

Abstract
Humans carrying mutations in neurokinin B (NKB) or the NKB receptor fail to undergo puberty due to decreased secretion of GnRH. Despite this pubertal delay, many of these patients go on to achieve activation of their hypothalamic-pituitary-gonadal axis in adulthood, a phenomenon termed reversal, indicating that NKB signaling may play a more critical role for the timing of pubertal development than adult reproductive function. NKB receptor-deficient mice are hypogonadotropic but have no defects in the timing of sexual maturation. The current study has performed the first phenotypic evaluation of mice bearing mutations in Tac2, the gene encoding the NKB ligand, to determine whether they have impaired sexual development similar to their human counterparts. Male Tac2−/− mice showed no difference in the timing of sexual maturation or fertility compared with wild-type littermates and were fertile. In contrast, Tac2−/− females had profound delays in sexual maturation, with time to vaginal opening and first estrus occurring significantly later than controls, and initial abnormalities in estrous cycles. However, cycling recovered in adulthood and Tac2−/− females were fertile, although they produced fewer pups per litter. Thus, female Tac2−/− mice parallel humans harboring NKB pathway mutations, with delayed sexual maturation and activation of the reproductive cascade later in life. Moreover, direct comparison of NKB ligand and receptor-deficient females confirmed that only NKB ligand-deficient animals have delayed sexual maturation, suggesting that in the absence of the NKB receptor, NKB may regulate the timing of sexual maturation through other tachykinin receptors.
The neurokinin B (NKB) signaling pathway was initially implicated in reproductive neuroendocrinology after observations that NKB expression is dramatically increased in estrogen receptor-expressing cells in the hypothalamus of postmenopausal women (1, 2). Further rodent work demonstrated that NKB expression in the arcuate nucleus of the hypothalamus (ARH) is negatively regulated by estradiol, as expression increased with ovariectomy and decreased with exogenous estrogen treatment (3). Evidence for NKB's role in reproduction was bolstered by findings of coexpression of NKB with 2 other reproductive neuropeptides, kisspeptin and dynorphin, in an ARH cell population now referred to as KNDy (Kisspeptin/Neurokinin B/Dynorphin) neurons (4–7). Kisspeptin had already been implicated as a key gatekeeper of puberty with the finding in 2003 that mutations in the kisspeptin signaling pathway cause hypogonadotropic hypogonadism, a condition characterized by absent pubertal development due to abnormal GnRH secretion from the hypothalamus (8–10). In 2009, mutations in the genes encoding NKB and the NKB receptor, neurokinin receptor 3 (NK3R), were also identified in Turkish patients with hypogonadotropism (11), demonstrating that NKB also plays a critical role for human sexual maturation.
Hypogonadotropic patients are typically counseled that they will require lifelong treatment (12). However, multiple reports have clearly established that a subset of hypogonadotropic patients unexpectedly develop recovery of reproductive function in adulthood after clear pubertal deficiency (13–25), including normalization of sex steroids, increases in testicular volume, and paternity/maternity without the use of fertility medications, a phenomenon termed “reversal.” The overall lifetime incidence of reversal of hypogonadotropic hypogonadism was found to be 22% in a recent retrospective analysis (26). Notably, examination of a group of hypogonadotropic patients with NKB pathway mutations for whom follow-up data was available revealed that 83% of these patients had evidence for activation of the hypothalamic-pituitary-gonadal (HPG) axes (23). The association of NKB pathway mutations with reversal suggests that NKB signaling may be critical for the timing of sexual development but dispensable for adult reproductive function.
In contrast to these intriguing phenotypes in humans, early studies examining mice with mutations in the gene encoding NK3R (Tacr3) (27) reported them to be reproductively normal, leading to confusion in the field as to what role NKB signaling played in reproductive regulation in the mouse. However, detailed phenotyping by our group subsequently revealed that Tacr3−/− mice did in fact have reproductive deficits (28). Specifically, mice with NK3R mutations had low uterine and testicular weights, and female mice had abnormal estrous cycles with prolonged time spent in diestrus. However, markers of sexual maturation (time to preputial separation, vaginal opening, and first estrus) were not different between Tacr3−/− and wild-type (WT) littermates. Moreover, both male and female Tacr3−/− mice were fertile. These observations demonstrate that the phenotypes of NKB receptor-deficient humans and mice are closer than previously appreciated but that the impairments in the rodent are observed in adulthood and not during sexual maturation. Phenotypes of mice lacking the NKB ligand have not been previously described. Thus, the current study has performed the first reproductive phenotyping, from early life through adulthood, in mice homozygous for mutations in Tac2, the gene encoding NKB in rodents. In addition, animals deficient for either the NKB ligand or the NKB receptor were directly compared to investigate similarities and differences in their reproductive phenotypes.
Materials and Methods
Reproductive phenotypes and brain neuropeptide expression were examined in transgenic mice with mutations in either the NKB ligand or receptor, which are encoded by Tac2 and Tacr3, respectively. Tac2+/− breeding pairs were generated by the Texas A&M Institute for Genomic Medicine (College Station, TX). Briefly, exons 1 and 2 were replaced with an IRES-lacz-neo cassette by homologous recombination (Supplemental Figure 1). NKB deficiency was verified by a complete lack of NKB staining in the ARH nucleus (Supplemental Figure 1). Genotyping of offspring from Tac2+/− breedings was carried out with forward primers designed against the endogenous region of exon 2 (5′-GCACCAGAAAGGAGATCAG-3′) and the transgenic cassette (5′-CGTTGGCTACCCGTGATATT-3′) and used in combination with a common downstream reverse primer (5′-ACCAGATATCCAAGACACAG-3′). Tacr3+/− breeding pairs were generated by Deltagen and have been described previously (27, 28). Exon 1 of Tacr3 was removed by homologous recombination, which was confirmed by Southern blot analysis (27). Genotyping was performed similarly as above, with forward primers designed against the WT region of exon 1 (5′-AGAAACTTACAAAGAGCCACCCACC-3′) and the transgenic cassette (5′-ACTTACATGCTTCCAGGCACAATAG-3′) and a common downstream reverse (5′-CCTGGGTGGGATTAGATAAATGCCTGCTCT-3′).
All mice were generated and maintained on a Sv129/C57BL/6 hybrid background and group housed (3–5 per cage) at the Massachusetts General Hospital Center for Comparative Medicine in a temperature- and light-controlled environment with lights on from 6 am to 6 pm, and food and water were provided ad libitum. The Massachusetts General Hospital Institutional Animal Care and Use Committee approved all procedures.
Male reproductive phenotypes
To assess male sexual maturation and fertility, male WT and Tac2 knockout mice (Tac2−/−) mice were examined daily for the presence of preputial separation and weekly for anogenital distance beginning at postnatal day (PND)25. Blood was collected from adult animals (PND56–PND90) by submandibular cheek bleed for serum RIA of LH, FSH, and testosterone levels (University of Virginia Research in Reproduction Ligand Assay and Analysis Core). In addition, testes were dissected, weighed, and prepared for histological examination (performed by the Harvard Rodent Histopathology Core). WT and Tac2−/− males were mated with adult WT C57BL/6 female mice for 12 weeks, and the latency to first litter, total number of litters as well as the average number of pups per litter was assessed.
Female reproductive phenotypes
WT and Tac2−/− female mice were examined daily for the presence of vaginal opening beginning on PND24. After vaginal opening was observed, vaginal lavage was performed daily to determine the day of first E and, in a subset of 3 WT and 4 Tac2−/− females, for continued monitoring of estrous cycling. In adult animals (PND100–PND140), uterine and ovarian weights were collected after euthanasia by CO2 inhalation, and ovaries were subsequently prepared for histological examination (performed by the Harvard Rodent Histopathology Core). Fertility in females was assessed both at the time of first E (WT PND31, Tac2−/− PND42) and in adulthood (PND81–PND100) to determine whether potential defects in fertility are age specific. Blood was collected from submandibular cheek bleed from all females before pairing with WT male and LH and FSH values were determined by RIA (UVA Center for Research in Reproduction Ligand Assay and Analysis Core). Females were paired with males for 12 weeks and the latency to the first litter, total number of litters, and number of pups per litter was assessed in both young and adult groups. These outcomes as well as the 12-week mating paradigm were chosen because they previously revealed a subfertility phenotype in Tacr3−/− female mice (28).
Immunohistochemistry (IHC)
Because ARH kisspeptin is thought to mediate sex steroid negative feedback in rodents (29), adult WT and Tac2−/− female mice aged between 4 and 5 months of age underwent bilateral ovariectomy under isoflurane sedation to allow for maximum ARH kisspeptin expression. Ten days after ovariectomy, female animals were sedated with a ketamine:xylazine cocktail (236:23.6 mg/kg) and perfused transcardially with saline and 4% paraformaldehyde. Brains were removed and postfixed in 4% paraformaldehyde overnight, followed by a 24-hour incubation in 30% sucrose. Brains were subsequently frozen on dry ice, cut by cryostat into 25μM sections, and stored in cryoprotectant. For free-floating IHC, sections were rinsed in potassium phosphate buffered saline, preincubated with 2% normal donkey serum, and incubated in primary antibody overnight at room temperature. Tissue was then incubated in donkey antirabbit biotinylated secondary antibody (711–065-152; Jackson ImmunoResearch), followed by incubation in A/B solution (PK-6100; Vector Laboratories), and finally nickel-diaminobenzidine (Sk-4100; Vector Laboratories). Primary antibodies were as follows: kisspeptin (1:30,000, number 564, a generous gift of Dr Alain Caraty) (30), NKB (1:1000, NB300–201; Novus Biological), and GnRH (1:10,000, PA1–120; Thermo Scientific). The kisspeptin and GnRH antibodies have been previously characterized (30–32), and the NKB antibody showed no immunoreactivity when used at a concentration of 1:1000 in the brain of Tac2−/−-deficient mice (Supplemental Figure 1).
For cell count and fiber density analysis, images were taken at a constant exposure with a total of 4 ARH and 2 anteroventral periventricular nucleus (AVPV) and 6 preoptic area sections analyzed per animal. The total number of AVPV kisspeptin and preoptic area GnRH cells was counted across these sections. ARH kisspeptin fiber density analysis was performed using Fiji (33) by converting to a 16-bit image and measuring the percent of pixels in a fixed region of interest with immunoreactivity above a common detection threshold as described previously (34).
Comparison of Tac2−/− and Tacr3−/− female mice
Given apparent discrepancies in the phenotypes of female Tac2−/− and those previously described for Tacr3−/− mice, a separate cohort of Tac2−/− females were assessed alongside Tacr3−/− females to compare the timing of sexual maturation between the 2 genotypes. Time to vaginal opening and first E were measured as described above for Tac2−/−, Tacr3−/−, and WT littermates from both transgenic lines.
Radioimmunoassays
All hormone assays were performed by the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core. Blood samples were spun at 2500 rpm for 20 minutes, and serum was collected and frozen until assayed. The sandwich LH assay has a reportable range of 0.02–37.4 ng/mL with inter- and intraassay coefficients of variability of 8.0% and 6.4%, respectively. Mouse LH (AFP5306A) and FSH (AFP5308D) reference prep, provided by Dr A.F. Parlow and the National Hormone and Peptide program, are used for assay standards.
The FSH assay has a reportable range of 2–75 ng/mL with inter- and intraassay coefficients of variability of 7.5% and 6.9%, respectively. For LH values that were below detectability of 0.02-ng/mL values were set at 0.02 ng/mL for analysis. The testosterone assay has a reportable range of 10–800 ng/dL with inter- and intraassay coefficients of variability of 6.4% and 4.4%, respectively, and standards were provided with the RIA kit (TKTT2; Siemens Healthcare Diagnostics).
Statistical analysis
Date to preputial separation, vaginal opening, and first E were analyzed by survival curve analysis and log-rank (Mantel Cox) test. Differences in uterine, testicular, and ovarian weights, as well as hormone levels and fertility in males, were analyzed by 2-tailed, unpaired t tests. Estrous cycle parameters, female fertility, and hormone levels were compared by two-way ANOVA and Bonferroni post hoc tests to determine whether fertility parameters differed between young and adult mice. To assess changes in time spent in diestrus/metestrus (D/M), cycle length, and bodyweight over time, two-way repeated measures ANOVA and Bonferroni post hoc tests were used. Analysis of total IHC cell counts and averaged percent of area were compared by 2-tailed t tests. All analysis was performed using Prism GraphPad Software, values are reported as mean ± SEM, and P < .05 was considered significant, with the exception of post hoc power calculations, which were computed in Sigma plot.
Results
Male reproductive phenotypes
To fully characterize reproductive phenotypes, several markers were examined to assess both the timing of sexual maturation and reproductive function in adulthood. Comparison between Tac2−/− male mice and their WT littermates revealed relatively few differences during development (Table 1). The average date of preputial separation, a marker of increased testosterone levels during sexual maturation, was not significantly different in Tac2−/− compared with WT (WT 31.67 ± 0.3 d, Tac2−/− 32.67 ± 0.5 d, log-rank survival curve comparison P = .09). Anogenital distance, another marker of testosterone levels, was modestly but significantly decreased at PND63 in Tac2−/− compared with WT littermates (WT 16.62 ± 0.3 mm, Tac2−/− 15.54 ± 0.3 mm, t test P = .02) mice. There was no statistically significant difference in bodyweight or testicular weights between Tac2−/− and WT animals (Table 1), and histology revealed the presence of mature sperm in both genotypes (Figure 1). Testosterone and LH levels were also not significantly different between genotypes (Table 1). However, FSH levels were significantly decreased in Tac2−/− compared with WT mice (WT 32.66 ± 3.6 ng/mL, Tac2−/− 14.73 ± 2.7 ng/mL, t test P < .001). Fertility was also assessed in adult males, and there was no difference between Tac2−/− and WT animals with regard to latency to first litter (WT 28.6 ± 3.3, Tac2−/− 26 ± 1.9, t test P = .48), average number of litters (WT 3.0 ± 0, Tac2−/− 2.4 ± 0.2, t test P = .11), or the average number pups born per litter (WT 8.0 ± 1.0, Tac2−/− 7.5 ± 0.43, t test P = .61) (Figure 1).
Table 1.
Male Reproductive Phenotypes
| WT |
Tac2−/− |
|||
|---|---|---|---|---|
| Mean ± SE | N | Mean ± SE | N | |
| Body weight | 27.2 ± 1.2 | 16 | 25.2 ± 1.7 | 16 |
| Testicular weight (g) | 196.5 ± 9.0 | 16 | 218 ± 42 | 16 |
| Testicular weight (% of body weight) | 0.73 ± 0.03 | 16 | 1.01 ± 0.23 | 16 |
| PND63 anogenital distance (mm) | 16.62 ± 0.3 | 10 | 15.54 ± 0.3a | 10 |
| Day of preputial separation | 31.7 ± 0.3 | 11 | 32.7 ± 0.5 | 10 |
| LH (ng/mL) | 0.06 ± 0.01 | 10 | 0.16 ± 0.06 | 11 |
| FSH (ng/mL) | 32.66 ± 3.65 | 10 | 14.73 ± 2.68a | 12 |
| Testosterone (ng/dL) | 30.6 ± 10 | 8 | 39.3 ± 13 | 8 |
Testicular weights as well as serum LH, FSH, and testosterone values were assayed between PND56–PND90. LH values below assay detectability of 0.02 ng/mL were set at this value for analysis.
P < .05 by unpaired t test.
Figure 1. Reproductive phenotyping in Tac2−/− male mice.
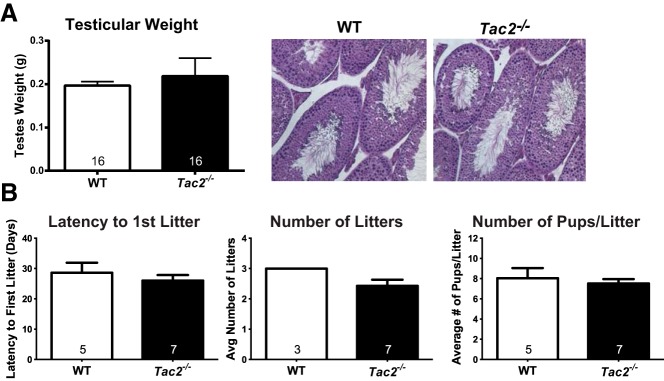
A, Testicular weight was similar between Tac2−/− (black bar) and WT (white bar) males, and the presence of mature spermatozoa was present in the testes of both genotypes. B, Fertility was also not significantly different based on the latency to the first litter after pairing with a WT female, the average number of litters in a 12-week period and the average number of pups per litter. Sample sizes are indicated within the bars.
Female reproductive phenotypes
In contrast to Tac2−/− males, Tac2−/− female mice demonstrated significant delays in sexual maturation (Table 2). Vaginal opening, a marker of estradiol levels and sexual maturation in mice, was significantly delayed in Tac2−/− females compared with their WT littermates (WT 29.11 ± 0.63 d, Tac2−/− 32.33 ± 1.62 d, log-rank survival curve comparison P = .03) (Figure 2). The average day of first E was delayed by 6 days in Tac2−/− females compared with WT littermates (WT 34.67 ± 1.24 d, Tac2−/− 40.55 ± 2.30 d, log-rank survival curve comparison P = .03) (Figure 2). Consistent with estradiol's known anorexigenic effects, Tac2−/− females weighed modestly but significantly more than their WT littermates beginning at PND28 (WT 12.85 ± 3.8 g, Tac2−/− 14.78 ± 3.9 g, Bonferroni post hoc test P < .05) (Figure 2).
Table 2.
Female Reproductive Phenotypes
| WT |
Tac2−/− |
|||
|---|---|---|---|---|
| Mean ± SE | N | Mean ± SE | N | |
| Body weight (g) | 17.6 ± 0.5 | 12 | 20.6 ± 0.5a | 12 |
| Ovarian weight (mg) | 10.0 ± 1.8 | 5 | 5.3 ± 0.9a | 5 |
| Ovarian weight (% of body weight) | 0.03 ± 0.01 | 5 | 0.02 ± 0.00a | 5 |
| Uterine weight (mg) | 71.1 ± 9.5 | 8 | 50.8 ± 9.6 | 8 |
| Uterine weight (% of body weight) | 0.30 ± 0.04 | 8 | 0.23 ± 0.04 | 8 |
| Day of vaginal opening | 29.11 ± 0.63 | 9 | 32.33 ± 1.62 | 9 |
| Day of first E | 34.67 ± 1.24 | 9 | 40.55 ± 2.3a | 9 |
| LH (ng/mL) | 0.24 ± 0.10 | 5 | 0.40 ± 0.34 | 7 |
| FSH (ng/mL) | 6.50 ± 2.04 | 5 | 5.94 ± 0.81 | 7 |
LH values below assay detectability of 0.02 ng/mL were set at this value for analysis.
P < .05 by unpaired t test (body weight, ovarian weight) or log-rank survival curve analysis (day of first E).
Figure 2. Delayed sexual maturation in Tac2−/− female mice.
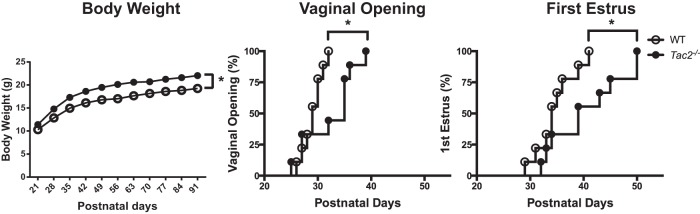
Tac2−/− female mice (filled circle) weigh significantly more than WT littermates (open circles) beginning at PND28 (left panel). Tac2−/− female mice have significant delays in the time to vaginal opening, with vaginal opening occurring on average 3 days later than WT littermates (middle panel). First E was also significantly later in Tac2−/− females occurring on average 6 days after WT animals (right panel). Sample size was n = 12 for body weight and n = 9 for both vaginal opening and first E. *, P < .05 by two-way repeated measures ANOVA for body weight and by log-rank survival analysis for vaginal opening and first E.
Estrous cycles were assessed to determine whether Tac2−/− female mice also had deficits in adult reproductive function. Tac2−/− female mice had abnormal estrous cycles from PND25–PND59 (Figure 3), with significantly fewer cycles than WT littermates (WT 5.33 cycles, Tac2−/− 1.25 cycles, Bonferroni multiple comparisons P < .05). Tac2−/− female mice also spent significantly more time in metestrus/diestrus (M/D) than WT animals (WT 40% of time in D/M, Tac2−/− 60% of time in D/M, Bonferroni multiple comparisons P < .05). However, from PND60 to PND94, cycling differences between Tac2−/− and WT animals appeared to have diminished and were no longer statistically significant. Post hoc analysis revealed that the current experiment had 80% power (α = 0.0125, to allow for Bonferroni four-way comparison) to detect a 20% difference in time spent in M/D and 2.15 cycle number difference at the PND60–PND94 time point; therefore, it is possible that smaller differences were not detected by this experiment (Figure 3). Ovarian weight was significantly decreased compared with WT littermates (WT 10.02 ± 1.76 mg, Tac2−/− 5.28 ± 0.93 mg, t test P = .04) (Table 1 and Figure 4), consistent with a hypogonadal state in adulthood. Corpora lutea were only present in 3 out of 5 ovaries from adult Tac2−/− females, compared with 5 out of 5 ovaries from WT females (Figure 4).
Figure 3. Impaired estrous cyclicity in Tac2−/− female mice recovers over time.

Individual estrous cycles for 2 WT (left panel) and 2 Tac2−/− (middle panel) females indicate impaired estrous cycles in Tac2−/− females. However, cycling appeared to normalize between PND60 and PND90. Comparison of cycling between PND25–PND59 and PND60–PND94 in 3 WT and 4 Tac2−/− females revealed increased time spent in D/M and a lower number of total cycles in Tac2−/− females during PND25–PND59 but no significant difference from WT littermates during PND60–PND94. E, estrus; P, proestrus; D, diestrus; and M, metestrus.
Figure 4. Fertility and gonadal weights in Tac2−/− female mice.

A, Ovarian weight was significantly decreased in adult Tac2−/− females, and although corpora lutea (CL) were observed in both genotypes, the total number of animals with CL was lower in Tac2−/− females compared with WT littermates. *, P < .05 by unpaired t test for ovarian weight. B, Fertility was assessed in 2 separate cohorts of females paired with a male either at the time of first E (PND31–PND42) or later in adulthood (adult; PND80–PND100). Latency to first litter and number of litters was not significantly different between WT and Tac2−/− females regardless of age. The average number of pups per litter was overall significantly lower for Tac2−/− animals when both age groups were combined (P < .05; two-way ANOVA). *, P < .05 for genotype effect by two-way ANOVA.
Given improvement in estrous cycling later in life, both fertility and gonadotropin levels were assessed at the time of first E (WT PND31 and Tac2−/− PND42) as well as later in adulthood (PND80–PND110). Neither LH nor FSH levels were significantly different between Tac2−/− and WT animals regardless of age (two-way ANOVA; P > .05). There was evidence of overall subfertility in Tac2−/− females: the average number of pups per litter was lower in these females compared with WT littermates when both age groups were combined (two-way ANOVA; P < .05). The latency to first litter and total number of litters born during 12 weeks of pairing with a WT male was not significantly different between WT and Tac2−/− after mating either at the time of first E or adulthood (Figure 4). However, it is possible that examination over a longer time period could reveal subtle defects in these measures.
Immunohistochemistry
To assess whether defects in sexual maturation and estrous cycling may be due to altered kisspeptin or GnRH content in the brains of Tac2−/− mice, IHC was performed to determine protein content in brains from ovariectomized females (Figure 5). The number of GnRH-immunoreactive neurons observed between WT and Tac2−/− females were not significantly different in the sections analyzed (WT 52.6 ± 10.6 cells, Tac2−/− 33 ± 4.4 cells, t test P = .17). Kisspeptin cell number could not be determined due to the density of immunoreactive fibers in the ARH. However, overall kisspeptin-immunoreactivity in the ARH was also not significantly different between Tac2−/− and WT (WT 44.9 ± 4.8% of area, Tac2−/− 32.6 ± 5.1% area, t test P = .12). Post hoc analysis revealed that the current study was powered (80%, α = 0.05) to detect mean differences of more than 36 GnRH cells and more than 23% change in kisspeptin-immunoreactive density, indicating that larger sample sizes would be needed to detect smaller differences. However, Tac2−/− females had a significant decrease in the total number of kisspeptin-immunoreactive neurons in the AVPV compared with WT littermates (WT 39.2 ± 6.6 cells, Tac2−/− 16.7 ± 3.5 cells, t test P = .03).
Figure 5. GnRH- and kisspeptin-immunoreactivity in Tac2−/− females.

The total number of GnRH-immunoreactive cells (1:10 000, PA1–120; Thermo Scientific) was not significantly different between WT and Tac2−/− ovariectomized adult females (top panel). There was no significant difference in the percent area of a sampling region positive for kisspeptin-immunoreactive (1:30 000; Caraty 564) fibers in the ARH between WT and Tac2−/− females (middle panel). The total number of kisspeptin-immunoreactive cells was significantly decreased in the AVPV in Tac2−/− females compared with WT (bottom panel). Sample sizes are indicated within the bars; *, P < .05 by unpaired t test.
Comparison of Tac2−/− and Tacr3−/− female mice
Female mice bearing deletions of the gene encoding the NKB receptor (Tacr3) were previously shown to have normal timing of sexual maturation but to have hypogonadotropism in adulthood. Side-by-side comparison of Tac2−/− and Tacr3−/− females confirmed that the time of vaginal opening is significantly delayed in Tac2−/− females (WT 30.42 ± 0.48 d, Tac2−/− 34.75 ± 0.75 d, log-rank P = .0001) but not in Tacr3−/− females (WT 27.25 ± 0.55 d, Tacr3−/− 29.33 ± 1.32 d, log-rank P = .09) (Figure 6). Similarly, delays in the timing of first E were observed in Tac2−/− females (WT 31.25 ± 1.3 d, Tac2−/− 39.75 ± 2.0 d, log-rank P = .002) but not in Tacr3−/− animals (WT 32.08 ± 0.62 d, Tacr3−/− 32.83 ± 0.89 d, log-rank P = .24) (Figure 6).
Figure 6. Comparison of sexual maturation in Tac2−/− and Tacr3−/− females.

A separate cohort of Tac2−/− females were examined side-by-side with Tacr3−/− females to determine whether there were any differences in timing of sexual maturation between the 2 genotypes. Survival curve analysis of time to vaginal opening (top panels) and first E (bottom panels) revealed significant delays compared with WT (open circles) in a separate cohort of Tac2−/− females (filled circles; left panels). Tacr3−/− females (filled squares) had no significant delay in either time to vaginal opening or first E compared with WT littermates (open squares; right panels). Sample size, 12 females per group.
Discussion
Previous characterization of NKB receptor-deficient mice revealed reproductive defects that occurred only later in life, after sexual maturation occurred normally. In contrast, human patients with NKB pathway mutations have arrested pubertal development due to hypogonadotropism. The current study aimed to examine NKB ligand-deficient mice to determine whether these animals provide a closer model of NKB deficiency in humans. Indeed, absence of NKB resulted in a profound delay in sexual maturation as demonstrated by an almost 1-week delay in the time to first E as well as abnormal estrous cycling in early adulthood in female mice. Therefore, in contrast to the NKB receptor-deficient mice, absence of NKB in female mice appears to more closely recapitulate the pubertal defect observed in humans. Moreover, although NKB-deficient mice displayed abnormal estrous cycles in early adulthood, this defect ameliorated over time as evidenced by fertility in females. This recovery of HPG activity parallels the high incidence of reversal of hypogonadotropism seen in patients with NKB pathway mutations. Thus, data from both humans and mice support the hypothesis that NKB is more critical for the timing of sexual maturation than for adult reproductive function.
Although the delay in sexual maturation was profound in female mice lacking NKB, it was not as severe as the delays previously observed in mice with kisspeptin pathway deficiencies. Kisspeptin receptor- and ligand-deficient animals showed an average 10-day delay in vaginal opening, with the range extending to as long as 13 weeks (8, 35). Data from both primate and mouse models suggest that NKB acts upstream of kisspeptin, because both pharmacological blockade and physiological desensitization of the kisspeptin receptor block the ability of NKB to stimulate GnRH release (36, 37). The current finding that NKB deficiency results in a less severe sexual maturation phenotype compared with kisspeptin-deficient mice is concordant with this relative hierarchy. In contrast to this proposed pathway, there is also evidence that NKB directly regulates GnRH given the NKB receptor is found on GnRH terminals and NKB can depolarize these terminals in electrophsyiological recordings (38, 39). Regardless of the site of action, the observation that these Tac2−/− animals have deficiencies in sexual maturation indicates that NKB is critical for the timing sexual maturation.
Despite their delayed sexual maturation NKB-deficient mice appeared similar to their WT littermates in measures of adult reproductive function. Although early estrous cycles in Tac2−/− females were longer, with disproportionately more time spent in M/D, these animals began to cycle normally after PND60 and were also fertile. This evidence indicates that there was improvement in coordination of the HPG axis over time similar to reversal observed in human patients. The recent development of ultrasensitive gonadotropin assays (40), which could allow for frequent blood sampling in mice, may soon make it possible to determine directly whether LH pulsatility improves over time in NKB-deficient mice as observed in human reversal patients. Our group previously observed that kisspeptin-deficient mice also improve over time, going from constant diestrus in early adulthood to long periods of both diestrus and estrus in later adulthood (41). Although this cycling is improved, it is still markedly abnormal, and importantly, this fluctuation between diestrus and estrus was not associated with ovulation. Therefore, the improvement observed in reproductive function observed in kisspeptin-deficient mice appears relatively minor compared with the improvement in cycling observed in NKB-deficient mice, which become indistinguishable from WT animals over time. These findings highlight a more temporally restricted role of NKB for the activation of GnRH neurons during sexual maturation versus a sustained requirement of kisspeptin for GnRH physiology.
NKB's critical role for the regulation of sexual maturation compared with its more dispensable role for adult reproductive function in female mice is mirrored in reversal of hypogonadotropism in patients bearing mutations in the NKB pathway. A retrospective comparison between patients with clear evidence of reversal with those who had sustained hypogonadotropism found the reversal cohort was enriched for mutations in the NKB pathway, consistent with the high rates of reversal previously reported in patients with these mutations (23). Not all reports examining patients with NKB pathway mutations have observed evidence of reversal (42). This could be due to differences in the frequency and age at which patients were examined off of steroid and gonadotropin treatments because reversal can occur within a wide age-range with one example of an individual reversing at 41 years of age (26). Unfortunately, no other clinical features appear enriched in reversal patients, making it difficult to predict who will reverse and why. If reversal of hypogonadotropism is dependent on GnRH release, then the ability to reverse indicates that some number of GnRH neurons underwent proper migration during neonatal development and are present in the adult hypothalamus. What remains unclear is why individuals with NKB pathway mutations fail to show GnRH neuronal activation at the appropriate time, given the high proportion of these individuals who achieve reversal. One potential trigger for reversal may be steroid hormone treatment, which is the most common therapy used to treat hypogonadism in hypogonadotropic patients. Steroid hormone treatments are also prescribed for children with central delayed puberty and appear to increase the probability of puberty onset in this group (43, 44). Conversely, the current finding that Tac2−/− mice recovered reproductive function without any intervention may be a hint that reversal is an intrinsic process.
Although humans with NKB pathway mutations and NKB-deficient female mice both show abnormalities in the timing of sexual maturation, this process appears unaffected in male and female NKB receptor-deficient mice (28). This finding was verified by a side-by-side comparison of the genotypes confirming discordant phenotypes between the ligand and receptor knockouts. In other pathways (GnRH, kisspeptin), ligand deficiency has been shown to cause similar or even less severe phenotypes than deficiency of the corresponding receptor. However, these are thought to be more simple signaling systems with a single ligand and a single receptor (35, 45, 46). The finding of a more severe phenotype in NKB-deficient mice than NKB receptor-deficient mice could be due to the previously demonstrated promiscuity of NKB for the other 2 tachykinin receptors, neurokinin receptor 1 (NK1R) and 2 (NK2R) (47, 48). Counter to this hypothesis are recent data demonstrating that pharmacological blockade specifically of NK3R using the antagonist SB222200 is sufficient to delay puberty onset in rats (49). The discordance between this finding and normal timing of puberty in Tacr−/− mice (28 and current study) could be due to developmental compensation in the genetic knockout model. However, only pharmacological blockade of all 3 receptors (as opposed to blockade of any one receptor) decreased basal LH release (50) and blocked NKB-induced KNDy neuronal depolarization (51), consistent with the proposed hypothesis that all 3 tachykinin receptors may contribute to reproductive regulation. If this hypothesis is correct, NKB signaling through NK1R and NK2R may be sufficient to achieve normal timing of sexual maturation in NK3R-deficient mice. Conversely, given that there are also 2 additional ligands in the tachykinin family, substance P and neurokinin A, a more complicated imbalance of this larger network may account for phenotypic differences between Tac2−/− and Tacr3−/− mice. In any case, current models of NKB regulation of reproduction may need to be revised to incorporate the emerging complexity of the entire tachykinin system.
Given evidence that NKB may regulate GnRH indirectly via kisspeptin, the current study examined whether NKB deficiency affects kisspeptin expression in the hypothalamus. Although ARH kisspeptin protein levels were similar between a small cohort of female WT and NKB-deficient and WT mice, there were significantly fewer kisspeptin-expressing cells in the AVPV of NKB-deficient mice. This difference in AVPV kisspeptin protein was observed in ovariectomized animals when expression is already expected to be low. Several studies have investigated the role of NKB in the autoregulation of ARH KNDy cells (4, 52, 53). However, little is known about how NKB regulates kisspeptin cells in the AVPV. For example, it is currently unknown whether AVPV kisspeptin cells express tachykinin receptors or depolarize in response to NKB stimulation. Although the current study indicates NKB may regulate AVPV kisspeptin in the ovariectomized state, what role, if any, NKB has in regulating AVPV kisspeptin in the intact state remains unclear. Interestingly, male mice have a much smaller population of AVPV kisspeptin cells compared with females, and the current study found that Tac2−/− male mice had normal timing of sexual maturation and adult reproductive function. However, ARH NKB expression also appears lower in male sheep and mice (54, 55); therefore, sexual dimorphism in NKB expression, and not kisspeptin, may underlie the gender differences in Tac2−/− mice. Both men and women with NKB pathway mutations have delays in puberty, indicating an apparent species difference in the role of NKB for the timing of male sexual development. Notably, NKB expression and action in the brain is widespread (56, 57); therefore, it is also possible that NKB deficiency delays sexual maturation through kisspeptin-independent pathways in humans and female mice.
Consistent with this widespread expression of NKB in the brain, there is evidence that NKB deficiency results in phenotypes beyond the current focus of reproduction (27). It was noted in the current study that Tac2−/− females have significant increases in body weight compared with WT littermates. Estradiol is thought to inhibit food intake and increase metabolism; therefore, it is possible that the hypogonadism in NKB-deficient mice may contribute to the higher body weight. Lack of a reliable mouse estradiol assay prevented direct analysis of estradiol levels in the current study. However, weight differences have not been observed in other hypogonadal female mice, including GnRH-deficient “hpg” mice (35, 58), indicating that NKB may play a more direct role in energy metabolism, such as food intake and/or metabolic rate. This hypothesis is consistent with previous work revealing NKB expression is regulated by energy availability (59–61), and thus, NKB might provide a link between metabolic and reproductive neuroendocrine circuits. Ablation of ARH NKB cells was recently shown to block ovariectomy-induced weight gain, indicating that it may play an important role specifically in mediating estradiol negative feedback to metabolic circuits (62). Although these data are in contrast to the current findings of increased weight with absence of NKB expression, differences in the mechanism of deficiency, adult ablation vs genetic deficiency, complicate direct comparison of these studies.
Data from both humans and mice with NKB pathway deficiencies reveal that disordered timing of sexual maturation does not preclude the possibility of normal HPG function in adulthood. These data suggest that the modulation of the HPG cascade may differ at sexual maturation vs adult life, but the physiologic differences between these processes are currently unknown. The mechanism by which NKB modulates the timing of sexual maturation is unclear but points to critical window of NKB action for the proper timing of GnRH activation. The close phenocopy between NKB-deficient humans and mice indicates these animals could serve as the first model of human reversal and could be used to investigate the mechanisms by which a stalled HPG system can eventually become activated.
Acknowledgments
We thank the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core for serum LH and FSH assays and the Harvard Rodent Histopathology Core for pathology services.
This work was supported by the National Institutes of Health Grant U54 HD028138.
Disclosure Summary: The authors have nothing to disclose.
For News & Views see page 1207
- ARH
- arcuate nucleus of the hypothalamus
- AVPV
- anteroventral periventricular nucleus
- D/M
- diestrus/metestrus
- first E
- first estrus
- HPG
- hypothalamic-pituitary-gonadal
- IHC
- immunohistochemistry
- KNDY
- Kisspeptin/Neurokinin B/Dynorphin
- M/D
- metestrus/diestrus
- NKB
- neurokinin B
- neurokinin receptor 3
- NK3R
- PND
- postnatal day
- WT
- wild type.
References
- 1. Rance NE, McMullen NT, Smialek JE, Price DL, Young WS., 3rd Postmenopausal hypertrophy of neurons expressing the estrogen receptor gene in the human hypothalamus. J Clin Endocrinol Metab. 1990;71:79–85. [DOI] [PubMed] [Google Scholar]
- 2. Rance NE, Young WS., 3rd Hypertrophy and increased gene expression of neurons containing neurokinin-B and substance-P messenger ribonucleic acids in the hypothalami of postmenopausal women. Endocrinology 1991;128:2239–2247. [DOI] [PubMed] [Google Scholar]
- 3. Rance NE, Bruce TR. Neurokinin B gene expression is increased in the arcuate nucleus of ovariectomized rats. Neuroendocrinology. 1994;60:337–345. [DOI] [PubMed] [Google Scholar]
- 4. Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci. 2009;29:11859–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goodman RL, Lehman MN, Smith JT, et al. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology. 2007;148:5752–5760. [DOI] [PubMed] [Google Scholar]
- 6. Burke MC, Letts PA, Krajewski SJ, Rance NE. Coexpression of dynorphin and neurokinin B immunoreactivity in the rat hypothalamus: morphologic evidence of interrelated function within the arcuate nucleus. J Comp Neurol. 2006;498:712–726. [DOI] [PubMed] [Google Scholar]
- 7. Ramaswamy S, Seminara SB, Ali B, Ciofi P, Amin NA, Plant TM. Neurokinin B stimulates GnRH release in the male monkey (Macaca mulatta) and is colocalized with kisspeptin in the arcuate nucleus. Endocrinology. 2010;151:4494–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. [DOI] [PubMed] [Google Scholar]
- 9. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Funes S, Hedrick JA, Vassileva G, et al. The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system. Biochem Biophys Res Commun. 2003;312:1357–1363. [DOI] [PubMed] [Google Scholar]
- 11. Topaloglu AK, Reimann F, Guclu M, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Whitcomb RW, Crowley WF., Jr Clinical review 4: diagnosis and treatment of isolated gonadotropin-releasing hormone deficiency in men. J Clin Endocrinol Metab. 1990;70:3–7. [DOI] [PubMed] [Google Scholar]
- 13. Bauman A. Markedly delayed puberty or Kallmann's syndrome variant. J Androl. 1986;7:224–227. [DOI] [PubMed] [Google Scholar]
- 14. Finkelstein JS, Spratt DI, O'Dea LS, et al. Pulsatile gonadotropin secretion after discontinuation of long term gonadotropin-releasing hormone (GnRH) administration in a subset of GnRH-deficient men. J Clin Endocrinol Metab. 1989;69:377–385. [DOI] [PubMed] [Google Scholar]
- 15. Kadva A, Di WL, Djahanbakhch O, Monson J, Silman R. Evidence for the Bauman variant in Kallmann's syndrome. Clin Endocrinol (Oxf). 1996;44:103–110. [DOI] [PubMed] [Google Scholar]
- 16. Quinton R, Cheow HK, Tymms DJ, Bouloux PM, Wu FC, Jacobs HS. Kallmann's syndrome: is it always for life? Clin Endocrinol (Oxf). 1999;50:481–485. [DOI] [PubMed] [Google Scholar]
- 17. Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF, Jr, Hayes FJ. The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab. 2001;86:2470–2475. [DOI] [PubMed] [Google Scholar]
- 18. Dewailly D, Boucher A, Decanter C, Lagarde JP, Counis R, Kottler ML. Spontaneous pregnancy in a patient who was homozygous for the Q106R mutation in the gonadotropin-releasing hormone receptor gene. Fertil Steril. 2002;77:1288–1291. [DOI] [PubMed] [Google Scholar]
- 19. Pitteloud N, Acierno JS, Jr, Meysing AU, Dwyer AA, Hayes FJ, Crowley WF., Jr Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab. 2005;90:1317–1322. [DOI] [PubMed] [Google Scholar]
- 20. Ribeiro RS, Vieira TC, Abucham J. Reversible Kallmann syndrome: report of the first case with a KAL1 mutation and literature review. Eur J Endocrinol. 2007;156:285–290. [DOI] [PubMed] [Google Scholar]
- 21. Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–873. [DOI] [PubMed] [Google Scholar]
- 22. Sinisi AA, Asci R, Bellastella G, et al. Homozygous mutation in the prokineticin-receptor2 gene (Val274Asp) presenting as reversible Kallmann syndrome and persistent oligozoospermia: case report. Hum Reprod. 2008;23:2380–2384. [DOI] [PubMed] [Google Scholar]
- 23. Gianetti E, Tusset C, Noel SD, et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95:2857–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tornberg J, Sykiotis GP, Keefe K, et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci USA. 2011;108:11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One. 2012;7:e39450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sidhoum VF, Chan YM, Lippincott MF, et al. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab. 2014;99:861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Siuciak JA, McCarthy SA, Martin AN, et al. Disruption of the neurokinin-3 receptor (NK3) in mice leads to cognitive deficits. Psychopharmacology (Berl). 2007;194:185–195. [DOI] [PubMed] [Google Scholar]
- 28. Yang JJ, Caligioni CS, Chan YM, Seminara SB. Uncovering novel reproductive defects in neurokinin B receptor null mice: closing the gap between mice and men. Endocrinology. 2012;153:1498–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005;146:3686–3692. [DOI] [PubMed] [Google Scholar]
- 30. Franceschini I, Lomet D, Cateau M, Delsol G, Tillet Y, Caraty A. Kisspeptin immunoreactive cells of the ovine preoptic area and arcuate nucleus co-express estrogen receptor α. Neurosci Lett. 2006;401:225–230. [DOI] [PubMed] [Google Scholar]
- 31. Bosch MA, Xue C, Rønnekleiv OK. Kisspeptin expression in guinea pig hypothalamus: effects of 17β-estradiol. J Comp Neurol. 2012; 520:2143–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chung WC, Pak TR, Suzuki S, Pouliot WA, Andersen ME, Handa RJ. Detection and localization of an estrogen receptor β splice variant protein (ERβ2) in the adult female rat forebrain and midbrain regions. J Comp Neurol. 2007;505:249–267. [DOI] [PubMed] [Google Scholar]
- 33. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Q, Symes AJ, Kane CA, et al. A novel role for Wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS One. 2010;5:e10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lapatto R, Pallais JC, Zhang D, et al. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology. 2007;148:4927–4936. [DOI] [PubMed] [Google Scholar]
- 36. Ramaswamy S, Seminara SB, Plant TM. Evidence from the agonadal juvenile male rhesus monkey (Macaca mulatta) for the view that the action of neurokinin B to trigger gonadotropin-releasing hormone release is upstream from the kisspeptin receptor. Neuroendocrinology. 2011;94:237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grachev P, Li XF, Lin YS, et al. GPR54-dependent stimulation of luteinizing hormone secretion by neurokinin B in prepubertal rats. PLoS One. 2012;7:e44344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krajewski SJ, Anderson MJ, Iles-Shih L, Chen KJ, Urbanski HF, Rance NE. Morphologic evidence that neurokinin B modulates gonadotropin-releasing hormone secretion via neurokinin 3 receptors in the rat median eminence. J Comp Neurol. 2005;489:372–386. [DOI] [PubMed] [Google Scholar]
- 39. Gaskins GT, Glanowska KM, Moenter SM. Activation of neurokinin 3 receptors stimulates GnRH release in a location-dependent but kisspeptin-independent manner in adult mice. Endocrinology. 2013;154:3984–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Steyn FJ, Wan Y, Clarkson J, Veldhuis JD, Herbison AE, Chen C. Development of a methodology for and assessment of pulsatile luteinizing hormone secretion in juvenile and adult male mice. Endocrinology. 2013;154:4939–4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan YM, Broder-Fingert S, Wong KM, Seminara SB. Kisspeptin/Gpr54-independent gonadotrophin-releasing hormone activity in Kiss1 and Gpr54 mutant mice. J Neuroendocrinol. 2009;21:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Francou B, Bouligand J, Voican A, et al. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One. 2011;6:e25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soliman AT, Khadir MM, Asfour M. Testosterone treatment in adolescent boys with constitutional delay of growth and development. Metabolism. 1995;44:1013–1015. [DOI] [PubMed] [Google Scholar]
- 44. Rosenfeld RG, Northcraft GB, Hintz RL. A prospective, randomized study of testosterone treatment of constitutional delay of growth and development in male adolescents. Pediatrics. 1982;69:681–687. [PubMed] [Google Scholar]
- 45. Cattanach BM, Iddon CA, Charlton HM, Chiappa SA, Fink G. Gonadotrophin-releasing hormone deficiency in a mutant mouse with hypogonadism. Nature. 1977;269:338–340. [DOI] [PubMed] [Google Scholar]
- 46. Wu S, Wilson MD, Busby ER, Isaac ER, Sherwood NM. Disruption of the single copy gonadotropin-releasing hormone receptor in mice by gene trap: severe reduction of reproductive organs and functions in developing and adult mice. Endocrinology. 2010;151:1142–1152. [DOI] [PubMed] [Google Scholar]
- 47. Ingi T, Kitajima Y, Minamitake Y, Nakanishi S. Characterization of ligand-binding properties and selectivities of three rat tachykinin receptors by transfection and functional expression of their cloned cDNAs in mammalian cells. J Pharmacol Exp Ther. 1991;259:968–975. [PubMed] [Google Scholar]
- 48. Helke CJ, Krause JE, Mantyh PW, Couture R, Bannon MJ. Diversity in mammalian tachykinin peptidergic neurons: multiple peptides, receptors, and regulatory mechanisms. FASEB J. 1990;4:1606–1615. [PubMed] [Google Scholar]
- 49. Li SY, Li XF, Hu MH, et al. Neurokinin B receptor antagonism decreases luteinising hormone pulse frequency and amplitude and delays puberty onset in the female rat. J Neuroendocrinol. 2014;26:521–527. [DOI] [PubMed] [Google Scholar]
- 50. Noritake K, Matsuoka T, Ohsawa T, et al. Involvement of neurokinin receptors in the control of pulsatile luteinizing hormone secretion in rats. J Reprod Dev. 2011;57:409–415. [DOI] [PubMed] [Google Scholar]
- 51. de Croft S, Boehm U, Herbison AE. Neurokinin B activates arcuate kisspeptin neurons through multiple tachykinin receptors in the male mouse. Endocrinology. 2013;154:2750–2760. [DOI] [PubMed] [Google Scholar]
- 52. Navarro VM, Castellano JM, McConkey SM, et al. Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am J Physiol Endocrinol Metab. 2011;300:E202–E210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Navarro VM, Gottsch ML, Wu M, et al. Regulation of NKB pathways and their roles in the control of Kiss1 neurons in the arcuate nucleus of the male mouse. Endocrinology. 2011;152:4265–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goubillon ML, Forsdike RA, Robinson JE, Ciofi P, Caraty A, Herbison AE. Identification of neurokinin B-expressing neurons as an highly estrogen-receptive, sexually dimorphic cell group in the ovine arcuate nucleus. Endocrinology. 2000;141:4218–4225. [DOI] [PubMed] [Google Scholar]
- 55. Ruiz-Pino F, Navarro VM, Bentsen AH, et al. Neurokinin B and the control of the gonadotropic axis in the rat: developmental changes, sexual dimorphism, and regulation by gonadal steroids. Endocrinology. 2012;153:4818–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Marksteiner J, Sperk G, Krause JE. Distribution of neurons expressing neurokinin B in the rat brain: immunohistochemistry and in situ hybridization. J Comp Neurol. 1992;317:341–356. [DOI] [PubMed] [Google Scholar]
- 57. Porter KL, Hileman SM, Hardy SL, Nestor CC, Lehman MN, Goodman RL. Neurokinin-3 receptor activation in the retrochiasmatic area is essential for the full pre-ovulatory LH surge in ewes. J Neuroendocrinol. 2014;26:776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rajendren G, Zhou H, Moonga BS, Zaidi M, Sun L. Restoration of bone mass in hpg mouse by preoptic area grafting. Ann NY Acad Sci. 2006;1068:341–347. [DOI] [PubMed] [Google Scholar]
- 59. True C, Kirigiti M, Ciofi P, Grove KL, Smith MS. Characterisation of arcuate nucleus kisspeptin/neurokinin B neuronal projections and regulation during lactation in the rat. J Neuroendocrinol. 2011;23:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. True C, Kirigiti MA, Kievit P, Grove KL, Smith MS. Leptin is not the critical signal for kisspeptin or luteinising hormone restoration during exit from negative energy balance. J Neuroendocrinol. 2011;23:1099–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Navarro VM, Ruiz-Pino F, Sánchez-Garrido MA, et al. Role of neurokinin B in the control of female puberty and its modulation by metabolic status. J Neurosci. 2012;32:2388–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mittelman-Smith MA, Williams H, Krajewski-Hall SJ, et al. Arcuate kisspeptin/neurokinin B/dynorphin (KNDy) neurons mediate the estrogen suppression of gonadotropin secretion and body weight. Endocrinology. 2012;153:2800–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]