Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466. [DOI] [PubMed] [Google Scholar]
- 6. Burnichon N, Cascon A, Schiavi F, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. 2012;18(10):2828–2837. [DOI] [PubMed] [Google Scholar]
- 7. Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42(3):229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhuang Z, Yang C, Lorenzo F, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367(10):922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jochmanova I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst. 2013;105(17):1270–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14(2):108–119. [DOI] [PubMed] [Google Scholar]
- 11. Rana HQ, Rainville IR, Vaidya A. Genetic testing in the clinical care of patients with pheochromocytoma and paraganglioma. Curr Opin Endocrinol Diabetes Obes. 2014;21(3):166–176. [DOI] [PubMed] [Google Scholar]
- 12. Favier J, Gimenez-Roqueplo AP. Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab. 2010;24(6):957–968. [DOI] [PubMed] [Google Scholar]
- 13. Arias-Stella J, Valcarcel J. Chief cell hyperplasia in the human carotid body at high altitudes; physiologic and pathologic significance. Hum Pathol. 1976;7(4):361–373. [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez-Cuevas H, Lau I, Rodriguez HP. High-altitude paragangliomas diagnostic and therapeutic considerations. Cancer. 1986;57(3):672–676. [DOI] [PubMed] [Google Scholar]
- 15. Saldana MJ, Salem LE, Travezan R. High altitude hypoxia and chemodectomas. Hum Pathol. 1973;4(2):251–263. [DOI] [PubMed] [Google Scholar]
- 16. Sharabi Y, Grotto I, Huerta M, Grossman E. Susceptibility of the influence of weight on blood pressure in men versus women: lessons from a large-scale study of young adults. Am J Hypertens. 2004;17(5 Pt 1):404–408. [DOI] [PubMed] [Google Scholar]
- 17. Opotowsky AR, Siddiqi OK, Webb GD. Trends in hospitalizations for adults with congenital heart disease in the U.S. J Am Coll Cardiol. 2009;54(5):460–467. [DOI] [PubMed] [Google Scholar]
- 18. Thompson RJ. Current understanding of the O2-signalling mechanism of adrenal chromaffin cells. Cell Biol Chromaffin Cells. 2004;95–106. [Google Scholar]
- 19. Watanabe H, Burnstock G, Jarrott B, Louis WJ. Mitochondrial abnormalities in human phaeochromocytoma. Cell Tissue Res. 1976;172(2):281–288. [DOI] [PubMed] [Google Scholar]
- 20. Cornog JL, Wilkinson JH, Arvan DA, Freed RM, Sellers AM, Barker C. Extra-adrenal pheochromocytoma. Some electron microscopic and biochemical studies. Am J Med. 1970;48(5):654–660. [DOI] [PubMed] [Google Scholar]
- 21. Smith P, Jago R, Heath D. Anatomical variation and quantitative histology of the normal and enlarged carotid body. J Pathol. 1982;137(4):287–304. [DOI] [PubMed] [Google Scholar]
- 22. Astrom K, Cohen JE, Willett-Brozick JE, Aston CE, Baysal BE. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum Genet. 2003;113(3):228–237. [DOI] [PubMed] [Google Scholar]
- 23. Nolting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol. 2012;23(1):21–33. [DOI] [PubMed] [Google Scholar]
- 24. Gruber H, Metson R. Carotid body paraganglioma regression with relief of hypoxemia. Ann Intern Med. 1980;92(6):800–802. [DOI] [PubMed] [Google Scholar]
- 25. Folger GM, Jr, Roberts WC, Mehrizi A, et al. Cyanotic malformations of the heart with pheochromocytoma: a report of five cases. Circulation. 1964;29:750–757. [DOI] [PubMed] [Google Scholar]
- 26. Reynolds JL, Gilchrist TF. Congenital heart disease and pheochromocytoma. Am J Dis Child. 1966;112(3):251–255. [DOI] [PubMed] [Google Scholar]
- 27. Nissenblatt MJ. Cyanotic heart disease: “low altitude” risk for carotid body tumor? Johns Hopkins Med J. 1978;142(1):18–22. [PubMed] [Google Scholar]
- 28. Bockelman HW, Arya S, Gilbert EF. Cyanotic congenital heart disease with malignant paraganglioma. Cancer. 1982;50(11):2513–2517. [DOI] [PubMed] [Google Scholar]
- 29. Rutter TW, Mullin V. Pheochromocytoma in a patient with Eisenmenger's complex. Anesthes Analg. 1991;73(4):496–498. [DOI] [PubMed] [Google Scholar]
- 30. Medlicott SAC, Harder JR, Denmark LN. Double inlet left ventricle associated with a subpulmonary tissue tag, dissection of the pulmonary trunk and pheochromocytoma. Cardiol Young. 1997;7(03):334–336. [Google Scholar]
- 31. Juneja R, Krishnamani NC, Kothari SS, Guleria S, Mahawar RM. Pheochromocytoma and congenital cyanotic heart disease. Indian Heart J. 2000;52(4):452–454. [PubMed] [Google Scholar]
- 32. Kita T, Imamura T, Date H, et al. Two cases of pheochromocytoma associated with tetralogy of Fallot. Hypertens Res. 2003;26(5):433–437. [DOI] [PubMed] [Google Scholar]
- 33. Sparks JW, Seefelder C, Shamberger RC, McGowan FX. The perioperative management of a patient with complex single ventricle physiology and pheochromocytoma. Anesthes Analg. 2005;100(4):972–975. [DOI] [PubMed] [Google Scholar]
- 34. Bellingham GA, Dhir AK, Luke PP. Case report: retroperitoneoscopic pheochromocytoma removal in an adult with Eisenmenger's syndrome. Can J Anaesth. 2008;55(5):295–301. [DOI] [PubMed] [Google Scholar]
- 35. Yoshihara A, Tanabe A, Saito H, et al. A case of malignant pheochromocytoma with Holt-Oram syndrome. Endocr J. 2008;55(1):153–159. [DOI] [PubMed] [Google Scholar]
- 36. Cheung YW, Spevack DM. Single left ventricle and pheochromocytoma. Congen Heart Dis. 2008;3(5):355–358. [DOI] [PubMed] [Google Scholar]
- 37. Chung SJ, Lee AL, Shin CH, Yang SW, Bae EJ, Noh JI. Pheochromocytoma associated with cyanotic congenital heart disease. Korean J Pediatr. 2008;51(1):93–97. [Google Scholar]
- 38. Gabhane SK, Gangane NM, Sinha RT. Pentalogy of Fallot and cardiac paraganglioma: a case report. Cases J. 2009;2:9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Filgueiras-Rama D, Oliver JM, Ruiz-Cantador J, et al. Pheochromocytoma in Eisenmenger's syndrome: a therapeutic challenge. Rev Port Cardiol. 2010;29(12):1873–1877. [PubMed] [Google Scholar]
- 40. Hwang BH, Kim HY, Jung SE, Park KW. Extra-adrenal pheochromocytoma after operation of congenital heart disease: a case report of 18-year-old boy. J Korean Surg Soc. 2012;83(1):65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rich BS, Moo TA, Mark S, et al. Sympathetic paraganglioma in a patient with unrepaired tetralogy of Fallot: a case report and review of the literature. J Clin Endocrinol Metab. 2013;98(1):7–12. [DOI] [PubMed] [Google Scholar]
- 42. Balakrishnan G, Ravikumar R, Rao S, Balakrishnan KR. Tetralogy of Fallot with pheochromocytoma: an unusual therapeutic challenge. Asian Cardiovasc Thorac Ann. 2013;21(4):464–466. [DOI] [PubMed] [Google Scholar]
- 43. Kraayenbrink MA, Steven CM. Anaesthesia for carotid body tumour resection in a patient with the Eisenmenger syndrome. A case report. Anaesthesia. 1985;40(12):1194–1197. [DOI] [PubMed] [Google Scholar]
- 44. Hirsch JH, Killien FC, Troupin RH. Bilateral carotid body tumors and cyanotic heart disease. AJR Am J Roentgenol. 1980;134(5):1073–1075. [DOI] [PubMed] [Google Scholar]
- 45. Jewkes AJ, Ward RS, Black J, et al. Bilateral carotid body tumors in a patient with cyanotic congenital heart disease–a case report. Vasc Endovasc Surg. 1990;24(2):127. [Google Scholar]
- 46. Wilmshurst P, Newbegin C, Paes R. Chemodectoma in a patient with a single ventricle. Heart. 1997;77(4):385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pecorari G, Roccia F, Nadalin J, Giordano C, Garzaro M. Combined endovascular and surgical treatment of carotid body tumor in a patient with thoracic situs solitus. Head Neck. 2008;30(11):1523–1526. [DOI] [PubMed] [Google Scholar]
- 48. Eisenhofer G, Walther MM, Huynh TT, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001;86(5):1999–2008. [DOI] [PubMed] [Google Scholar]
- 49. Eisenhofer G, Lenders JW, Timmers H, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57(3):411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohuchi H, Takasugi H, Ohashi H, Yamada O, Watanabe K, Yagihara T, et al. Abnormalities of neurohormonal and cardiac autonomic nervous activities relate poorly to functional status in Fontan patients. Circulation. 2004;110(17):2601–2608. [DOI] [PubMed] [Google Scholar]
- 51. Oechslin E, Kiowski W, Schindler R, Bernheim A, Julius B, Brunner-La Rocca HP. Systemic endothelial dysfunction in adults with cyanotic congenital heart disease. Circulation. 2005;112(8):1106–1112. [DOI] [PubMed] [Google Scholar]
- 52. Bolger AP, Sharma R, Li W, et al. Neurohormonal activation and the chronic heart failure syndrome in adults with congenital heart disease. Circulation. 2002;106(1):92–99. [DOI] [PubMed] [Google Scholar]