

Abstract
Death-associated protein 3 (DAP3) is a molecule with a significant role in the control of both apoptosis and anoikis. Apoptosis is the predominant type of programmed cell death (PCD) which may occur in response to irreparable damage to DNA, or in response to induction by inflammatory cells. Anoikis is subset of apoptosis which occurs in epithelial cells in response to detachment from the surrounding matrix. Both apoptosis and anoikis are of interest in the context of carcinogenesis. In this review, we shall discuss apoptosis and anoikis, and the recent literature regarding the role of DAP3 in both these pathways.
Keywords: Apoptosis, Review, Death-associated protein 3, Anoikis, Cancer, Carcinogenesis, Oncogenesis, Breast cancer
Background
Apoptosis is one of the several mechanisms of programmed cell death (PCD) which may occur in response to irreparable damage to DNA, or when induced by inflammatory cells. It is an active, ATP dependent process, as opposed to necrosis, which is a passive process of cell destruction. It is distinct from necrosis, which may also be referred to as accidental cell death (ACD). Histologically, necrosis is characterised by karyolysis and cell swelling, whilst the stigmata of apoptosis include shrinkage of the nucleus (pyknosis) and cytosol, with a virtual absence of inflammation [1,2].
The existence of programmed cell death was initially proposed by Gluckman (1951) [3]. Further investigation into cell death discerned at least two distinct patterns. In addition to the better known necrosis, histological studies in hepatic ischaemia were suggestive of a so-called ‘shrinking necrosis’, which was distinct from the more typical necrosis [4]. Kerr et al. recognised this to be distinct from canonical necrosis, coined the term ‘apoptosis’ for this process, and noted its role in embryogenesis, teratogenesis, tumour response, and normal tissue turnover in response to hormonal factors [5].
It is suggested that apoptosis and necrosis may be two extremes of a continuum of cell demise. Deleterious stimuli of insufficient magnitude to cause necrosis may cause involution of tissue by way of apoptosis. As noted above, this was the original context in which apoptosis was identified [6,7]. The essential distinction between the two processes has been stated to be that, whilst necrosis is typically a passive process, apoptosis is an energy-dependent phenomenon requiring an expenditure of ATP [8].
The processes underlying apoptosis have been implicated in carcinogenesis and have thus been the subject of continued research in the context of cancer. The earliest and most well − documented apoptosis gene is the so-called ‘guardian of the genome’, tumour protein 53 (p53). It was initially identified in animal models in 1979 in association with SV40 large T antigen, and was dubbed p53 reflecting its molecular weight of 53 kDa [9,10]. Subsequent studies found it to be over expressed in cells transformed by chemical carcinogens or ultra-violet radiation [11-13]. Wild type p53 was found to be a tumour suppressor, with a role in cell cycle progression, apoptosis, and the cell response to DNA damage. Mutation of p53 have been implicated in neoplastic transformation in a number of models [11].
Since then, a number of other genes have been studied for their role in programmed cell death including, but not limited to, B-cell lymphoma 2 protein (BCL2), the BCL2 associated X protein (BAX) [14] and those encoding caspases 3, 6, 8 and 9 [15].
Anoikis is a relatively novel area of research in the field of PCD that has also been of great interest in the context of carcinogenesis. Specifically, it is the invocation of apoptosis in response to cell detachment from the extra cellular matrix (ECM), especially in the context of parenchymal cells [16].
The ability to survive detachment from the ECM is prerequisite for the transition to oncogenesis. This makes anoikis a significant area of interest for cancer research [17].
Our understanding of the apoptosis pathway continues to evolve. Two major canonical pathways have been identified, which terminate in a common execution pathway. In addition, a further pathway bypassing the common execution cascade is also discussed below. Finally, we shall also review the literature regarding anoikis and the specific programmes within apoptosis that contribute to it.
Extrinsic pathway
This pathway is mediated by the activation of trans-membranous receptors and is commonly induced by inflammatory cells in response to viral infection or oncogenesis. These receptors are part of the tumour necrosis factor receptor (TNFR) super family, six of whom have been characterised as death receptors (DR). These receptors have been characterised as type I transmembrane protein, with an N-terminal cytoplasmic terminus. This end of the protein contains an active motif termed a ‘death domain’ (DD), which interacts with adapter proteins, thus initiating intra-cellular pathways. In vivo, the ligands for these receptors have been identified as TNF-α, the Fas ligand (Fas-L), TNF related apoptosis inducing ligand (TRAIL), as well as other members of the TNF family. The best documented of these receptors are TNFR1 and Fas (DR1 and DR2). Others include TRAIL-R1 and 2 (DR 4 and DR5), and DR3 and DR6 [18].
Death Inducing Signalling Complex (DISC)
The intra-cellular signalling complex for DRs has been designated as the death inducing signalling complex (DISC). Broadly speaking, the DR mediates its actions when activated by interaction with its respective ligand. The DR typically undergoes trimerisation when thus activated. The DD of the receptor interacts with the DD on the FAS Associated Death Domain (FADD) protein, which is normally localised in the nucleus. In turn, the FADD interacts with pro-caspase 8, which is also known as FADD-like interleukin-1 beta-converting enzyme (FLICE). These interactions are mediated by corresponding death effector domains (DED) on FADD and FLICE, leading to the activation of the apical caspase 8 [19]. This interaction is inhibited by FLICE-like inhibitory protein (FLIP), which competes with FLICE by interacting with FADD [20].
The DISCs of the aforementioned six DRs have small but significant variations between themselves whilst retaining the general plan described above. It would be reasonable to surmise that these variations enable variations in the downstream effects of each of the six DRs.
The most significant variation can be seen in the context of FAS and the two TRAIL receptors. In addition to pro-caspase 8, they also activate pro-caspase 10. The downstream effects of caspase 10 are believed to be largely similar to those of caspase 8, thus providing a redundant signalling pathway [21,22].
Furthermore, the TNFR DISC requires a DD-bearing adaptor protein in addition to FADD, referred to as TNFRSF1A-associated via death domain (TRADD). TRADD is required for FADD-mediated apoptosis. In addition, TRADD also mediates interactions with TNF receptor-associated factor 2 (TRAF2), which mediates immune effects of TNF. Specifically, it activates the nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (NFKB1), which is involved in the control of genes responsible for inflammation and immune response [23].
DAP3 and DRs
A similar role in coordinating downstream effects of DRs is seen in the context of death-associated protein 3 (DAP3). DAP3 is a highly conserved GTP-binding protein of 4 kDa, encoded by the DAP3 gene located in chromosome 1 q21 [24]. It is normally kept inactive as phosphoprotein by the action of protein kinase B (AKT/PKB). When activated, it co-localises with FADD and participates in the formation of the DISC [25]. Initially evidence suggested the DAP3 is involved in the actions of TNF-α, Fas-L, and TRAIL. However, more recent studies are suggestive that its association is strongest with the Fas receptor related DISC. In this context its actions are enabled by binding with death ligand signal enhancer (DELE), a relatively novel protein whose knockdown in HeLa cells was found to inhibit apoptosis due to TNF- α, Fas or TRAIL (Figure 1) [26].
Figure 1.
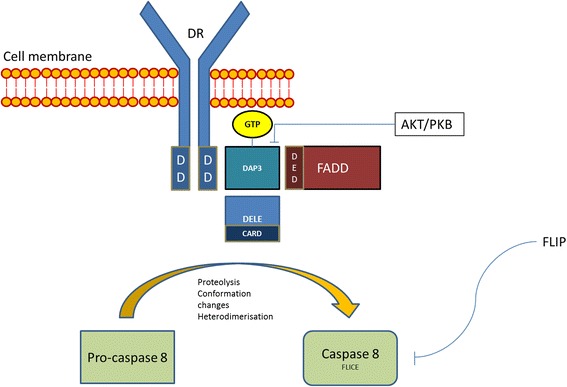
Schematic representation of the relation of DAP3 and DELE with Fas receptor related DISC. AKT/PKB: protein kinase B, CARD: caspase activation and recruitment domain, DAP3: death-associated protein 3, DD: death domains, DED: death effector domains, DELE: death ligand signal enhancer, DR: death receptor, DISC: death inducing signalling complex, FADD: Fas Associated Death, FLICE: FADD-like interleukin-1 beta-converting enzyme, FLIP: FLICE inhibitory protein.
In a similar fashion, DAP3 was found to mediate interactions between FADD and LKB1 interacting protein 1 (LIP1) in human osteosarcoma cells, which anchors LKB1 to the cytoplasm. This study characterised the expression of LKB1 to be critical for TRAIL-induced apoptosis mediated by DAP3 on the basis of knock-down studies in osteosarcoma cells [27]. LKB1 has been previously studied with regards to its role in Peutz-Jeghers syndrome. It is characterised as a serine-threonine kinase, with a predominantly tumour-suppressant role in contrast to the mTOR pathway [28,29].
Furthermore, DAP3 has been shown to be critical for the mediation of anoikis, which is to be discussed at greater length below [25].
Intrinsic pathway
This pathway is influenced by a large array of non-receptor factors which may positively or negatively influence the activation of apoptosis. These factors include but are not limited to toxins, hormones, viral infections, physical stimuli and free radicals. The major strand of this pathway involves the release of mitochondrial contents due to changes in mitochondrial outer membrane permeabilisation (MOMP) [30].
BCL2 family and control of mitochondrial outer membrane permeabilisation (MOMP)
MOMP is directly controlled by the BCL2 family of proteins. The BCL2 family of protein are defined by the presence of BCL2 homology motifs (BH1 to BH4). These domains were initially seen in BCL2, the archetypical member of this group of proteins. BCL2 was initially identified as an oncogene in B-cell lymphoma cells [30]. This was followed by the discovery of BAX, which was characterised as an anti-oncogenic protein [31]. Since then, around two dozen members of the BCL2 family have been identified, with variable tissue expression and differences in expression of BH domains [32].
Broadly speaking, BCL2 proteins can be classified as either pro-apoptotic or anti-apoptotic proteins. In turn the pro-apoptotic molecules are divided into multi-domain molecules, and BH3-only molecules. A precise model for the relationship between classes of BCL2 molecules is yet to be devised, with up to four enumerated by Shamus-din et al. The consensus seems to be that in normal conditions the multi-domain pro-apoptotic molecules (BAX, BAK, BOK) are sequestered in combination with anti-apoptotic molecules (BCL2-like, including BCL-xL, BCL-w and others). When this pathway is activated, BH3-only molecules (BID, PUMA, NOXA) bind with the BCL2-like molecules, allowing the multi-domain pro-apoptotic molecules to localise to the mitochondrial outer membrane. BAX/BAK oligomerise to form pores in the membrane, causing MOMP, which leads to disgorging of intra-mitochondrial contents [33].
The BH3 molecules have been identified as the control element of this process. Their expression and activation seems to be influenced by a variety of stimuli. p53 is the most significant up stream activator for the BH3 molecules [34,35].
The first wave of mitochondrial apoptogenic agents include cytochrome c, the second mitochondria-derived activator of caspase/direct IAP binding protein with low Pi (SMAC/DIABLO) and high temperature requirement A2/Omi (HTRA2/OMI).
Cytochrome c and the apoptosome recruitment complex
Cytochrome c interacts with apoptotic protease activating factor 1 (APAF1) in the presence of ATP or dATP to form a multimeric complex referred to as an apoptosome. APAF1 has a DD termed the caspase activation and recruitment domain (CARD). [36]. CARD enables interaction with and activation of pro-caspase 9, leading to the production of activated caspase 9 [37,38].
Inhibitors of apoptosis proteins (IAPs), SMAC/DIABLO and HTRA2/OMI
The activation of the caspase cascade has been characterised as an all or none outcome. However, the activation of caspases is normally prevented by the inhibitors of apoptosis proteins (IAPs). Upon invocation of the internal apoptotic pathway, factors sequestered in the mitochondria are released which include, SMAC/DIABLO [39]. SMAC/DIABLO inhibits the activity of IAPs, especially that of X-linked IAP (XIAP) by sequestering it [40]. Furthermore, Yu et al. suggest that SMAC/DIABLO sensitises cells to apoptosis induced by PUMA, thus providing a positive feedback for apoptosis [41]. This may be partially balanced by a recently described function of XIAP, in which it impairs the release of SMAC/DIABLO [42].
Similarly, MOMP also leads to the release of HTRA2/OMI, a serine protease which binds and cleaves various IAPs [43]. Additionally, a proteome-wide analysis suggests that HTRA2/OMI may also target cytoskeletal proteins [44].
Mitochondrial DNA-ases
Further mitochondrial contents are then released which contribute to DNA breakdown. These include apoptosis inducing factor (AIF) and endonuclease G (EndoG).
AIF binds to DNA in a sequence-nonspecific manner, inducing chromatin condensation and initiating DNA breakdown [45,46].
EndoG has been demonstrated to form a complex with histone H2B and human DNA topoisomerase II alpha (TOPO2a) after translocation to the cytosol. Whist the details are as yet undetermined, this complex is believed to mediate the apoptosis-associated breakdown of DNA [47].
Pro-survival role of DAP3 in mitochondrial function
Counter-intuitively, DAP3 has a pro-survival role in mitochondrial function. There are believed to be distinct intra-mitochondrial and cytosolic pools of DAP3, with the former resulting from the translocation of the latter through the mitochondrial membrane. The mitochondrial DAP3 forms an important portion of the 28 S subunit of the mitochondrial ribosome, thus essential for mitochondrial protein synthesis [48]. Furthermore, Kim et al. reported a deficiency of cytochrome c oxidase 1 in the mitochondria of Dap3 −/− mouse embryos. As this is a protein encoded in the mitochondrial genome, it could be inferred that DAP3 has an integral role in intra-mitochondrial protein synthesis [49].
The duality seen in the intra-mitochondrial and cytosolic roles of DAP3 is not unlike other mitochondrial proteins, such as cytochrome c and IAP.
PIDDsomes
p53 coordinates DNA repair and may induce apoptosis via various pathways. In addition to induction of BH3-only BCL2 proteins, p53 is known to induce the expression of the protein induced by p53 with a death domain (PIDD). PIDD forms a complex with the RIP associated ICH1/CED homologous protein with death domain (RAIDD). RAIDD is an adaptor protein which also possesses a CARD motif. This enables it to recruit pro-caspase 2. This complex is referred to as the PIDDsome, and is believed to mediate apoptosis in response to DNA damage. The active caspase 2 is formed by conformational changes in the pro-caspase induced by dimerization. Caspase 2 is an apical caspase, which activates further components of the caspase cascade [50,51].
Common execution pathway: the caspase cascade
Intra- and extra-cellular signals converge on a common pathway which mediates the catabolic processes that constitute apoptosis. This pathway consists of a cascade of cysteinyl aspartic acid-protease, commonly referred to as caspases. Up to 14 have been enumerated in human and mammalian species [15]. They broadly fall into three groups:
Apical (Initiators)
As detailed above in relation to the discussions regarding the relevant recruitment complexes, the apical caspases include caspases 2, 8, 9, and 10. These pro-enzymes have DDs, which coordinate the pro-caspases with DISC components with similar domains. This interaction induces dimerization and conformation change in the pro-caspases, leading to transformation to active caspase, as well as, auto-activation of the caspases [37,52].
With regards to caspase 8, Scaffidi et al. postulate that cells may be classified into two types regarding apoptosis. Type 1 have been characterised as cell which only require the action of caspase 8 to undergo apoptosis, whist type 2 cells which would also require MOMP to successfully undergo induced cell death [53].
When activated, they cleave downstream caspases, such as caspases 3 and 7. Also, they further stimulate upstream apoptosis processes. For instance, caspase 8 is known to cleave BH3 interacting domain death agonist (BID) to yield truncated BID (tBID). tBID increases MOMP, thus further committing the cell towards cell death [54]. Similarly, caspase 8 is known to cleave XIAP [55].
Effectors (Executioners)
Caspases 3, 6 and 7 are activated by the apical caspases, and mediate the effects of the caspase cascade.
Caspase 3 is the most significant of the effector caspases, and can be activated by caspases 8, 9 and 10. It shares a lot of substrates with caspase 7. When activated, it mediates breakdown of DNA and of the cytoskeleton, degradation of XIAP, activation of caspase 6, as well as activation of the rest of the caspase cascade [56].
Caspase activated DNA-ase (CAD), also known as DNA fragmentation factor, 40 kDa, beta polypeptide (DFFB), normally exists in complex with inhibitor of CAD (ICAD). When activated, caspase 3 cleaves ICAD, leaving CAD unimpeded to breakdown DNA [57,58]. Upon activation, caspase 3 is believed to participate in further activation of the other components in the apoptosis pathway. It cleaves BID, thus further activating the permeabilisation of the mitochondrial outer membrane [56].
Caspase 3 also inhibits XIAP, thus further potentiating the action of caspase 9 [59].
Furthermore, caspase 3 cleaves gelsolin, which has a role in actin polymerisation and signal transduction [60].
Aminophospholipid translocase normally mediates an ATP-dependent process in which phosphotidyl serine (PS) is transferred from the outer layer of cell membrane to the inner layer. An accumulation of PS on the outer surface which occurs in the course of apoptosis, marks the cell for phagocytosis [61]. Studies in erythrocytes implicate caspases 3 and 8 in inactivation of the aminophospholipid translocase [62].
On the other hand, caspase 6 has a distinct substrate specificity versus caspases 3 and 7 which is currently being delineated. Of note, caspase 6 is being studied regarding it avidity for lamin proteins of the nuclear envelop [63].
Inflammatory caspases
Caspase 1, 4, 5, and 11 have been found to have roles in immune response and innate immunity.
Specifically, they are activated by recruitment complexes composed of nucleotide-binding domain (NB) and leucine-rich repeat (LRR) containing receptors (NLR). Especially noteworthy of this group of proteins are NLRP1 and NLRP3, which form complexes with adaptor protein PYDCARD. These complexes are known as inflammosomes. These inflammosomes recruit caspases 1 and 5 respectively [64]. Inflammosomes have been implicated in immunity and more recently, metabolic syndrome [65].
Similarly, caspase 11 is also recruited by inflammosomes in the context of immune response to intracellular pathogens [66].
Caspase 4 has been implicated in NLR signalling and the activation of inflammosomes [67,68]. In addition, it also may be involved in mediation of endoplasmic reticulum stress related apoptosis [69-71].
Perforin/Granzyme pathway
This is a relatively novel pathway utilised by immune cells in addition to Fas-Fas ligand interactions. In this case the T and NK cells insert pore molecules into the cell membrane of the targeted cell, which have been termed perforins. The perforins interact with intracellular secretory granules (SGs) which release granule associated enzymes (granzymes) A and B [72].
Granzyme A activates DNAase NM23-H1, which destroy nuclear material. This DNAase is normally bound to the SET complex, which is cleaved by granzyme A leading to the release and activation of the said DNAase [73].
Granzyme B activates pro-caspases 3 and 10 and cleaves ICAD. In addition, it activates the intrinsic pathway by inducing cytochrome c release and by cleaving BID protein, a member of the BCL2 family [74,75].
Anoikis
Anoikis was initially described by Frisch et al. as apoptosis resulting from the detachment of epithelial cells from surrounding inter-cellular matrix and from surrounding cells. It is believed to be a cellular programme aimed preservation of normal tissue structure from disruption by inappropriate cell growth and migration. In the course of oncogenesis, neoplastic cells would inevitably have to overcome this cellular programme as part of the epidermal-mesenchymal transition [16].
Our understanding of this cellular programme is currently evolving. Different cell types require specific integrin to extracellular matrix (ECM) interactions. Furthermore, the internal signalling of these interactions into the apoptosis pathways may vary between cell types, and are currently being delineated. These signals are believed to be largely channelled through the internal apoptosis pathway, with some involvement of the external pathway, culminating in the common execution pathway (Figure 2) [76].
Figure 2.
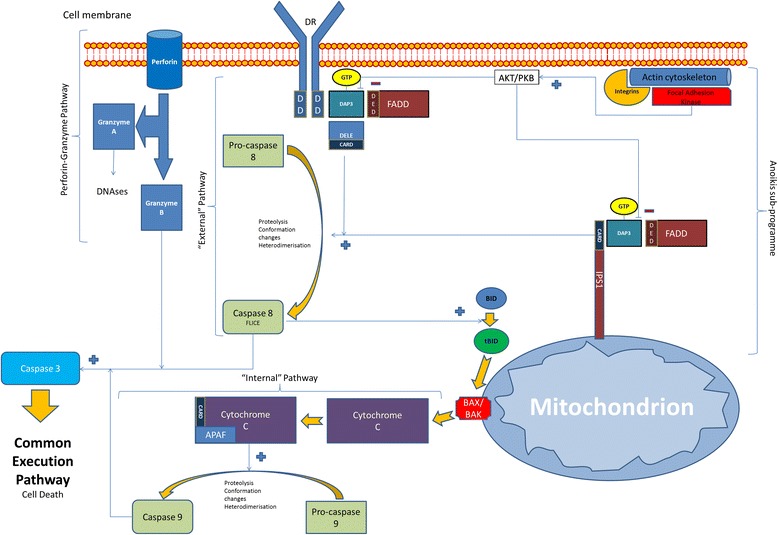
General scheme illustrating the internal, external and perforin/granzyme pathways within apoptosis, including the sub-programmes specific to anoikis and DAP3. AKT/PKB: protein kinase B, CARD: caspase activation and recruitment domain, DAP3: death-associated protein 3, DD: death domains, DED: death effector domains, DELE: death ligand signal enhancer, DR: death receptor, DISC: death inducing signalling complex, FADD: Fas Associated Death, FLICE: FADD-like interleukin-1 beta-converting enzyme, IPS: 1interferon-β promoter stimulator 1.
Role of the internal pathway in anoikis
The signalling from integrins requires mediation by focal adhesion kinase (FAK) which is believed to inhibit p53, which would induce apoptosis if the action of FAK or contact with ECM were lost. Ilic et al. have determined that the p53-induced apoptosis is susceptible to inhibition by BCL2 [77,78]. Studies by Chen et al. found that changes in cell morphology can also induce anoikis, and that the integrins, FAK and the actin cytoskeleton of the cell all play roles in anoikis as part of a focal adhesion complex [79].
Pro-survival signals, including those from anoikis, are believed to be passed by the extracellular-related kinase 1 and 2 (ERK1/2) and AKT/PKB are known to interact with BH3 BCL2 proteins, in particular BIM [80]. Similarly, the RAF pathway, the mitogen/extracellular kinase pathway (MEK) and MAP kinase pathway are also known to transmit such signals, as shown in studies in lung fibroblasts [81]. Puthalakath et al. have identified a role similar to that of BIM for BMF in the context of anoikis [82].
Idogawa et al. have characterised BAD as a specific BH3 molecule for anoikis. Whilst it did not initiate anokis, it did increase sensitivity to loss of AKT/PKB signalling [83].
Removal of such signals results in activation of the BH3 proteins, which sequester the anti-apoptotic BCL2 proteins, thus shift the balance towards BAX-BAK pore formation and initiation of MOMP, formation of the apoptosome, and activation of caspase 9 [84].
Role of the external pathway in anoikis, including that of DAP3
A role for the external pathway in anoikis was proposed initially by Frisch et al., and has been the subject of much study. The role of caspase 8 and Fas-DISCs in the propagation of the apoptosis cascade was identified earlier on [85]. In a study in colonic cells, Grossman et al. characterised the activation of caspase 8 to be a late event in anoikis, which was not likely to be the initiator of the cascade [86]. However, more recent studies in lymphocytes, oesophageal squamous cell and breast cancer cells characterise a FAK-caspase 8 axis central to anoikis signalling [87-89]. Studies in Jurkat cells have suggested that integrins, specifically β1 integrin, forms a complex with AKT/PKB, which inhibits pro-caspase 8 via PI3K. Loss of this inhibition is believed to be an initiator for anoikis [90].
The activation of caspase 8 in the context of anoikis has been associated with interactions with FADD of the Fas-related DISC. This interaction is inhibited by FLIP, which competes for FADD. This function of the Fas-related DISC appears to be ligand-independent [20]. DAP3 has been characterised as critical for anoikis. Upon the loss of the influence of AKT/PKB, it is dephosphorylated and interacts with FADD [25].
The FADD-DAP3 interaction requires the involvement of interferon-β promoter stimulator 1 (IPS1), which is a CARD bearing protein anchored on the mitochondrial outer membrane. It is a component of the innate immune response for viral infections, and communicates with inflammatory pathways. IPS1 localises the reaction to the mitochondrial membrane and recruits pro-caspase 8, thus triggering the caspase cascade (Figure 3) [91].
Figure 3.
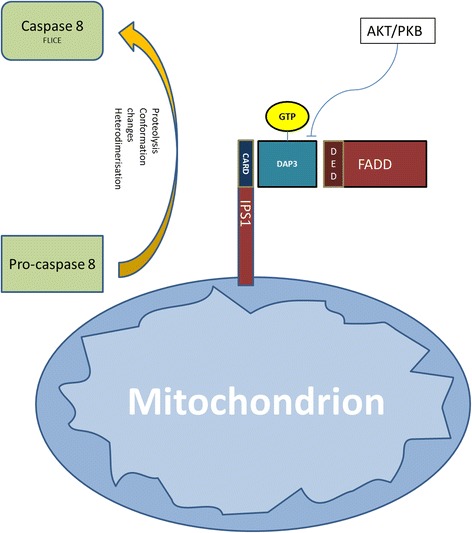
Schematic representation of the relation of DAP3 and IPS1 on mitochondrial surface. AKT/PKB: protein kinase B, CARD: caspase activation and recruitment domain, DAP3: death-associated protein 3, DD: death domains, DED: death effector domains, DELE: death ligand signal enhancer, DR: death receptor, DISC: death inducing signalling complex, FADD: Fas Associated Death, FLICE: FADD-like interleukin-1 beta-converting enzyme, FLIP: FLICE inhibitory protein, IPS: 1interferon-β promoter stimulator 1.
Conclusion
Death associated protein 3 and cancer
In conclusion, the current literature places DAP3 at the nexus of several highly significant and occasionally antagonistic pathways. However, there is scant literature with regards to its role in human neoplastic disease. It could be expected that DAP3 would have a tumour suppressant role predicated on its function as a pro-apoptotic molecule. However, DAP3’s countervailing pro-survival role in mitochondrial protein synthesis reduces such assumptions to the level of speculation.
Indeed, Mariani et al. reported increased expression levels of DAP3 in invasive glioblastoma tumour cells and in glioma cell lines with induced migratory phenotype [92]. These observations could be an instance of a neoplastic disease in which the mitochondrial maintenance prevails over the pro-apoptotic functions of the DAP3. Furthermore, this could be explained by the fact that P-loop mutant DAP3 is less effective in inducing apoptosis, and the COOH-terminal deleted protein (230 amino acids) acts in a dominant negative fashion, protecting cells from induced apoptosis [24].
On the other hand, our group has published a clinical study, in which we examined the mRNA expression of DAP3 in a breast cancer cohort of 127 patients, with a median follow-up of 10 years. Specifically we recorded a strong inverse correlation between mean copy number of DAP3 and tumour grade, Nottingham Prognostic Index, clinical stage, and clinical outcome. Furthermore, Kaplan-Meier analysis shown better survival in the high transcription group approaching significance [93].
Further investigations would be required to characterise the role of DAP3 in human breast cancer. We have presented our initial findings regarding studies into cell behaviour of knock-down sub-lines of MCF7 and MDA-MB-231 at the Scientific Meeting of British Association of Surgical Oncology in London, and have recently published our final results. We found that DAP3 knockdown sub-lines of MCF7 and MDA-MB‑231 had significantly increased adhesion and decreased growth when compared to the controls. Furthermore, invasion and migration were significantly increased in the MDA-MB-231DAP3kd cells when compared to controls. The results of the adhesion, migration and invasion assays are supportive of the hypothesis of DAP3 as a pro-apoptotic antioncogenic protein, whilst the countervailing effect on growth may be due to pro-survival role of DAP3 in mitochondrial protein synthesis [94].
In view of the locus of DAP3 within several cellular pathways of importance in oncogenesis, we believe that DAP3 would be a very fruitful avenue for further research. Such research would hopefully engender a better understanding of underlying pathways, enabling more discrete therapeutic targeting.
Acknowledgements
This study was funded by grants from the Breast Cancer Hope Foundation (London, UK).
Abbreviations
- ACD
Accidental cell death
- AIF
Apoptosis inducing factor
- AKT/PKB
Protein kinase B
- APAF1
Apoptotic protease activating factor 1
- BAX
BCL2 associated X protein
- BCL2
B-cell lymphoma 2 protein
- BID
BH3 interacting domain death agonist
- CAD
Caspase activated DNA-ase
- CARD
Caspase activation and recruitment domain
- caspases
Cysteinyl aspartic acid-proteases
- DAP3
Death-associated protein 3
- DD
Death domain
- DED
Death effector domains
- DELE
Death ligand signal enhancer
- DISC
Death inducing signalling complex
- DR
Death receptors
- ECM
Extra cellular matrix
- EndoG
Endonuclease G
- ERK1/2
Extracellular-related kinase 1 and 2
- FADD
FAS Associated Death Domain
- FAK
Focal adhesion kinase
- FLICE
FADD-like interleukin-1 beta-converting enzyme
- FLIP
FLICE-like inhibitory protein
- granzymes
Granule associated enzymes
- HTRA2/OMI
High temperature requirement A2/Omi
- IAPs
Inhibitors of apoptosis proteins
- ICAD
Inhibitor of CAD
- LIP1
LKB1 interacting protein 1
- IPS1
Interferon-β promoter stimulator 1
- MEK
Mitogen/extracellular kinase pathway
- MOMP
Mitochondrial outer membrane permeabilisation
- NFKB1
Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- NLR
Nucleotide-binding domain (NB) and leucine-rich repeat (LRR) containing receptors
- PCD
Programmed cell death
- PIDD
Protein induced by p53 with a Death Domain
- PS
Phosphotidyl serine
- RAIDD
RIP associated ICH1/CED homologous protein with death domain
- SMAC/DIABLO
Second mitochondria-derived activator of caspase/direct IAP binding protein with low Pi
- tBID
Truncated BH3 interacting domain death agonist
- TNF
Tumour necrosis factor
- TNFR
Tumour necrosis factor receptor
- TOPO2a
Topoisomerase II alpha
- TRAIL
TNF related apoptosis inducing ligand
- TRADD
TNFRSF1A-associated via death domain
- TRAF2
TNF receptor-associated factor 2
- XIAP
X-linked IAP
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
UW conceived the idea as part of his doctoral dissertation, did literature search and drafted the manuscript; MMAWO, ZSK, and AK did literature search and contributed in drafting and proof-reading the manuscript; WGJ, AKS and KM were involved in discussion and drafting the manuscript; AKS and KM supervised the project, made substantial contributions to the concept and design of manuscript. All authors read and approved the final manuscript.
Contributor Information
Umar Wazir, Email: umar.wazir@rcsed.ac.uk.
Mona MAW Orakzai, Email: monzir.awa@gmail.com.
Zubair S Khanzada, Email: zubairshabbir@gmail.com.
Wen G Jiang, Email: jiangw@cardiff.ac.uk.
Anup K Sharma, Email: noopysharma@hotmail.com.
Abdul Kasem, Email: abkasem@doctors.org.uk.
Kefah Mokbel, Email: kefahmokbel@hotmail.com.
References
- 1.Hacker G. The morphology of apoptosis. Cell Tissue Res. 2000;301(1):5–17. doi: 10.1007/s004410000193. [DOI] [PubMed] [Google Scholar]
- 2.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146(1):3–15. [PMC free article] [PubMed] [Google Scholar]
- 3.GlÜCksmann A. Cell deaths in normal vertebrate ontogeny. Biol Rev. 1951;26(1):59–86. doi: 10.1111/j.1469-185X.1951.tb00774.x. [DOI] [PubMed] [Google Scholar]
- 4.Kerr JF. Shrinkage necrosis: a distinct mode of cellular death. J Pathol. 1971;105(1):13–20. doi: 10.1002/path.1711050103. [DOI] [PubMed] [Google Scholar]
- 5.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE, et al. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol. 2000;182(1):41–9. doi: 10.1002/(SICI)1097-4652(200001)182:1<41::AID-JCP5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Papucci L, Formigli L, Schiavone N, Tani A, Donnini M, Lapucci A, et al. Apoptosis shifts to necrosis via intermediate types of cell death by a mechanism depending on c-myc and bcl-2 expression. Cell Tissue Res. 2004;316(2):197–209. doi: 10.1007/s00441-004-0872-z. [DOI] [PubMed] [Google Scholar]
- 8.Leist M. Intracellular Adenosine Triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185(8):1481–6. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang C, Simmons DT, Martin MA, Mora PT. Identification and partial characterization of new antigens from simian virus 40-transformed mouse cells. J Virol. 1979;31(2):463–71. doi: 10.1128/jvi.31.2.463-471.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979;31(2):472–83. doi: 10.1128/jvi.31.2.472-483.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.May P, May E. Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene. 1999;18(53):7621–36. doi: 10.1038/sj.onc.1203285. [DOI] [PubMed] [Google Scholar]
- 12.Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4(9):1689–94. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci U S A. 1979;76(5):2420–4. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basu A, Haldar S. The relationship between BcI2, Bax and p53: consequences for cell cycle progression and cell death. Mol Hum Reprod. 1998;4(12):1099–109. doi: 10.1093/molehr/4.12.1099. [DOI] [PubMed] [Google Scholar]
- 15.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124(4):619–26. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. Mol Aspects Med. 2010;31(2):205–14. doi: 10.1016/j.mam.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7(7):532–42. doi: 10.1038/nri2115. [DOI] [PubMed] [Google Scholar]
- 19.Screaton RA, Kiessling S, Sansom OJ, Millar CB, Maddison K, Bird A, et al. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: a potential link between genome surveillance and apoptosis. Proc Natl Acad Sci U S A. 2003;100(9):5211–6. doi: 10.1073/pnas.0431215100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aoudjit F, Vuori K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: a role for c-flip and implications for anoikis. J Cell Biol. 2001;152(3):633–43. doi: 10.1083/jcb.152.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002;21(17):4520–30. doi: 10.1093/emboj/cdf441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kischkel FC, Lawrence DA, Tinel A, LeBlanc H, Virmani A, Schow P, et al. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem. 2001;276(49):46639–46. doi: 10.1074/jbc.M105102200. [DOI] [PubMed] [Google Scholar]
- 23.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84(2):299–308. doi: 10.1016/S0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 24.Kissil JL, Kimchi A. Assignment of death associated protein 3 (DAP3) to human chromosome 1q21 by in situ hybridization. Cytogenet Cell Genet. 1997;77(3–4):252. doi: 10.1159/000134587. [DOI] [PubMed] [Google Scholar]
- 25.Miyazaki T, Shen M, Fujikura D, Tosa N, Kim HR, Kon S, et al. Functional role of death-associated protein 3 (DAP3) in anoikis. J Biol Chem. 2004;279(43):44667–72. doi: 10.1074/jbc.M408101200. [DOI] [PubMed] [Google Scholar]
- 26.Harada T, Iwai A, Miyazaki T. Identification of DELE, a novel DAP3-binding protein which is crucial for death receptor-mediated apoptosis induction. Apoptosis. 2010;15(10):1247–55. doi: 10.1007/s10495-010-0519-3. [DOI] [PubMed] [Google Scholar]
- 27.Takeda S, Iwai A, Nakashima M, Fujikura D, Chiba S, Li HM, et al. LKB1 is crucial for TRAIL-mediated apoptosis induction in osteosarcoma. Anticancer Res. 2007;27(2):761–8. [PubMed] [Google Scholar]
- 28.Duivenvoorden WC, Beatty LK, Lhotak S, Hill B, Mak I, Paulin G, et al. Underexpression of tumour suppressor LKB1 in clear cell renal cell carcinoma is common and confers growth advantage in vitro and in vivo. Br J Cancer. 2013;108(2):327–33. doi: 10.1038/bjc.2012.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A. 2013;110(32):E2950–7. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47(1):19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 31.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74(4):609–19. doi: 10.1016/0092-8674(93)90509-O. [DOI] [PubMed] [Google Scholar]
- 32.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2(3):183–92. doi: 10.1016/S1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 33.Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(4):a008714. doi: 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hikisz P, Kilianska ZM. PUMA, a critical mediator of cell death–one decade on from its discovery. Cell Mol Biol Lett. 2012;17(4):646–69. doi: 10.2478/s11658-012-0032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80(2):293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 36.Zou H. An APAF-1 Cytochrome c Multimeric complex is a functional Apoptosome that activates Procaspase-9. J Biol Chem. 1999;274(17):11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 37.Pop C, Timmer J, Sperandio S, Salvesen GS. The apoptosome activates caspase-9 by dimerization. Mol Cell. 2006;22(2):269–75. doi: 10.1016/j.molcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 38.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 39.Zhou LL, Zhou LY, Luo KQ, Chang DC. Smac/DIABLO and cytochrome c are released from mitochondria through a similar mechanism during UV-induced apoptosis. Apoptosis. 2005;10(2):289–99. doi: 10.1007/s10495-005-0803-9. [DOI] [PubMed] [Google Scholar]
- 40.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408(6815):1008–12. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 41.Yu J, Wang P, Ming L, Wood MA, Zhang L. SMAC/Diablo mediates the proapoptotic function of PUMA by regulating PUMA-induced mitochondrial events. Oncogene. 2007;26(29):4189–98. doi: 10.1038/sj.onc.1210196. [DOI] [PubMed] [Google Scholar]
- 42.Flanagan L, Sebastia J, Tuffy LP, Spring A, Lichawska A, Devocelle M, et al. XIAP impairs Smac release from the mitochondria during apoptosis. Cell Death Dis. 2010;1:e49. doi: 10.1038/cddis.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17(12):1487–96. doi: 10.1101/gad.1097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vande Walle L, Van Damme P, Lamkanfi M, Saelens X, Vandekerckhove J, Gevaert K, et al. Proteome-wide Identification of HtrA2/Omi substrates. J Proteome Res. 2007;6(3):1006–15. doi: 10.1021/pr060510d. [DOI] [PubMed] [Google Scholar]
- 45.Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, et al. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9(9):680–4. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- 46.Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, et al. Regulation of AIF expression by p53. Cell Death Differ. 2006;13(12):2140–9. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- 47.Varecha M, Potesilova M, Matula P, Kozubek M. Endonuclease G interacts with histone H2B and DNA topoisomerase II alpha during apoptosis. Mol Cell Biochem. 2012;363(1–2):301–7. doi: 10.1007/s11010-011-1182-x. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki T, Terasaki M, Takemoto-Hori C, Hanada T, Ueda T, Wada A, et al. Proteomic analysis of the mammalian mitochondrial ribosome. Identification of protein components in the 28 S small subunit. J Biol Chem. 2001;276(35):33181–95. doi: 10.1074/jbc.M103236200. [DOI] [PubMed] [Google Scholar]
- 49.Kim HR, Chae HJ, Thomas M, Miyazaki T, Monosov A, Monosov E, et al. Mammalian dap3 is an essential gene required for mitochondrial homeostasis in vivo and contributing to the extrinsic pathway for apoptosis. FASEB J. 2007;21(1):188–96. doi: 10.1096/fj.06-6283com. [DOI] [PubMed] [Google Scholar]
- 50.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304(5672):843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 51.Bouchier-Hayes L, Green DR. Caspase-2: the orphan caspase. Cell Death Differ. 2012;19(1):51–7. doi: 10.1038/cdd.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pennarun B, Meijer A, de Vries EG, Kleibeuker JH, Kruyt F, de Jong S. Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim Biophys Acta. 2010;1805(2):123–40. doi: 10.1016/j.bbcan.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17(6):1675–87. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta. 2011;1813(4):558–63. doi: 10.1016/j.bbamcr.2011.01.026. [DOI] [PubMed] [Google Scholar]
- 55.Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell Death Differ. 2012;19(1):42–50. doi: 10.1038/cdd.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walsh JG, Cullen SP, Sheridan C, Luthi AU, Gerner C, Martin SJ. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc Natl Acad Sci U S A. 2008;105(35):12815–9. doi: 10.1073/pnas.0707715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391(6662):96–9. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 58.Tsuruta T, Oh-Hashi K, Ueno Y, Kitade Y, Kiuchi K, Hirata Y. RNAi knockdown of caspase-activated DNase inhibits rotenone-induced DNA fragmentation in HeLa cells. Neurochem Int. 2007;50(4):601–6. doi: 10.1016/j.neuint.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Denault JB, Eckelman BP, Shin H, Pop C, Salvesen GS. Caspase 3 attenuates XIAP (X-linked inhibitor of apoptosis protein)-mediated inhibition of caspase 9. Biochem J. 2007;405(1):11–9. doi: 10.1042/BJ20070288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boccellino M, Giuberti G, Quagliuolo L, Marra M, D’Alessandro AM, Fujita H, et al. Apoptosis induced by interferon-alpha and antagonized by EGF is regulated by caspase-3-mediated cleavage of gelsolin in human epidermoid cancer cells. J Cell Physiol. 2004;201(1):71–83. doi: 10.1002/jcp.20058. [DOI] [PubMed] [Google Scholar]
- 61.Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem. 1997;272(42):26159–65. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- 62.Mandal D, Mazumder A, Das P, Kundu M, Basu J. Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J Biol Chem. 2005;280(47):39460–7. doi: 10.1074/jbc.M506928200. [DOI] [PubMed] [Google Scholar]
- 63.Mintzer R, Ramaswamy S, Shah K, Hannoush RN, Pozniak CD, Cohen F, et al. A whole cell assay to measure caspase-6 activity by detecting cleavage of lamin A/C. PLoS One. 2012;7(1):e30376. doi: 10.1371/journal.pone.0030376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dagenais M, Skeldon A, Saleh M. The inflammasome: in memory of Dr. Jurg Tschopp. Cell Death Differ. 2012;19(1):5–12. doi: 10.1038/cdd.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14(1):10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 66.Vigano E, Mortellaro A. Caspase-11: the driving factor for noncanonical inflammasomes. Eur J Immunol. 2013;43(9):2240–5. doi: 10.1002/eji.201343800. [DOI] [PubMed] [Google Scholar]
- 67.Nickles D, Falschlehner C, Metzig M, Boutros M. A genome-wide RNA interference screen identifies caspase 4 as a factor required for tumor necrosis factor alpha signaling. Mol Cell Biol. 2012;32(17):3372–81. doi: 10.1128/MCB.06739-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sollberger G, Strittmatter GE, Kistowska M, French LE, Beer HD. Caspase-4 is required for activation of inflammasomes. J Immunol. 2012;188(4):1992–2000. doi: 10.4049/jimmunol.1101620. [DOI] [PubMed] [Google Scholar]
- 69.Yamamuro A, Kishino T, Ohshima Y, Yoshioka Y, Kimura T, Kasai A, et al. Caspase-4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J Pharmacol Sci. 2011;115(2):239–43. doi: 10.1254/jphs.10217SC. [DOI] [PubMed] [Google Scholar]
- 70.Li C, Wei J, Li Y, He X, Zhou Q, Yan J, et al. Transmembrane Protein 214 (TMEM214) mediates endoplasmic reticulum stress-induced caspase 4 enzyme activation and apoptosis. J Biol Chem. 2013;288(24):17908–17. doi: 10.1074/jbc.M113.458836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165(3):347–56. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, Tschopp J, et al. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med. 1991;173(5):1099–109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112(5):659–72. doi: 10.1016/S0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- 74.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trapani JA. Granzymes, cytotoxic granules and cell death: the early work of Dr. Jurg Tschopp. Cell Death Differ. 2012;19(1):21–7. doi: 10.1038/cdd.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76(11):1352–64. doi: 10.1016/j.bcp.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 77.Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9(5):701–6. doi: 10.1016/S0955-0674(97)80124-X. [DOI] [PubMed] [Google Scholar]
- 78.Ilic D, Almeida EA, Schlaepfer DD, Dazin P, Aizawa S, Damsky CH. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143(2):547–60. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276(5317):1425–8. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 80.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278(21):18811–6. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 81.Le Gall M, Chambard JC, Breittmayer JP, Grall D, Pouyssegur J, Van Obberghen-Schilling E. The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol Biol Cell. 2000;11(3):1103–12. doi: 10.1091/mbc.11.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293(5536):1829–32. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- 83.Idogawa M, Adachi M, Minami T, Yasui H, Imai K. Overexpression of BAD preferentially augments anoikis. Int J Canc Suppl J Int Canc Suppl. 2003;107(2):215–23. doi: 10.1002/ijc.11399. [DOI] [PubMed] [Google Scholar]
- 84.Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5(8):733–40. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- 85.Frisch SM. Evidence for a function of death-receptor-related, death-domain-containing proteins in anoikis. Curr Biol. 1999;9(18):1047–9. doi: 10.1016/S0960-9822(99)80455-2. [DOI] [PubMed] [Google Scholar]
- 86.Grossmann J, Walther K, Artinger M, Kiessling S, Scholmerich J. Apoptotic signaling during initiation of detachment-induced apoptosis (“anoikis”) of primary human intestinal epithelial cells. Cell Growth Differ. 2001;12(3):147–55. [PubMed] [Google Scholar]
- 87.Walker TN, Cimakasky LM, Coleman EM, Madison MN, Hildreth JE. Antibody against integrin lymphocyte function-associated antigen 1 inhibits HIV type 1 infection in primary cells through caspase-8-mediated apoptosis. AIDS Res Hum Retroviruses. 2013;29(2):371–83. doi: 10.1089/aid.2011.0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fanucchi S, Veale RB. Delayed caspase-8 activation and enhanced integrin beta1-activated FAK underpins anoikis in oesophageal carcinoma cells harbouring mt p 53–R175H. Cell Biol Int. 2011;35(8):819–26. doi: 10.1042/CBI20100894. [DOI] [PubMed] [Google Scholar]
- 89.Lauricella M, Ciraolo A, Carlisi D, Vento R, Tesoriere G. SAHA/TRAIL combination induces detachment and anoikis of MDA-MB231 and MCF-7 breast cancer cells. Biochimie. 2012;94(2):287–99. doi: 10.1016/j.biochi.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 90.Estrugo D, Fischer A, Hess F, Scherthan H, Belka C, Cordes N. Ligand bound beta1 integrins inhibit procaspase-8 for mediating cell adhesion-mediated drug and radiation resistance in human leukemia cells. PLoS One. 2007;2(3):e269. doi: 10.1371/journal.pone.0000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li HM, Fujikura D, Harada T, Uehara J, Kawai T, Akira S, et al. IPS-1 is crucial for DAP3-mediated anoikis induction by caspase-8 activation. Cell Death Differ. 2009;16(12):1615–21. doi: 10.1038/cdd.2009.97. [DOI] [PubMed] [Google Scholar]
- 92.Mariani L, Beaudry C, McDonough WS, Hoelzinger DB, Kaczmarek E, Ponce F, et al. Death-associated protein 3 (Dap-3) is overexpressed in invasive glioblastoma cells in vivo and in glioma cell lines with induced motility phenotype in vitro. Clinical Canc Res. 2001;7(8):2480–9. [PubMed] [Google Scholar]
- 93.Wazir U, Jiang WG, Sharma AK, Mokbel K. The mRNA expression of DAP3 in human breast cancer: correlation with clinicopathological parameters. Anticancer Res. 2012;32(2):671–4. [PubMed] [Google Scholar]
- 94.Wazir U, Sanders AJ, Wazir AM, Ye L, Jiang WG, Ster IC, et al. Effects of the knockdown of death-associated protein 3 expression on cell adhesion, growth and migration in breast cancer cells. Oncol Rep. 2015;33(5):2575–82. doi: 10.3892/or.2015.3825. [DOI] [PubMed] [Google Scholar]