

Abstract
In this study, we explored the effects of oocytic phosphoinositide 3-kinase (PI3K) activation on folliculogensis by generating transgenic mice, in which the oocyte-specific Cre-recombinase induces the expression of constitutively active mutant PI3K during the formation of primordial follicles. The ovaries of neonatal transgenic (Cre+) mice showed significantly reduced apoptosis in follicles, which resulted in an excess number of follicles per ovary. Thus, the elevation of phosphatidylinositol (3,4,5)-trisphosphate levels within oocytes promotes the survival of follicles during neonatal development. Despite the increase in AKT phosphorylation, primordial follicles in neonatal Cre+ mice remained dormant demonstrating a nuclear accumulation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN). These primordial follicles containing a high level of nuclear PTEN persisted in postpubertal females, suggesting that PTEN is the dominant factor in the maintenance of female reproductive lifespan through the regulation of primordial follicle recruitment. Although the oocytic PI3K activity and PTEN levels were elevated, the activation of primordial follicles and the subsequent accumulation of antral follicles with developmentally competent oocytes progressed normally in prepubertal Cre+ mice. However, mature Cre+ female mice were anovulatory. Because postnatal day 50 Cre+ mice released cumulus-oocyte complexes with developmentally competent oocytes in response to super-ovulation treatment, the anovulatory phenotype was not due to follicular defects but rather endocrine abnormalities, which were likely caused by the excess number of overgrown follicles. Our current study has elucidated the critical role of oocytic PI3K activity in follicular function, as well as the presence of a PTEN-mediated mechanism in the prevention of immature follicle activation.
The number of follicles that leave the ovarian reserve and are activated to grow during each reproductive cycle is tightly controlled so that the reproductive lifespan is maintained for months in mice and decades in humans (1–4). Mechanistically, the activation of the immature oocyte is triggered by an increase in the levels of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) within the oocyte (5), which reflects the balance between the activities of the class I phosphoinositide 3-kinase (PI3K) and the phosphatase and tensin homolog deleted on chromosome 10 (PTEN). PI3K converts phosphatidylinositol-4,5-biphosphate (PIP2) to PIP3, whereas PTEN catalyzes the opposite reaction. PIP3 promotes membrane binding and subsequent phosphorylation/activation of v-akt murine thymoma viral oncogene homolog (AKT) AKT (p-AKT) by phosphoinositide-dependent kinase 1 (PDK1). Activated p-AKT phosphorylates forkhead box O3 (FOXO3A), which prompts its translocation from the nucleus to the cytoplasm (6). Because nuclear FOXO3A in oocytes is essential for the maintenance of primordial follicle dormancy, its nuclear exclusion activates primordial follicles. This model of follicle activation was derived from the phenotypes of Foxo3 null (7, 8), oocyte-specific Pten null (9), oocyte-specific Tsc1, and oocyte-specific Tsc2 null mice (10, 11). In each case, the ovarian reserve was completely depleted by puberty (postnatal day [PD]28) through premature activation of primordial follicles, resulting in primary ovarian insufficiency. Furthermore, this mechanistic model for the PIP3-triggered primordial follicle activation has been confirmed in human oocytes: PTEN inhibitors, PI3K activator, and AKT stimulators can activate immature follicles within human ovarian cortical fragments (7–9, 11–14).
In addition to coordinating activation of primordial follicles, intracellular PIP3 levels in the oocytes are also critical for the survival of ovarian follicles. Oocyte-specific PDK1 null mice gradually lose ovarian follicles of all classes, but this phenotype can be reversed by simultaneous null mutation of PTEN (15). Moreover, an oocyte-specific null mutation in ribosomal protein S6, a downstream target of the PI3K-AKT-mTORC1 pathway, also causes follicular loss (15). Thus, it appears the fate of follicles to survive or die is also determined by the level of PI3K-AKT-mTOR signaling activities within the oocyte. However, the actual molecular mechanism that controls oocyte survival during folliculogenesis has not been elucidated.
To explore the role of oocytic PI3K in follicle activation, survival, growth, and function, we generated transgenic mice in which PI3K is constitutively activated in immature oocytes. Here, we describe the phenotype of these mice, which has highlighted the critical role of oocytic PI3K activity in follicle function, as well as the presence of a PTEN-mediated mechanism in the prevention of immature follicle activation.
Materials and Methods
Animals
All procedures described in this study involving the use of mice were approved by the Northwestern University Animal Care and Use Committee. Mice were housed and bred in a barrier facility within Northwestern University's Center for Comparative Medicine and were provided with food and water ad libitum. Temperature, humidity, and photoperiod (14-h light, 10-h dark cycle) were kept constant. Transgenic mice with oocyte-specific expression of constitutively active PI3K were generated by crossing homozygous female mice for Cre-inducible knock-in allele for Pik3ca* (C57BL/6-Gt(ROSA)26Sortm7(Pik3ca*,EGFP)Rsky/J) (The Jackson Laboratory) (16) with heterozygous GDF9 (growth differentiation factor-9)-iCre male mice (Tg(Gdf9-cre)5092Coo) (The Jackson Laboratory) (17). GDF9-iCre mice carried the iCre (Codon-improved Cre recombinase) gene driven by GDF9 promoter. Accordingly, all mice used here have one copy of the Cre-inducible Pik3ca* knock-in allele. The expression of PIK3CA*, a constitutively active form of PI3K, was perinatally induced in oocytes as iCre removed the floxed stop cassette on the upstream of Pik3ca* in the ROSA26 locus. The presence/absence of GDF9-iCre gene was determined by PCR. Female mice positive and negative for GDF9-iCre were referred as Cre+ and Cre−, respectively, and the Cre− females were used as littermate controls. The presence of GDF9-iCre transgene itself did not affect the ovarian phenotypes, which has been demonstrated in our previous study (18). Cre-mediated gene activation was also monitored by the expression of enhanced green fluorescent protein (Supplemental Figure 1).
The onset of puberty was examined daily by the presence of vaginal opening from PD21. The fertility was tested by housing PD35 Cre− and Cre+ female mice with mature C57BL/6J males for 40 days, at which point the experiment was terminated due to health problems of Cre+ female mice caused by an unregulated proliferation of ovarian tissue.
Follicle counting
Ovaries from at least 3 mice per genotype were collected at PD8, PD14, PD19, PD28, and PD35. The ovaries were fixed with Modified Davidson's fixative (Electron Microscopy Science, Inc) for 24 hours at 4°C and then processed into paraffin blocks. Entire murine ovaries were sectioned through at 5-μm thickness. Follicles were counted in every 5th sections for both PD8 Cre− and Cre+ ovaries, every 10th sections for PD35 and PD50 Cre− ovaries. For PD35 and PD50 Cre+ ovaries, which were more than 20 and more than 5 times larger than age-matched Cre− ovaries, follicles were counted in every 50th and 29th sections, respectively. Follicles with a single layer of squamous granulosa cells (pregranulosa cells) were counted as primordial follicles. Follicles with a single layer of squamous granulosa cells (pregranulosa cells) and an enlarged oocyte (with area >300 μm2) were defined as primordial follicles with activated oocytes (PFAOs). Dormant primordial follicles were counted when the nucleus was present within the section. The follicles of other classes, primary, secondary, and antral follicles, were counted only when the oocyte nucleus contained nucleoli. To compare the follicle number per ovary, the average follicle number/section (follicle density) was multiplied by the total section number, which reflects the size of the ovary. The average number of sections in which a single follicle appears depends on the distribution (size and shape) of the target structure (nucleoli or nucleus) and the thickness of the sections. Because the thickness of each section cannot be accurately measured in the standard paraffin sections, the term “follicle number” herein represents only a relative but not the actual follicle number per ovary. We also expect error in comparison of follicle numbers between different classes of follicles because the distribution of nucleoli or nuclei should slightly differ between classes of follicles. These are general and fundamental issues for any study that estimates the number of ovarian follicles by counting oocytes in histology sections. Size (area) was measured using ImageJ 64 software (NIH) on digital images captured by an E600 Leica microscope. The oocyte size of primordial follicles (area) was measured in more than 100 oocytes/group (>3 mice/group). Sizes of antral follicles, antrum, and oocyte were measured in more than 24 antral follicles from 3 mice/group.
Histological and immunohistochemical analyses
H&E staining was performed using standard methods. Immunofluorescence (IF) and 3,3′-diaminobenzidine (DAB) staining were performed as previously described (18). The concentrations of primary antibodies used for IF were as follows: p63 (4A4) (1:200, sc-8431; Santa Cruz Biotechnology, Inc), forkhead in rhabdomyosarcoma-like 1/FOXO3A (1:50, sc-11351; Santa Cruz Biotechnology, Inc), ZP1 (zona pellucida 1) (1:100, sc-23708, Santa Cruz Biotechnology, Inc), ZP3 (1:100, sc-25802; Santa Cruz Biotechnology, Inc), Connexin 43 (1:50, C8093; Sigma-Aldrich), rabbit anti-inhibin-α (1:100, kindly provided by Dr Wylie Vale, The Salk Institute, La Jolla, CA) (19), anti-Müllerian hormone (AMH) (1:50, sc-6886; Santa Cruz Biotechnology, Inc), Bax (BCL2-associated X protein) (1:50, PC66; EMD Millipore), PTEN (1:50, catalog number 9188; Cell Signaling Technology), phospho-histone H3 Ser10 (1:100, catalog number 3377; Cell Signaling Technology), α-Laminin (1:100, L9393; Sigma-Aldrich), MSY2 (germ cell Y-box protein) (1:4000, kindly provided by Dr Richard Schultz, University of Pennsylvania, Philadelphia, PA), and steroidogenic acute regulatory protein (STAR) (1:100, kindly provided by Dr Douglas Stocco, Texas Tech University Health Sciences Center, Lubbock, TX). For IF detection, Alexa Fluor 488 goat antimouse or antirabbit IgG (A11001 and A11008, respectively; Life Technologies) were used for the secondary antibody. For DAB staining, biotinylated antirabbit IgG was used (Vector) (see Supplemental Table 1 for more details). The specificity of secondary antibody binding was confirmed by the absence of signal in negative controls incubated without primary antibody. The DAB Peroxidase Substrate kit (SK-4100; Vector) was used for DAB staining. Apoptotic cells were detected by the terminal deoxynucleotidyl transferase 2′-Deoxyuridine, 5′-Triphosphate nick end labeling (TUNEL) assay using the DeadEnd Fluorometric TUNEL System (G3250; Promega) following the manufacturer's instructions. For comparison of p63 intensity in both Cre− and Cre+ ovaries, serial dilution test for anti-p63 antibody (18) was performed with concentrations at 1:50, 1:100, 1:200, 1:400, 1:800, and 1:1600. The positive signal became undetectable at 1:1600 in both Cre− and Cre+ ovaries.
Isolation of oocytes from postnatal mouse ovaries and cumulus-oocyte complexes (COCs)
Oocytes were isolated from the ovaries of PD35 mice by puncturing the ovaries with 27-gauge needles in L15 media (Life Technologies) containing milrinone (20). Alternatively, oocytes were isolated from PD14 mouse ovaries by incubating pieces of ovarian tissues in Ca2+, Mg2+-free CZB medium (21) containing 1-mg/mL collagenase and 2-mg/mL deoxyribonuclease I at 37°C until oocytes were released from the ovaries. Isolated oocytes were washed with PBS and stored at −80°C until they were used for immunoblotting analysis.
Immunoblot analysis
Ovaries were lysed in cold lysis buffer (22) supplemented with phosphatase inhibitors and Complete Protease Inhibitor cocktail (Roche). Alternatively, oocytes isolated from PD14 mice were directly lysed with loading buffer (β-mercaptoethanol and NuPAGE lithium dodecyl sulfate Sample buffer [NP0008; Life Technologies]). Ovarian proteins (500 ng) or oocytes (n = 50) per lane were electrophoresed in a 4%–12%-gradient sodium dodecyl sulfate-polyacrylamide gel and transferred onto a nitrocellulose membrane (ovarian protein) or a polyvinylidene difluoride membrane (oocytes) (23). The membranes were blocked for 1 hour at room temperature in 2% enhanced chemiluminescence prime blocking reagents (GE Healthcare Biosciences) and incubated at 4°C overnight in Tris-buffered saline (pH 7.6) (TBS) containing 2% skim milk and the next antibodies: p63 (1:5000, sc-8431; Santa Cruz Biotechnology, Inc), MSY2/Y box binding protein 2 (1:5000, LS-C155449; LSBio), p-AKT (Ser473, 1:5000, catalog number 9271; Cell Signaling Technology), AKT (1:5000, catalog number 9272; Cell Signaling Technology), PTEN (1:5000, catalog number 9188; Cell Signaling Technology), GAPDH (glyceraldehyde 3-phosphate dehydrogenase) (1:10 000, catalog number 2118; Cell Signaling Technology), Foxl2 (1:5000, ab5096; Abcam), Cyp17 (cytochrome P450 17α hydroxylase/17,20 lyase) (1:5000, kindly provided by Dr Alan Conley, University of California, Davis, CA), Cyp19a (H-300) (1:5000, sc-30086; Santa Cruz Biotechnology, Inc), inhibin-α (1:5000, kindly provided by Dr Wylie Vale, The Salk Institute, La Jolla, CA), and activin-βA (1:5000). After overnight incubation, the membranes were washed with 1% Tween-TBS for 5 minutes × 3 times and incubated for 1 hour at room temperature with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare Biosciences) diluted 1:10, 000 in 2% milk 1% Tween-TBS. The ECL Prime kit (GE Healthcare Biosciences) was used for protein detection.
In vitro maturation (IVM) of isolated COCs
Collected COCs were transferred to prewarmed α-MEM, nucleosides, GlutaMAX supplement media (32571-036; Life Technologies) containing 10μM milrinone and then incubated at 37°C in 5% CO2 for 12–14 hours in α-MEM, which contains 10% fetal bovine serum, 10-ng/mL epidermal growth factor, and 1.5-IU/mL human chorionic gonadotropin (hCG) (24). After IVM, oocytes were denuded from the cumulus cells by treatment with 0.3% hyaluronidase, and the meiotic progression was examined under light microscopy. The presence of intact germinal vesicle (GV) indicated that oocytes were arrested in prophase of meiosis I. Polar body extrusion indicated that the cell had progressed to metaphase of meiosis II (MII). The absence of both a GV and the polar body indicated that the oocyte had resumed meiosis but had not reached MII; these oocytes were categorized as GV breakdown (GVBD).
Superovulation treatment
The pregnant mare's serum gonadotropin (catalog 367222; CalBiochem) was given to female mice at 5 IU/mouse by ip injection, followed by ip injection of 5 IU hCG/mouse (C1063; Sigma-Aldrich) 46–48 hours later. Ovaries and COCs from oviducts were harvested for analyses at 12–14 hours after hCG injection.
Hormone measurement
Blood was collected from PD35 and PD50 Cre− and Cre+ mice by cardiac puncture. Because the Cre+ female did not show regular estrus cycle, the blood was collected at exact ages without considering the stage of estrus cycle. The serum levels were determined using ELISA kits for AMH (LOD, limit of detection; 0.006 ng/mL; Beckman Coulter Corp), inhibin A (LOD; 5.45 pg/mL; Anshlab), and inhibin B (LOD; 1.6 pg/mL; Anshlab). FSH, 17β-estradiol, and testosterone measurements were performed by the University of Virginia Ligand Core Facility (Charlottesville, VA). FSH was measured as previously described (25). Serum 17β-estradiol and testosterone levels were measured utilizing E2 ELISA kit (catalog number ES180S-100; CalBiotech, Inc) and T RIA kit (catalog number TKTT2; Siemens Healthcare Diagnostics), respectively. The technical details are available on the website (http://www.medicine. virginia.edu/research/institutes-and-programs/crr/lab-facilities/assay-methods-page). The measurement of hormones was repeated by using sera from 4 different mice per group except FSH and inhibins, for which sera from 3 mice per group were analyzed.
Statistical analysis
All statistical analyses (two-way ANOVA) were performed using Prism 4.0 software (GraphPad Software). P < .05 was considered statistically significant. Statistical significance was marked with *, P < .05; **, P < .01; and ***, P < .001). All data for follicle counting and analysis for meiotic progression were analyzed with two-way ANOVA. P values were Bonferroni-corrected for multiple comparisons. Two-tailed and unpaired t test was used for statistical analyses in the serum hormone level and ovary weight. Statistical significance was marked with *, P < .05; **, P < .01; and ***, P < .001). P values were Gaussian approximation for comparisons.
Results
Cell autonomous activation of PI3K in oocytes is not sufficient to initiate global follicle activation
The ovarian phenotypes of Cre− and Cre+ mice were compared at PD8, PD14, and PD35 to determine whether the constitutive activation of PI3K initiates the activation of primordial follicles. Expression of PIK3CA*, a constitutively active form of PI3K, was perinatally induced in oocytes as Cre recombinase driven by the GDF9-promoter removed the floxed stop cassette on the upstream of Pik3ca* in the ROSA26 locus (Supplemental Figure 1). Morphological differences were striking; specifically, ovaries from Cre+ mice displayed a higher follicle density (Figure 1A). The number of primordial follicles at PD8 was 2 times higher in Cre+ ovaries compared with Cre− ovaries (Figure 1B). Despite this marked increase in primordial follicles, the numbers of primary, secondary and antral follicles in PD8 Cre+ female mice were identical to the numbers in Cre− female mice, indicating that constitutive activation of PI3K in oocytes was not sufficient to initiate activation of premature follicles. By PD35, active (primary, secondary, and antral) follicles accumulated in both Cre+ and Cre− ovaries (Figure 1C). Accordingly, Cre+ ovaries dramatically increased in weight 7-fold higher than Cre− ovaries (Figure 1, D and E). Concurrent with the enlargement of ovaries, the serum level of AMH/Müllerian-inhibiting substance was 10-fold higher in Cre+ mice, corresponding with the higher number of active follicles in the Cre+ ovary (Figure 1F). Furthermore, the expression pattern of AMH in PD35 ovaries was identical between Cre− and Cre+ ovaries as assessed by immunostaining (Figure 1G): AMH expression, which was undetectable in primordial follicles (Figure 1Gii, red arrow), became weakly detectable in primary follicles and reached its peak in the secondary follicles with multilayered granulosa cells (Figure 1G, white arrows). In antral follicles, AMH expression diminished (Figure 1G, yellow arrows) (26) and became undetectable in the large antral follicles (Figure 1G, asterisk). The IF assay confirmed that the high serum AMH level in PD35 Cre+ mice was due to the increase in the number of active follicles. Although the number of active follicles was significantly higher in Cre+ ovaries at PD35, the number of dormant primordial follicles was comparable (no statistically significant difference) in Cre− and Cre+ ovaries (Figure 1C). Thus, the phenotype of transgenic mice with oocyte-specific constitutive activation of PI3K is fundamentally different from previous null mutant mouse models for oocytic Pten, Foxo3, Tsc1, or Tsc2, in which primordial follicles were entirely lost by PD28 through premature activation (7, 9, 10).
Figure 1. Mice with constitutive PI3K activity in oocytes develop enlarged ovaries.

A, Morphological analysis of ovaries from Cre− and Cre+ littermates at PD8, PD14, and PD35. Activation of PI3K in oocytes dramatically increased the overall size of ovary. Insets indicate the presence of normal primordial follicles. H&E staining revealed a higher density of follicles in the Cre+ than Cre− ovaries at each developmental stage. Scale bar, 100 μm. B and C, Follicle numbers per ovary. At PD8, the number of primordial follicles was significantly higher in Cre+ than Cre− mice, whereas the number of activated follicles is comparable. At PD35, the primordial follicle number became comparable in Cre− and Cre+ mice, meanwhile the number of secondary and antral follicles significantly increased in Cre+ ovaries. Pr, dormant primordial; Pm, primary; Sec, secondary; Ant, antral follicles. The numbers for the PFAOs are not included (see Figure 4). D, Gross phenotype of Cre− and Cre+ ovaries at PD35. E, Ovary weight. Cre+ ovaries were significantly heavier than Cre− ovaries at PD35. ***, P < .001. Horizontal bar, mean weight; vertical bar, SD. F, Serum AMH levels were significantly higher in PD35 Cre+ than PD35 Cre− mice; ***, P < .001. G, AMH immunohistochemistry in PD35 Cre− and Cre+ ovaries. Red, pink, white, and yellow arrows indicate primary, early secondary, multilayer secondary, and small antral follicles, respectively. Large antral follicles showing negative expression of AMH were labeled with *. Scale bar, 100 μm.
PI3K activity in oocytes promotes survival of follicles during development
The increase in the number of primordial follicles in the PD8 Cre+ mice (Figure 1B) suggests that constitutive PI3K activity in the oocyte overrides the apoptotic selection that normally occurs during folliculogenesis (27–29). Therefore, apoptotic activities in PD5 ovaries were assessed by TUNEL assay and IF analysis for BAX, which is activated in apoptotic oocytes (30, 31). The TUNEL signal was detected in granulosa cells and oocytes of primordial follicles in PD5 Cre− but not in Cre+ follicles (Figure 2A, top). Likewise, the cytoplasmic expression of BAX was highly detected in Cre− but not Cre+ oocytes, confirming the reduced apoptosis in Cre+ ovaries at PD5 (Figure 2A, bottom). The PI3K activity and relative oocyte numbers were further examined by immunoblotting analysis of PD5 Cre+ and Cre− ovaries (Figure 2B). The higher p-AKT levels confirmed the increased PIP3 levels in the Cre+ ovaries. Moreover, higher levels of p63 (immature oocyte marker) (32) and MSY2 (oocyte marker) (33) indicated more oocytes. These observations confirmed that cell autonomous PI3K activity in oocytes promotes the survival of follicles by inhibiting apoptosis in oocytes and granulosa cells during early folliculogenesis. Because p63 level within the oocyte nucleus determines the sensitivity of immature oocyte to apoptotic signals (18), the p63 level per oocyte nucleus was assessed through a serial antibody dilution test. Despite dramatic reduction in apoptosis in Cre+ ovaries, there was no detectable difference in the expression levels of p63 in the PD5 Cre− and Cre+ oocytes (Figure 2C, inset). Thus, inhibition of apoptosis in Cre+ ovaries was not mediated by a reduction in the nuclear p63 level.
Figure 2. Constitutive PI3K activation in oocytes promotes survival of immature follicles.

A, TUNEL and BAX-IF assays in Cre− and Cre+ ovaries at PD5. Although Cre− ovaries contained a high number of TUNEL-positive oocytes (inset) and granulosa cells, PD5 Cre+ ovaries were almost negative for TUNEL. Likewise, the signal for BAX was significantly reduced in Cre+ ovaries. Oocyte (oo) and granulosa cells (GC) were labeled respectively. Scale bar, 100 μm. B, Immunoblotting analysis on PD5 Cre− and Cre+ ovaries. Each lane was loaded with protein extract from 1 whole ovary. The signal intensity for oocyte markers, p63 (nucleus) and MSY2 (cytoplasm), indicated a higher number of oocytes in Cre+ ovaries compared with Cre− ovaries. The increased intensity of p-AKT in Cre+ mice confirmed the activation of PI3K signaling. C, IF assay for p63 (green) in PD5 ovaries. Insets show the comparison of IF signal intensity for anti-p63 antibody (1:800 dilution) in PD5 Cre− and Cre+ ovaries. Scale bar, 100 μm.
PTEN is up-regulated in the primordial oocytes of Cre+ mice
Although oocytic p-AKT level was elevated, a significant number of primordial follicles remained dormant displaying nuclear FOXO3A expression (34) in Cre+ ovaries at PD14 (Figure 3A). These primordial follicles in Cre+ mice demonstrated an accumulation of PTEN in the oocyte nucleus (Figure 3A, inset). These observations suggest that the dormant state of immature follicles is maintained by a negative feedback mechanism that attenuates the PIP3/p-AKT levels in the nucleus through an up-regulation of nuclear PTEN. This model was further examined through immunoblotting analysis of oocytes isolated from PD14 Cre+ and Cre− ovaries (Figure 3B). Consistent with immunohistochemical analyses, oocyte-specific expression of constitutively active PI3K led to increased PTEN levels in Cre+ oocytes.
Figure 3. Activation of PI3K induces the nuclear accumulation of PTEN in the primordial oocytes.
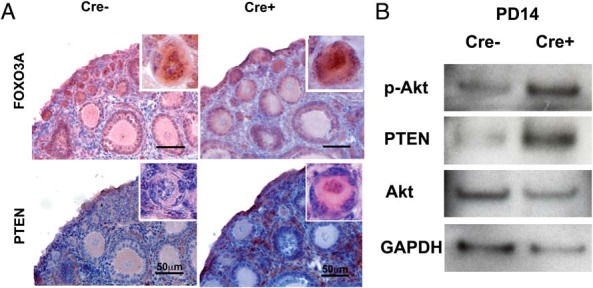
A, Expression of FOXO3A and PTEN in primordial follicles at PD14. Although the nuclear localization of FOXO3A was equally observed in the Cre− and Cre+ ovaries, every primordial oocyte in Cre+ ovaries showed higher nuclear PTEN expression than the primordial oocyte of Cre− ovaries. Scale bar, 50 μm. B, Immunoblotting analysis of isolated oocytes. The increase in p-AKT confirms the elevated PI3K activity in Cre+ oocytes. Moreover, a higher PTEN level in PD14 Cre+ oocytes indicated that the PI3K activity was counteracted by the up-regulation of PTEN.
Cell autonomous activation of PI3K drives oocyte growth independently of granulosa cell maturation
At PD8, the size of primordial follicles, as defined by the squamous morphology of granulosa cells (pregranulosa cells), was generally uniform in both Cre+ and Cre− ovaries. However, enlarged oocytes with pregranulosa cells became prominent in the ovaries of Cre+ mice by PD35 (Figure 4A). The enlarged oocytes were activated as assessed by the negative nuclear expression of FOXO3A, p63, and the positive signals for ZP1 and ZP3 (35, 36). Thus, we define this class of follicles as PFAOs. The pregranulosa cells in PFAOs were negative for inhibin-α and Connexin 43, which are typically expressed in mature granulosa cells (Figure 4A). Thus, PFAOs reflect an asynchronous activation of oocytes with no commensurate granulosa cell development.
Figure 4. Characterization of PFAOs.

A, Follicle classes and their marker expression pattern. Each panel is in the same magnification (scale bar, 50 μm; unless it is marked). The follicle is marked with an arrow in each panel. IF detection (green) for FOXO3A, which is expressed in the nucleus of dormant primordial follicles; p63, which is expressed in the oocyte nucleus from primordial to early secondary stages; ZP1 and ZP3, which are expressed in the zona pellucida of activated oocyte; and Connexin 43 and inhibin-α, which are expressed in the granulosa cells of activated follicles. PFAOs were either positive or negative for p63 depending on the size, and small PFAOs were positive (left panel) similarly to the primary and early secondary follicles. PFAOs with an intermediate size oocyte (between 320 and 500 μm2) did not show nuclear FOXO3A expression (*, oocyte is outlined by dotted line). Blue, DAPI. Scale bar, 50 μm. B, Oocyte size distribution of primordial follicles. The 2-dimensional size (area) of primordial follicles was assessed in histology sections. The cluster of blue columns corresponded to histologically normal (dormant) primordial follicles, whereas the yellow and red columns corresponded to the primordial follicles with enlarged oocytes. The yellow cluster appears to represent the activated oocyte in the early stage. The size of oocytes in normal primordial follicles (blue) in Cre+ ovaries was comparable with that in Cre− ovaries. The average size ± SD in each group is indicated on the top of each cluster. C, Developmental changes in the number of PFAO and primordial follicles. Blue, dormant follicles; red, PFAOs; Cre−, dotted lines; Cre+, solid lines. At PD8, Cre+ ovaries contained a high number of normal primordial follicles with a smaller number of PFAO. As PFAO increased from PD8 to PD19, the number of normal primordial follicles significantly dropped in Cre+ ovaries. Consequently, the number of dormant primordial follicles in Cre− and Cre+ ovaries became comparable after PD19. *, P < .05; ***, P < .001.
A clear division between dormant and activated oocytes (blue and red, respectively, in Figure 4B) within primordial follicles was readily observed through the size distribution of oocytes. A small number of oocytes with an intermediate size (320–540 μm2) (Figure 4B, yellow columns) showed reduced FOXO3A level (Figure 4A, marked with an asterisk), indicating that they were activated and in the transient state. As the number of PFAOs increased from PD8 to PD28, the number of dormant primordial follicles in Cre+ ovaries proportionally dropped (Figure 4C). The excess number of primordial follicles in Cre+ ovaries was depleted to values comparable with Cre− ovaries by PD28. Based on this observation, we hypothesize that there is an intrinsic mechanism to maintain a proper size of the ovarian reserve by retaining only a certain number of dormant oocytes. Accordingly, the formation of PFAOs may be explained as follows: the excess primordial follicles are removed by apoptosis in normal development. However, Cre+ oocytes were resistant to selective death and prone to immature activation due to an elevated PIP3 level. Thus, they underwent abnormal activation as PFAOs and were removed from the ovarian reserve.
Constitutive PI3K activation in oocytes affects somatic cell function
Histological analyses indicated that the dramatic enlargement of ovaries in PD35 Cre+ mice (Figure 1A) was due to the growth of excess follicles (Figure 1, B and C). Particularly, increase in the number of antral follicles was prominent (Figure 1C). In addition, each antral follicle was significantly larger in Cre+ ovaries (Figure 5A), to which both antrum volume and number of granulosa cells contributed. Interestingly, the size of oocytes within antral follicles was identical between Cre− and Cre+ ovaries (Figure 5A). The increase in the granulosa cell number was due to the reduction in follicular apoptosis rather than an increase in proliferation as indicated by the reduction in TUNEL-positive as well as phospho-Ser10 histone H3-positive granulosa cells in PD35 Cre+ ovaries (Figure 5B). As assessed by IF, expression of enhanced green fluorescent protein/transgene was undetectable in the somatic cells within follicles (Supplemental Figure 1B). Moreover, in our previous studies using the same GDF9-iCre male mice, Cre activity was not detected in granulosa cells by a more sensitive assay; crossing GDF9-iCre male mice with Rosa mT/mE dual-reporter transgenic female mice (18). Thus, we conclude the overgrowth of follicles was not a result of unexpected expression of mutant PI3K in granulosa cells. Surprisingly, the level of p-AKT was up-regulated in the granulosa cells of the enlarged antral follicles, indicating that PI3K activity within oocytes also affects the physiology of granulosa cells in Cre+ ovaries (Figure 5C). Due to the high number of large follicles in the Cre+ ovaries, follicles were tightly packed and the interfollicular stroma, which usually contains fibroblast layers and vasculatures, was scarce with reduced space between the basement membranes (stained with laminin) of follicles (Figure 5D). Nevertheless, markers for the endocrine function of theca (CYP17) and granulosa cells (CYP19A, inhibin-α, and activin-βA), which were absent at PD8, were detected at PD35 in both Cre+ and Cre− ovaries (Figure 5E), indicating that the endocrine function of follicles was normal in PD35 Cre+ ovaries.
Figure 5. Constitutive PI3K activation in oocytes induces the growth of active follicles.

A, Antral follicle size at PD35. The drawings indicate the relative areas (μm2) for antrum (blue), oocyte (yellow), and granulosa cell (green). The areas for all structures except oocytes were significantly larger in Cre+ mice. B, Oocytic PI3K activity reduces apoptosis in the granulosa cells of active follicles. TUNEL-positive (green) cells were significantly reduced in PD35 Cre+ ovaries compared with Cre− ovaries. However, positive granulosa cells for phospho-histone H3 (p-H3) (brown) were also reduced in Cre+ ovaries, suggesting that the increase in granulosa cell number in Cre+ antral follicles was due to reduced apoptosis rather than increased proliferation. Scale bar, 100 μm. C, IF assay for p-AKT. p-AKT signal was essentially undetectable in the follicles from Cre− ovaries. In Cre+ ovaries, oocytes showed strong membrane staining for p-AKT, indicating elevated PIP3 levels. In addition to oocytes, granulosa cells contained a higher level of p-AKT in Cre+ ovaries. Oo, oocyte. Scale bar, 100 μm. D, IF assay for Laminin (basement membrane marker) and MSY2 (oocyte marker) in PD35 ovaries highlighting an extremely high density of follicles in Cre+ ovaries. Scale bar, 100 μm. E, Immunoblotting assay for the endocrine activities of ovarian follicles. The expression levels of the next markers were tested on the whole ovarian extract from PD8 and PD35 Cre− and Cre+ mice. Expression of each marker indicates, Foxl2, amount of granulosa cell; CYP17, steroidogenesis in theca cells; CYP19a, steroid genesis in granulosa cell; inhibin-α and activin-βA, endocrine activity of granulosa cells. Loading was normalized by total protein amount. F, H&E staining of MOFs at PD35. Inset shows 2 oocytes separately surrounded by granulosa cells within a single follicle. Scale bar, 100 μm. G, Comparison of average number of MOFs per section in Cre− and Cre+ ovaries at PD35. MOF was never detected in Cre− ovaries.
Formation of multioocytic follicles (MOFs)
Although most follicles were functionally normal in Cre+ mouse ovaries, perinatal expression of constitutively active PI3K in oocytes increased formation of MOFs (Figure 5, F and G). Most of the MOFs appeared as 2 or more oocytes surrounded by granulosa cells within a single follicle. The prevalence of MOFs was likely caused by a disproportionally high ratio of oocyte to somatic cells in Cre+ ovaries during folliculogenesis. Occasionally, MOFs contained multiple oocytes enclosed within the same zona pellucida. This type of MOF was presumably caused by incomplete oocyte nest breakdown. The signaling involved in the oocyte nest breakdown and follicle formation is not fully understood. Nonetheless, a controlled interaction between germ cells and somatic cells is likely essential for proper nest breakdown and separation of follicles. Thus, oocytic PI3K activation may cause MOF through the disruption of oocyte-somatic cell communications during the formation of primordial follicles by skewing the cellular ratio or altering the oocytic intracellular signaling.
Oocytes from Cre+ ovaries are capable of resuming meiosis
To test the competence of the oocytes, COCs were collected from PD35 Cre− and Cre+ mice by puncturing the ovaries. More than 400 COCs per ovary were recovered from PD35 Cre+ mice (Figure 6A), which was 13 times the number of those recovered from Cre− mice. After IVM the cumulus cells expanded in both Cre− and Cre+ COCs (Figure 6B), and oocytes from both Cre− and Cre+ mice progressed to the MII stage as indicated by the presence of polar bodies (Figure 6C). In both Cre− and Cre+ groups, more than 70% of the oocytes progressed to the MII stage (Figure 6D). Therefore, constitutive activation of PI3K in oocytes did not impair the competency of the oocyte to resume meiosis in vitro.
Figure 6. Expression of PIK3CA* does not affect resumption and progression of the meiotic division.

A, Number of COCs recovered from Cre− and Cre+ at PD35. Cre+ ovary released a high number of COCs. B, Cumulus expansion test. COC data were shown before (left) and after (right) IVM, respectively. Both Cre− (top) and Cre+ (bottom) COCs demonstrated normal cumulus expansion. Scale bar, 50 μm. C, Mature oocytes with polar body exclusion. Polar bodies are indicated by arrows. Oocytes from PD35 Cre− and Cre+ ovaries resumed meiosis with IVM. Scale bar, 50 μm. D, Meiotic stage distribution of oocytes after IVM. GV, arrested stage; GVBD, meiotic division has progressed to GVBD, which is the intermediate stage between GV and MII; Deg, degenerated. There were no significant differences between the 2 groups in the stage distribution of oocytes after IVM.
Constitutive PI3K activity in oocytes resulted in anovulation
Vaginal opening was first observed at PD27 ± 1 in both Cre− and Cre+ female mice, suggesting comparable ovarian activity at peripuberty. However, when Cre− (n = 8) and Cre+ (n = 6) females were subjected to mating test for 40 days (from PD35 to PD75), none of the 6 Cre+ female mice (0/6, 0%) produced pups, whereas all 8 Cre− control females (8/8, 100%) produced a litter with average pup number of 7.1 ± 1.7 (±SD). To identify the mechanism of sterility, the ovarian phenotype was assessed at PD50. All ovaries from PD50 Cre+ mice (n = 10) demonstrated extensive hemorrhage (Figure 7B, black arrow), which appeared to have resulted from the degradation of overgrown follicles. In agreement with this observation, the number of TUNEL-positive cells increased from PD30 to PD50 in Cre+ ovaries (Supplemental Figure 2). Although all classes of follicles persisted, including primordial follicles (Figure 7B, inset, and C), the number of follicles that contained healthy oocytes significantly decreased in all follicle classes from PD35 to PD50 in the Cre+ ovaries (Figure 7D). These observations suggest that the effect of oocytic PI3K activity on ovarian physiology dramatically changes before and after puberty. The possibility that the recruitment of primordial follicles was accelerated in postpubertal Cre+ mice could not be completely excluded as a mechanism of primordial follicle loss in PD50 mice. Nonetheless, this hypothesis could not be confirmed by the follicle numbers as all classes of follicles were significantly reduced from PD35 to PD50. The presence of corpus luteum (CL) was confirmed only in the ovaries of PD50 Cre− mice (5/5, 100%), but not PD50 Cre+ female mice (0/5, 0%), in H&E stained sections (Figure 7B) and IF assay for STAR (Figure 7E). Thus, there was no histological evidence of ovulation from Cre+ ovaries, plausibly the primary cause of infertility.
Figure 7. Constitutive PI3K activity in oocytes results in anovulation.

A, Gross morphology of Cre− and Cre+ ovaries at PD50. An extensive hemorrhage was observed in Cre+ ovaries. B, Histological characteristics of Cre+ ovaries at PD50. Although the CLs were prominent in Cre− ovaries, there was no CL in Cre+ ovaries at PD50. Instead, large hemorrhagic cysts were observed throughout the Cre+ ovaries (black arrow). Inset, Immunohistochemistry for p63 indicated the presence of normal primordial follicles in the ovaries of PD50 Cre+ mice. Scale bar, 100 μm. C, Relative follicle numbers of each class in PD50 ovary; **, P < .01; ***, P < .001. D, Follicle numbers per Cre+ ovary at PD50 was significantly lower than that at PD35; ***, P < .001. E, IF assay for STAR on PD50 Cre− and Cre+ ovaries. Scale bar, 100 μm. F, Serum levels of estradiol (pg/mL), FSH (ng/mL), testosterone (pg/mL), inhibin A (pg/mL), and inhibin B (pg/mL) at PD50. Sera from 4 mice each for Cre− and Cre+ groups were used for the analysis of estradiol and testosterone levels. For the analysis of serum FSH and inhibin levels, sera from 3 mice each for Cre+ and Cre− groups were used. ns, nonsignificant; **, P < .01; ***, P < .001. Serum testosterone and 17β-estradiol levels were not significantly different between Cre− and Cre+ mice. Level of serum FSH in Cre+ mice was significantly lower than that of Cre− mice, whereas the serum levels of inhibins A and B in Cre+ mice were significantly higher than those of Cre− mice. Two-tailed, unpaired t test was used for statistical analysis. G, The number of superovulated COCs from Cre− (n = 5) and Cre+ (n = 3) at PD50. H, Meiosis resumption in Cre− and Cre+ oocytes from PD50 mice. Polar bodies (black arrow) were excluded from the oocytes collected from PD50 Cre− and Cre+ mice. Oocytes from superovulated COCs in both Cre− and Cre+ were healthy and resumed meiosis as shown in insets (arrows mark polar body). Scale bar, 50 μm. I, Formation of CL in Cre+ mice after superovulation. H&E staining and the IF assay for STAR detected the CL in the ovaries of PD50 Cre− and Cre+ mice subjected to superovulation test. Scale bar, 100 μm.
To test whether the anovulatory phenotype reflected defects in the endocrine control, serum levels of FSH, testosterone, 17β-estradiol, inhibin A, and inhibin B were tested (Figure 7F). Although the difference in serum testosterone and 17β-estradiol levels did not reach a statistically significant level, the levels of FSH was significantly lower in Cre+ than Cre− female mice at PD50 confirming the alteration of the endocrine profile in oocyte-specific PI3K transgenic mice. Furthermore, the serum concentrations of both inhibin A and B were significantly increased in the PD50 Cre+ female mice, reflecting the overgrowth of excess follicles. Thus, the anovulation in Cre+ mice was likely due to the lowered FSH levels caused by the high serum levels of inhibin A and B. To test whether the antral follicles in Cre+ ovaries were responsive to exogenous ovulatory stimuli, PD50 Cre+ and Cre− female mice were subjected to superovulation treatment. The treatment recovered comparable numbers of oocytes from both Cre+ and Cre− mice (12.7 ± 1.5 per ovary for Cre+ mice vs 11.8 ± 2.6 per ovary for Cre− mice) (Figure 7G). The presence of polar bodies indicated the Cre+ oocytes were competent (Figure 7H, arrows). Furthermore, the superovulation treatment induced a high expression of STAR in the postovulatory follicles in Cre+ female mice (Figure 7I), indicating that the anovulation of PD50 Cre+ female mice was not due to the incompetent follicles or oocytes. Hence, we theorize that a high number of active follicles in Cre+ female mice disrupted the endocrine system resulting in follicular degradation, hemorrhage and anovulation, causing sterility of the Cre+ female. Importantly, the oocytic PI3K activity did not inhibit the normal maturation of meiosis-competent oocytes.
Discussion
It is believed that signaling, which tilts the balance between PIP2 and PIP3 towards PIP3, triggers the activation of oocytes. Our current study demonstrates that oocyte-specific constitutive activation of the PI3K does not tilt the PIP2-PIP3 balance sufficiently to activate the entire pool of primordial follicles, contrasting to the premature ovarian failure phenotype of null mutant mice for PTEN, TSC1 (tuberous sclerosis 1), TSC2, or FOXO3A (7, 10, 11, 13, 34, 37, 38). These studies clearly indicate that many negative regulators of PI3K signaling must be continuously expressed to repress the activation of primordial follicles. In contrast, cell autonomous activation of PI3K in the oocyte did not induce a global activation of primordial follicles; instead, the phenotype indicates the presence of a PTEN-mediated mechanism that protects the ovarian reserve from premature activation. These observations further suggest that activation of the oocyte is triggered by a loss of inhibition rather than a mere activation of the PI3K signaling pathway. Accordingly, PTEN activity within the oocyte appears to be the dominant regulator of follicle activation. In addition, the kinetics of PFAO development in Cre+ ovaries suggests the presence of an intrinsic mechanism that maintains a proper size of the ovarian reserve.
The prevalence of PFAOs in Cre+ mice indicates that the activation of oocytes does not require concomitant activation of granulosa cells. The asynchronous activation of oocytes and granulosa cells suggests that follicular activation does not occur solely through nuclear exclusion of FOXO3A in oocytes. This model is supported by the presence of the enlarged primordial follicles in Foxo3 and Pten null mutant mice. The intra- and extracellular signals coordinating activation of oocytes and granulosa cells still remain unclear. Accordingly, we propose that activation of primordial follicles is triggered through an attenuation of PTEN by an external signal(s) that acts on pregranulosa cells (and oocytes). Alternatively, the external activating signal may act solely on the oocyte to down-regulate PTEN activity, although it may also require a proper permissive stromal environment, which is absent in the neonatal ovaries, to coordinate activation of pregranulosa cells.
In the normal mouse ovary, germ cell nests are partitioned into granulosa cell-enclosed oocytes between PD1 and PD3 (2, 39). A large number of germ cells are lost through apoptosis during and after this period (27–29). In this study, Cre+ ovaries contained a smaller number of apoptotic cells and a higher number of total follicles than the Cre− ovaries at PD5, indicating that an increase in intraoocytic PIP3 level inhibits innate selective loss of primordial follicles. The importance of PI3K signaling in oocyte survival and maintenance is supported by the phenotype of oocyte-specific PDK1 null mice, which gradually lose ovarian follicles. Moreover, apoptosis in PDK1 null oocytes can be attenuated by the simultaneous deletion of PTEN (15). These observations suggest that the fate of the oocyte to die or survive is determined by the balance between PIP2 and PIP3. We have previously shown that the ovaries of oocyte-specific Trp63 conditional knockout mice contained significantly higher numbers of primordial follicles than the ovaries of normal mice at PD5 (18). Studies by Kerr et al demonstrated that a null mutation in NADPH oxidase activator and Bcl binding component 3 (40) as well as bcl-2-lke protein 11 and Bcl-2 modifying factor (41) increases the number of surviving oocytes at PD10. Thus, the selective loss of oocytes in neonatal mouse ovaries involves p63-regulated apoptosis, in which these mitochondrial proteins play critical roles. However, the expression level of p63 appears unchanged between Cre− and Cre+ oocytes, thus constitutive PI3K activity did not inhibit apoptosis of the oocytes through direct down-regulation of p63. Accordingly, we hypothesize that the reduced PI3K-PDK1-AKT signaling within oocyte induces selective loss of oocytes in neonatal mice by activating p63 and the downstream mitochondrial proteins.
It has been shown that the overactivation of FSH signaling causes follicular hemorrhages in mouse models (42). Therefore, the presence of follicular hemorrhage in the Cre+ mice, in which the serum FSH levels is reduced, is intriguing. In our current study, oocytic PIP3 overproduction somehow activated the PI3K-AKT signaling in the granulosa cells. Because the PI3K-AKT signaling is downstream of FSH action in granulosa cells (43), follicular hemorrhage caused by the oocytic PI3K activation and the FSH overstimulation may be mediated through the same downstream molecular mechanisms. Accordingly, we propose the next mechanisms that underlie the postpubertal phenotypes of Cre+ mice: at puberty, primordial follicles that contain high levels of oocytic PIP3 are recruited to grow by the pulsatile gonadotropin secretion from the pituitary. In the growing follicles after puberty, the oocytic PI3K activity promotes growth and hormone production independently of FSH through the activation of AKT in the granulosa cells. Consequently, the gonadotropin-independent activation of granulosa cells causes uncontrolled follicular growth and hormone production, which results in follicular degradation and hemorrhage. At the same time, the high levels of follicular hormones produced by the excessive number of active follicles suppress the pituitary secretion of gonadotropins through the negative feedback, resulting in anovulation. Further studies are needed to confirm this hypothesis.
The phenotype of oocyte-specific mutant PI3K transgenic mice revealed the critical role of oocytic PIP3 levels in follicular growth. Most importantly, this mouse model revealed the presence of a PTEN-mediated mechanism in the prevention of immature follicle activation. The mechanisms of PTEN up-regulation, as well as the factors that mediate the activation of AKT in the granulosa cells, are yet to be identified. The molecular mechanisms inducing such strong phenotypes in the mutant mice will be further examined in our future studies.
Acknowledgments
We thank the late Dr Wylie Vale (The Salk Institute, La Jolla, CA) for providing inhibin-α, Dr Douglas Stocco (Texas Tech University Health Sciences Center, Lubbock, TX) for STAR, Dr Richard Schultz (University of Pennsylvania, Philadelphia, PA) for MSY2, and Dr Alan Conley (University of California, Davis, CA) for CYP17 antibodies. We also thank The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core (Charlottesville, VA) for the serum FSH, testosterone, and 17β-estradiol level analyses and Lu Bai and Catherine Nguyen for the serum inhibin A and B level analyses.
This work was supported by the National Institutes of Health Grants RL1HD058295, UL1DE19587, P50HD076188, and P01HD021921 (to S.K., K.E., M.H.C., M.R., J.Z., K.A.W., and T.K.W.), P50HD076188, R01CA154358, and R01HD064402 (to V.A.S. and T.K.), and U54 HD28934 (to The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Akt
- v-akt murine thymoma viral oncogene homolog
- AMH
- anti-Müllerian hormone
- Bax
- BCL2-associated X protein
- CL
- corpus luteum
- COC
- cumulus-oocyte complex
- Cyp17
- cytochrome P450 17α hydroxylase/17,20 lyase
- DAB
- 3,3′-diaminobenzidine
- FOXO3A
- forkhead box O3
- GDF9
- growth differentiation factor-9
- GV
- germinal vesicle
- GVBD
- GV breakdown
- hCG
- human chorionic gonadotropin
- IF
- immunofluorescence
- IVM
- in vitro maturation
- LOD
- limit of detection
- MII
- metaphase of meiosis II
- MOF
- multioocytic follicle
- MSY2
- germ cell Y-box protein
- p-AKT
- phosphorylation/activation of AKT
- PD
- postnatal day
- PFAO
- primordial follicles with activated oocyte
- PDK1
- phosphoinositide-dependent kinase 1
- PI3K
- phosphoinositide 3-kinase
- PIP2
- phosphatidylinositol-4,5-biphosphate
- PIP3
- phosphatidylinositol (3,4,5)-trisphosphate
- PTEN
- phosphatase and tensin homolog deleted on chromosome 10
- STAR
- steroidogenic acute regulatory protein
- TBS
- Tris-buffered saline (pH 7.6)
- TSC
- tuberous sclerosis 1
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- ZP1
- zona pellucida 1.
References
- 1. Fortune JE. The early stages of follicular development: activation of primordial follicles and growth of preantral follicles. Anim Reprod Sci. 2003;78(3–4):135–163. [DOI] [PubMed] [Google Scholar]
- 2. Fortune JE, Cushman RA, Wahl CM, Kito S. The primordial to primary follicle transition. Mol Cell Endocrinol. 2000;163(1–2):53–60. [DOI] [PubMed] [Google Scholar]
- 3. McGee EA, Hsueh AJ. Initial and cyclic recruitment of ovarian follicles. Endocr Rev. 2000;21(2):200–214. [DOI] [PubMed] [Google Scholar]
- 4. Henderson SA, Edwards RG. Chiasma frequency and maternal age in mammals. Nature. 1968;218(5136):22–28. [DOI] [PubMed] [Google Scholar]
- 5. Liu K, Rajareddy S, Liu L, et al. Control of mammalian oocyte growth and early follicular development by the oocyte PI3 kinase pathway: new roles for an old timer. Dev Biol. 2006;299(1):1–11. [DOI] [PubMed] [Google Scholar]
- 6. Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117(4):421–426. [DOI] [PubMed] [Google Scholar]
- 7. Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301(5630):215–218. [DOI] [PubMed] [Google Scholar]
- 8. Reddy P, Shen L, Ren C, et al. Activation of Akt (PKB) and suppression of FKHRL1 in mouse and rat oocytes by stem cell factor during follicular activation and development. Dev Biol. 2005;281(2):160–170. [DOI] [PubMed] [Google Scholar]
- 9. Reddy P, Liu L, Adhikari D, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319(5863):611–613. [DOI] [PubMed] [Google Scholar]
- 10. Adhikari D, Flohr G, Gorre N, et al. Disruption of Tsc2 in oocytes leads to overactivation of the entire pool of primordial follicles. Mol Hum Reprod. 2009;15(12):765–770. [DOI] [PubMed] [Google Scholar]
- 11. Adhikari D, Zheng W, Shen Y, et al. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum Mol Genet. 2010;19(3):397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawamura K, Cheng Y, Suzuki N, et al. Hippo signaling disruption and Akt stimulation of ovarian follicles for infertility treatment. Proc Natl Acad Sci USA. 2013;110(43):17474–17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li J, Kawamura K, Cheng Y, et al. Activation of dormant ovarian follicles to generate mature eggs. Proc Natl Acad Sci USA. 2010;107(22):10280–10284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adhikari D, Gorre N, Risal S, et al. The safe use of a PTEN inhibitor for the activation of dormant mouse primordial follicles and generation of fertilizable eggs. PLoS One. 2012;7(6):e39034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reddy P, Adhikari D, Zheng W, et al. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum Mol Genet. 2009;18(15):2813–2824. [DOI] [PubMed] [Google Scholar]
- 16. Srinivasan L, Sasaki Y, Calado DP, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139(3):573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lan ZJ, Xu X, Cooney AJ. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod. 2004;71(5):1469–1474. [DOI] [PubMed] [Google Scholar]
- 18. Kim SY, Cordeiro MH, Serna VA, et al. Rescue of platinum-damaged oocytes from programmed cell death through inactivation of the p53 family signaling network. Cell Death Differ. 2013;20(8):987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu J, Lin SJ, Zou C, Makanji Y, Jardetzky TS, Woodruff TK. Inhibin α-subunit N terminus interacts with activin type IB receptor to disrupt activin signaling. J Biol Chem. 2012;287(11):8060–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu M, Kreeger PK, Shea LD, Woodruff TK. Tissue-engineered follicles produce live, fertile offspring. Tissue Eng. 2006;12(10):2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chatot CL, Ziomek CA, Bavister BD, Lewis JL, Torres I. An improved culture medium supports development of random-bred 1-cell mouse embryos in vitro. J Reprod Fertil. 1989;86(2):679–688. [DOI] [PubMed] [Google Scholar]
- 22. Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate–polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112(2):195–203. [DOI] [PubMed] [Google Scholar]
- 23. Ma P, Pan H, Montgomery RL, Olson EN, Schultz RM. Compensatory functions of histone deacetylase 1 (HDAC1) and HDAC2 regulate transcription and apoptosis during mouse oocyte development. Proc Natl Acad Sci USA. 2012;109(8):E481–E489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu M, West E, Shea LD, Woodruff TK. Identification of a stage-specific permissive in vitro culture environment for follicle growth and oocyte development. Biol Reprod. 2006;75(6):916–923. [DOI] [PubMed] [Google Scholar]
- 25. Gay VL, Midgley AR, Jr, Niswender GD. Patterns of gonadotrophin secretion associated with ovulation. Fed Proc. 1970;29(6):1880–1887. [PubMed] [Google Scholar]
- 26. Münsterberg A, Lovell-Badge R. Expression of the mouse anti-müllerian hormone gene suggests a role in both male and female sexual differentiation. Development. 1991;113(2):613–624. [DOI] [PubMed] [Google Scholar]
- 27. Pepling ME, Spradling AC. Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev Biol. 2001;234(2):339–351. [DOI] [PubMed] [Google Scholar]
- 28. Coucouvanis EC, Sherwood SW, Carswell-Crumpton C, Spack EG, Jones PP. Evidence that the mechanism of prenatal germ cell death in the mouse is apoptosis. Exp Cell Res. 1993;209(2):238–247. [DOI] [PubMed] [Google Scholar]
- 29. Bristol-Gould SK, Kreeger PK, Selkirk CG, et al. Fate of the initial follicle pool: empirical and mathematical evidence supporting its sufficiency for adult fertility. Dev Biol. 2006;298(1):149–154. [DOI] [PubMed] [Google Scholar]
- 30. Perez GI, Robles R, Knudson CM, Flaws JA, Korsmeyer SJ, Tilly JL. Prolongation of ovarian lifespan into advanced chronological age by Bax-deficiency. Nat Genet. 1999;21(2):200–203. [DOI] [PubMed] [Google Scholar]
- 31. Tingen C, Kim A, Woodruff TK. The primordial pool of follicles and nest breakdown in mammalian ovaries. Mol Hum Reprod. 2009;15(12):795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kurita T, Cunha GR, Robboy SJ, Mills AA, Medina RT. Differential expression of p63 isoforms in female reproductive organs. Mech Dev. 2005;122(9):1043–1055. [DOI] [PubMed] [Google Scholar]
- 33. Gu W, Tekur S, Reinbold R, et al. Mammalian male and female germ cells express a germ cell-specific Y-Box protein, MSY2. Biol Reprod. 1998;59(5):1266–1274. [DOI] [PubMed] [Google Scholar]
- 34. John GB, Shirley LJ, Gallardo TD, Castrillon DH. Specificity of the requirement for Foxo3 in primordial follicle activation. Reproduction. 2007;133(5):855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fléchon JE, Pavlok A, Kopecný V. Dynamics of zona pellucida formation by the mouse oocyte. An autoradiographic study. Biol Cell. 1984;51(3):403–406. [DOI] [PubMed] [Google Scholar]
- 36. Haddad A, Nagai ME. Radioautographic study of glycoprotein biosynthesis and renewal in the ovarian follicles of mice and the origin of the zona pellucida. Cell Tissue Res. 1977;177(3):347–369. [DOI] [PubMed] [Google Scholar]
- 37. Jagarlamudi K, Liu L, Adhikari D, et al. Oocyte-specific deletion of Pten in mice reveals a stage-specific function of PTEN/PI3K signaling in oocytes in controlling follicular activation. PLoS One. 2009;4(7):e6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu L, Rajareddy S, Reddy P, et al. Infertility caused by retardation of follicular development in mice with oocyte-specific expression of Foxo3a. Development. 2007;134(1):199–209. [DOI] [PubMed] [Google Scholar]
- 39. Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol. 1991;124:43–101. [DOI] [PubMed] [Google Scholar]
- 40. Kerr JB, Hutt KJ, Michalak EM, et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell. 2012;48(3):343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kerr JB, Myers M, Anderson RA. Dynamics of primordial follicle reserve. Reproduction. 2013;146(6);R205–R215. [DOI] [PubMed] [Google Scholar]
- 42. Kumar TR, Palapattu G, Wang P, et al. Transgenic models to study gonadotropin function: the role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol Endocrinol. 1999;13(6):851–865. [DOI] [PubMed] [Google Scholar]
- 43. Park Y, Maizels ET, Feiger ZJ, et al. Induction of cyclin D2 in rat granulosa cells requires FSH-dependent relief from FOXO1 repression coupled with positive signals from Smad. J Biol Chem. 2005;280(10):9135–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]