

Abstract
Background. Rotavirus causes 500 000 deaths and millions of physician visits and hospitalizations per year, with worse outcomes and reduced vaccine efficacy in developing countries. We hypothesized that the gut microbiota might modulate rotavirus infection and/or antibody response and thus potentially play a role in such regional differences.
Methods. The microbiota was ablated via germ-free or antibiotic approaches. Enhanced exposure to microbiota was achieved via low-dose dextran sodium sulfate (DSS) treatment. Rotavirus infection and replication was assessed by enzyme-linked immunosorbent assay (ELISA) and quantitative reverse-transcription polymerase chain reaction. Diarrhea was scored visually. Humoral responses to rotavirus were measured by ELISA and enzyme-linked immunosorbent spot assay.
Results. Microbiota elimination delayed infection and reduced infectivity by 42%. Antibiotics did not alter ratios of positive-sense to negative-sense strands, suggesting that entry rather than replication was influenced. Antibiotics reduced the diarrhea incidence and duration, indicating that the reduction in the level of rotavirus antigen was biologically significant. Despite lowered antigen level, antibiotics resulted in a more durable rotavirus mucosal/systemic humoral response. Increased rotavirus antibody response durability correlated with increased small intestinal rotavirus-specific, immunoglobulin A–producing antibody-secreting cell concentration in antibiotic-treated mice. Conversely, DSS treatment impaired generation of rotavirus-specific antibodies.
Conclusions. Microbiota ablation resulted in reduced rotavirus infection/diarrhea and a more durable rotavirus antibody response, suggesting that antibiotic administration before rotavirus vaccination could raise low seroconversion rates that correlate with the vaccine's inefficacy in developing regions.
Keywords: vaccine, microbiota, antibiotics, germ-free, mucosal immunity
(See the editorial commentary by Bartelt and Guerrant on pages 167–70.)
Rotavirus (RV), a double-stranded, nonenveloped RNA virus that preferentially infects intestinal epithelial cells, is the world's leading cause of acute gastroenteritis in young children [1]. Before the recent introduction of RV vaccines, RV universally infected children younger than 5 years. Most cases resolve within 8 days, although some result in more-severe complications eventuating in 2–4 million hospitalizations per year globally [2, 3]. However, the RV disease burden is greatest in developing countries, where RV causes 500 000 deaths annually [4, 5]. Such disparity in the RV disease burden is generally assumed to reflect general health and nutritional status, access to supportive care, and/or potential coinfections [6].
Widespread introduction of RV vaccines has greatly reduced the RV disease burden in developing countries. For example, in Malawi, one of the world's least developed nations, administration of Rotarix has lowered RV-associated deaths by 43%. Yet, vaccine efficacy in developing countries is markedly lower than that observed in Europe and the Americas (49% in Malawi vs >95% in Europe and the Americas) [2]. Much of this difference appears to be attributable to vaccines eliciting lesser immune responses, as Malawi only exhibited 57% anti-RV immunoglobulin A (IgA) seropositivity after Rotarix vaccination, whereas RotaTeq induced 95% anti-RV IgA seropositivity in the Americas and Europe [7, 8].
Various hypotheses may explain why RV vaccines are less immunogenic in some regions. A lack of proper nutrition, which influences immune responses, may be responsible for decreased vaccine efficacy. Additionally, high titers of maternal-derived transplacental antibody and breast milk IgA has RV vaccine strain neutralization potential and may lower RV antigen exposure by preventing the infection-like state associated with the RV vaccine. To overcome the presence of neutralizing antibody, which blocks the infection-like state and subsequent protective immune responses, higher RV doses have been proposed [9]. Another possibility is that chronic infection, such as with helminths, suppresses the immune response to the RV vaccine [5, 10]. A more general form of the latter hypothesis is that the infection of, or immune response to, RV vaccines is influenced by the gut microbiota, which is thought to differ considerably between individuals in developed countries and those in developing countries. Indeed, the microbiota exerts broad and varied influence on immune system development and function [11]. For example, in models of influenza and lymphocytic choriomeningitis infection, virus-specific adaptive responses, including both T-cell and B-cell responses, were lessened with antibiotic treatment, owing to a decline in dendritic cell and macrophage function [12, 13]. In the case of enteric viruses, namely poliovirus and reovirus, ablation of microbiota resulted in relative resistance to infection, which might lessen the generation of protective immune responses [14]. Such ability of the microbiota to influence both viral infection, which is necessary for the immune response, and the immune response suggest that the microbiota may mediate disparities in RV disease severity and/or vaccine efficacy.
Our goal was to investigate the possibility that the microbiota might influence infection with and/or immune responses to RV. Neonatal mice served as a model of RV disease, and an adult model of RV infection was considered analogous to RV vaccination, wherein protection against infection best correlates with levels of intestinal anti-RV IgA [1]. Our results indicate that commensal microbiota promotes RV infection and influences RV-induced immune responses.
MATERIALS AND METHODS
Animals
All experiments, except those using neonatal or germ-free mice, used 6–8-week-old male C57BL/6 mice purchased from Jackson Laboratories (Bar Harbor, ME). Experiments involving neonatal mice used 6-day-old offspring (male and female) of C57BL/6J mice. Experiments involving germ-free mice used C57BL/6 mice derived via embryo transfer, as previously described [15]. Mice were maintained in sterile isolators at Georgia State University, which approved all procedures.
Virus and Inoculations
Mouse RV strain EC was provided by Mary Estes (Baylor College of Medicine). Adult mice received 105 50% shedding doses (SD50) of RV, preceded by 1.33% (w/v) sodium bicarbonate (Sigma-Aldrich) by oral gavage. Neonates received 2 50% diarrhea doses of RV by oral gavage.
Antibiotics and Antibiotic Regimens
Adult mice were administered ampicillin (Sigma-Aldrich) at 1 g/L and neomycin (Sigma-Aldrich) at 0.5 g/L in drinking water ad libitum 1 week before inoculation and 1, 7, or 11 weeks after inoculation. Neonates received 200 μg of ampicillin and 100 μg of neomycin in 100 µL of water by oral gavage 1 day before and 1 day after inoculation. Additionally, mothers received 1 g/L ampicillin and 0.5 g/L neomycin in their drinking water, beginning 1 week before giving birth and lasting until offspring were weaned.
Infection of Germ-Free Mice With RV
Male and female germ-free mice aged 6–8 weeks were inoculated and maintained in gnotobiotic isolators (Park Bioservices) during and up to 9 weeks after inoculation. Germ-free status was monitored by quantitative polymerase chain reaction (PCR) for 16S ribosomal DNA (rDNA) and fecal culture in brain-heart infusion broth (BD).
Bacterial Load Quantification
Fecal bacterial DNA was isolated using the QiAamp DNA Stool Kit (Qiagen), and 16S rDNA was amplified by quantitative PCR (Bio-Rad).
Fecal Rotavirus Antigen Detection
Supernatants of fecal homogenates (100 mg/mL) were frozen or immediately analyzed by enzyme-linked immunosorbent assay (ELISA), as described elsewhere [16].
Duodenal RV Genome Quantitative Reverse-Transcription PCR (qRT-PCR)
Duodenal samples were harvested, washed in phosphate-buffered saline, and homogenized in TRIzol (Ambion). Duodenal RNA was probed for RV genome, as described elsewhere [17].
Single-Stranded qRT-PCR for RV Replication
RV replication ability in duodenal RNA samples was determined by the ratio of RV positive-sense to negative-sense strands, as described elsewhere [17].
Antibody ELISAs
Fecal and serum relative anti-RV antibody production and titer was analyzed as described previously [18]. Total IgA levels were measures as described elsewhere [19].
Small Intestinal, RV-Specific, IgA-Producing, Antibody-Secreting Cell Enzyme-Linked Immunosorbent Spot Assay
Whole small intestines and lamina propria and Peyer's patch cells were harvested as described previously [20]. Cells were applied to filter plates (Millipore) coated with purified rhesus RV. RV-specific IgA was probed by anti-IgA secondary antibody (SouthernBiotech).
Dextran Sodium Sulfate (DSS) Administration
DSS (MP Biomedicals) was diluted in drinking water at 1% (w/v) and administered ad libitum 4 days before and 3 days after RV inoculation.
Lipocalin-2 ELISA
Fecal supernatants were made as above, and lipocalin-2 was assessed as described elsewhere [21].
Statistical Analysis
Except when indicated otherwise, data are from a single experiment (n = 5 mice per condition) that was performed multiple times and yielded a similar pattern of results. Statistical significance was evaluated via the Student t test.
RESULTS
Ablation of Microbiota Delays RV Infectivity and Ameliorates RV-induced Disease
Infection of adult mice with RV does not result in severe disease manifestations but serves as a well-defined infection model [1]. Accordingly, oral inoculation of 6–8-week-old C57BL/6 mice with 105 SD50 of murine RV strain EC resulted in RV antigen becoming detectable in feces 1–2 days after inoculation. Such RV shedding, which peaks 3–4 days after inoculation and lasts 6–8 days after inoculation, is proportional to infectivity (ie, the level of viral genome in intestinal lysates) [22]. To investigate the role of microbiota, we administered RV to mice that had been treated with a combination of ampicillin and neomycin 1 week before inoculation, which they continued to received throughout infection. PCR-based quantitation indicated such antibiotics reduced gut bacterial loads by 99% (Supplementary Figure 1A). Such reduction of microbiota levels consistently resulted in a 1-day delay in the appearance of fecal RV antigen and an approximate 40% decrease in total RV shedding (as determined by the area under the curve; Figure 1A). Considering that antibiotic-treated mice still had a significant bacterial load in their gut and that antibiotics can alter relative proportions of bacteria, potentially resulting in increases in some species, germ-free mice were also used. Germ-free C57BL/6 mice maintained in sterile isolators throughout the experiment and conventionally housed control mice were orally inoculated with filter-sterilized RV. Fecal culture and qPCR verified the absence of bacteria in these mice. Similar to antibiotic-treated mice, germ-free mice exhibited a 1-day delay in initial appearance of infection. Germ-free mice also exhibited delayed RV clearance, likely reflecting their immature intestinal adaptive immune system [11], which is important for RV clearance (Figure 1B) [18]. Thus, delay of RV infection may be a uniform consequence of microbiota ablation. To verify that reduced levels of fecal RV antigen reflected reduced infectivity, we quantitated the level of RV genomes in duodenal lysates by qRT-PCR. Antibiotic treatment resulted in a 10-fold reduction in RV genomes 2 and 3 days after inoculation (Figure 1C), thus confirming that antibiotics reduced RV infectivity. We next measured the ratio of positive sense to negative sense RV strands, which reflects the extent of active RV replication [17]. This parameter did not significantly differ between control and antibiotic-treated mice (Figure 1D), suggesting that microbiota ablation reduced viral entry rather than RV replication.
Figure 1.

Ablation of microbiota retards rotavirus (RV) infectivity. C57BL6 male mice aged 6–8 weeks were treated with ampicillin and neomycin 1 week before oral inoculation with 105 50% shedding doses (SD50) of mouse RV strain EC. A, Feces specimens were collected daily and assayed for RV antigens by enzyme-linked immunosorbent assay (ELISA). B, Male and female germ-free mice aged 6–8 weeks were infected with filter-sterilized RV. Feces specimens were collected daily and were assayed for RV antigen by ELISA. C, Total RNA from the duodenum was prepared, and a lysate of cells from antibiotic-treated mice was probed for NSP3 messenger RNA, representative of the RV genome, by quantitative reverse-transcription polymerase chain reaction (qRT-PCR). D, Duodenal cell lysate was prepared, and each sample was analyzed for ratios of positive-sense to negative-sense strands by single-stranded qRT-PCR. The ratio value positively correlates with RV replication. *P < .05.
Analogous to the case for humans, RV infection in neonatal mice induces secretory diarrhea arising 2–3 days after inoculation and lasting for 3–8 days. To determine whether the reduced RV infectivity resulting from antibiotic treatment impacted RV disease, neonatal mice were administered antibiotics via oral gavage 1 day before and 1 day after inoculation and then inoculated with RV on day 0. Mice were then monitored daily for diarrhea, as indicated by the presence of runny, profuse, yellow-colored feces upon application of light pressure to the abdomen (Figure 2A). Antibiotic treatment resulted in lower daily rates of diarrhea on days 4–8 after inoculation (Figure 2B) and a 34% reduction in total diarrhea incidence (Figure 2C). Among mice that developed diarrhea, those that received antibiotics had diarrhea for approximately 1 day less than untreated mice (Figure 2D). Thus, reducing RV infection via microbiota ablation resulted in reduced the duration of diarrheal disease.
Figure 2.

Antibiotic treatment reduces rotavirus (RV)–induced diarrhea in neonatal mice. C57BL6 pregnant dams were treated with ampicillin and neomycin in drinking water ad libitum 1 week before giving birth, and treatment was continued until offspring were weaned. Offspring were treated with 100 µg of neomycin and 200 µg of ampicillin in 100 µL of water by oral gavage 1 day before and 1 day after inoculation. Six-day-old mice were inoculated with 2 50% diarrhea doses of mouse RV and were visually observed for diarrhea daily. A, Typical RV diarrhea in neonates. B, Daily rates of observable diarrhea on days 0–9 after inoculation. C, Incidence of diarrhea, represented as both a fraction and percentage of total mice inoculated. D, Of the mice that showed evidence of diarrhea, the numbers of days each mouse had diarrhea was averaged. *P < .05.
Absence of Microbiota Results in a More Durable RV-Specific Mucosal Antibody Response
RV infection initiates robust adaptive immunity that clears primary infection and provides protection against future infection [1]. Such infection-induced immunity is the basis of currently used RV vaccines, which are live attenuated viruses. Such protective immunity best correlates with RV-specific fecal IgA, whose levels often parallel those of serum immunoglobulin G (IgG) and IgA, which are typically measured in clinical studies [1, 18]. The adult mouse model of RV infection can be considered a model of RV vaccination in that both are asymptomatic infections that do not result in diarrhea but provide protective immunity [1]. Considering that antibiotics can reduce antibody responses to systemically administered antigens and that reduced infectivity likely reduced exposure to antigen, we hypothesized that antibiotic treatment might reduce RV-specific antibodies [12, 13]. To investigate this possibility, we treated mice with antibiotics 1 week before and up to 11 weeks after inoculation, collected feces and serum weekly, and assayed samples for RV-specific IgG and IgA. Antibiotic treatment did not affect antibody production at early times following RV inoculation but enhanced levels of RV-specific antibodies, particularly serum and fecal IgA, 9 weeks after inoculation and beyond (Figure 3). Such enhancement was observed upon measuring RV immune reactivity at a single dilution of serum or fecal supernatant or by quantitating the titer following a range of dilutions. This increase was specific for RV in that, in accordance with other studies, antibiotic treatment resulted in a modest reduction in the total IgA level (Supplementary Figure 2A and 2B) [11].
Figure 3.

Antibiotic treatment enhances the durability of the antibody response to rotavirus (RV). C57BL6 mice were treated with antibiotics as described and remained on antibiotics until 11 weeks after inoculation. A, C, and E, Serum RV immunoglobulin G (IgG; A), serum RV immunoglobulin A (IgA; C), and fecal RV IgA (E) production, as measured by RV immune reactivity at a single dilution of serum or fecal supernatant, 0, 9, 10, and 11 weeks after inoculation. Findings are reflective of late systemic and mucosal RV antibody responses. B, D, and F, Serum RV IgG (B), serum RV IgA (D), and fecal RV IgA (F) titers, as measured by the sample dilution at which OD450 equaled 0.2 over blank, 11 weeks after inoculation. *P < .05.
An alternate approach to the use of antibiotics to study the microbiota is the use of germ-free mice, although a caveat is that the gut-associated lymphoid tissue that mediates adaptive immunity is lacking in these mice [11]. In accordance with this knowledge and the delayed clearance of RV observed in these mice, germ-free mice exhibited a marked delay in production of fecal anti-RV IgA. Despite the lack of GALT, this impairment of germ-free mice to produce fecal anti-RV IgA was overcome with time. Moreover, analogous to antibiotic-treated mice, germ-free mice exhibited a serum anti-RV antibody response that was initially similar to that of conventional mice but became greater several weeks after inoculation (Figure 4). We next examined the effect of antibiotics on acquisition of RV-specific antibodies following pathogenic (ie, diarrhea-causing) RV infection in neonatal mice. Pathogenic viral infection is typically a strong inducer of adaptive immunity. Yet, despite reducing the duration of RV-induced diarrhea, antibiotic treatment enhanced serum anti-RV IgA production, with the highest titers observed predominantly at later times following infection (Figure 5). Thus, in contrast to our initial prediction, microbiota ablation resulted in enhanced RV-specific systemic and mucosal antibody responses.
Figure 4.
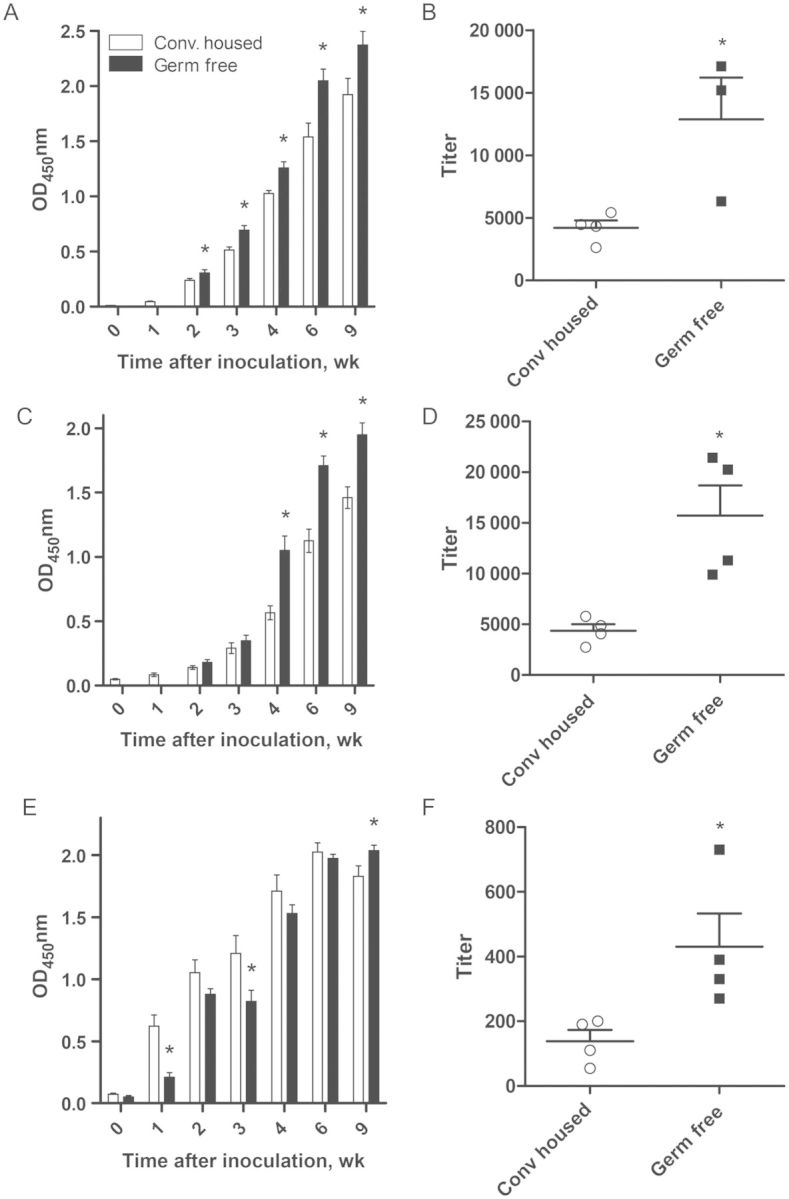
Germ-free mice exhibit enhanced serum antibody response to rotavirus (RV). Germ-free C57BL6 male and female mice were inoculated with filter-sterilized RV, feces and serum were collected weekly up to week 9 after inoculation, and samples were analyzed for the presence of RV antibody by enzyme-linked immunosorbent assay. Mice were monitored by fecal culture for germ-free status weekly until the experiment end point. A, C, and E, Serum RV immunoglobulin G (IgG; A), serum RV immunoglobulin A (IgA; C), and fecal RV IgA (E) production, as measured by RV immune reactivity at a single dilution of serum or fecal supernatant, 0–9 weeks after inoculation, reflective of late systemic and mucosal RV antibody responses. B, D, and F, Serum RV IgG (B), serum RV IgA (D), and fecal RV IgA (F) titers, as measured by the sample dilution at which OD450 equaled 0.2 over blank, at 9 weeks after inoculation. *P < .05. Abbreviation: Conv, conventionally.
Figure 5.

Antibiotic-treated neonatal mice exhibit enhanced serum immunoglobulin A (IgA) production following rotavirus (RV) inoculation. Neonates were treated with antibiotics as described in Materials and Methods and inoculated with RV. Feces and serum specimens were collected at various weeks after inoculation, and samples were probed for RV antibody. A and C, Serum RV immunoglobulin G (IgG; A) and serum RV immunoglobulin A (IgA; C) production, as measured by RV immune reactivity at a single dilution of serum, 4, 6, and 9 weeks after inoculation. B, Serum RV IgG titer, as measured by the sample dilution at which OD450 equaled 0.2 over blank, at 9 weeks after inoculation. D, Serum RV IgA titer, as measured by the serum dilution at which OD450 equals 0.2 over blank, at 9 weeks after inoculation. *P < .05.
Antibiotic enhancement of the duration of the antibody response to RV suggests the possibility of incorporating antibiotic treatment into vaccine campaigns. However, given the negative potential consequence of prolonged antibiotic administration, we next sought to determine whether briefer exposures might also enhance RV antibody generation. Thus, we compared the effects of a 2-week course of antibiotics (from 1 week before to 1 week after inoculation) to control conditions (ie, no antibiotics) and to antibiotic therapy continued throughout the experiment (from 1 week before to 7 weeks after inoculation). Mice receiving only 2 weeks of antibiotics displayed a return of fecal bacterial load within 2 weeks after antibiotic cessation, whereas mice that received antibiotics throughout the experiment continued to exhibit bacterial suppression (Supplementary Figure 1B and 1C). Whereas maximal enhancement of RV-specific antibody responses was observed with maintained antibiotic treatment, a 2-week course was sufficient to significantly enhance serum anti-RV IgG and IgA levels and titers (Figure 6A–D). A further reduced course of antibiotics, lasting from 2 days before to 3 days after RV inoculation, resulted in a trend toward increased fecal and serum anti-RV IgA titers that was not statistically significant (Supplementary Figure 3). We hypothesized that antibiotic-mediated enhancement of the durability of the mucosal antibody response involved more-sustained levels of RV-specific antibody-secreting cells (ASCs). Thus, we next performed ELISPOT analysis to quantitate levels of RV-specific IgA–producing ASCs from small intestinal lamina propria and Peyer's patches. In control mice, inoculation with RV resulted in RV-specific ASCs going from an undetectable level to a level of 4000 cells per million lamina propria and Peyer's patches cells, which then declined by about 40-fold by 7 weeks after inoculation. Antibiotic treatment did not significantly affect the generation of RV-specific IgA–producing ASCs at 2 weeks after inoculation. However, at 7 weeks after inoculation, antibiotic treatment markedly enhanced levels of these cells. The degree of enhancement (about 20-fold) was greatest in mice that received antibiotics throughout the experiment but was nonetheless robust (increase, 10-fold) in mice receiving the 2-week course of antibiotics (Figure 6E).
Figure 6.

Antibiotic treatment results in greater maintenance of rotavirus (RV)–specific antibody producing–cells in intestine. C57BL6 mice were treated with antibiotics as described in Materials and Methods. However, one group continued antibiotic therapy for only 1 week after inoculation (for 2 weeks total), whereas another group of mice continued antibiotic therapy throughout the duration of the experiment (for 8 weeks total). A and C, Serum RV immunoglobulin G (IgG; A) and serum RV immunoglobulin A (IgA; C) generation weekly until 7 weeks after inoculation, reflective of late systemic and mucosal RV antibody responses. B and D, Serum RV IgG titer (B) and serum RV IgA titer (D) as measured by the sample dilution at which OD450 equaled 0.2 over blank, at 7 weeks after inoculation. E, Small intestinal lamina propria (LP) and Peyer's patch (PP) cells were isolated and applied to coated plates at 2 weeks and 7 weeks after inoculation. The concentration of RV-specific, IgA-producing antibody-secreting cells (ASCs) were calculated in each group. *P < .05. Abbreviation: UnTx, untreated.
Impact on Microbiota on the Basal State of Innate Immune Activation May Regulate RV-Specific Adaptive Immunity
We hypothesized that lowering bacterial load through antibiotic treatment is reducing the extent of the basal state of innate immune activation, thus allowing for RV-specific danger signals to more readily activate GALT during infection; this should result in stronger activation of virus-specific cells and the persistence of ASC in the gut. The converse of this hypothesis is that inducing a greater extent of innate immune activation before virus administration might attenuate the antibody response. To test this possibility, mice were exposed to DSS via drinking water, which compromises the integrity of the gut epithelial barrier and results in increased exposure of immune cells to microbiota and its products. Although DSS treatment can result in robust life-threatening colitis, exposure to low levels of DSS, such as 1.0%, results in only modest histopathologic changes in the gut but still induces readily detectable activation of proinflammatory gene expression, which can be monitored by measuring levels of fecal lipocalin-2 [21]. Accordingly, exposure to 1.0% DSS for 1 week did not result in apparent symptoms of colitis but nonetheless induced robust expression of fecal lipocalin-2 (Figure 7A). DSS treatment did not alter the course of RV infectivity (Figure 7B). Moreover, DSS-induced low-grade inflammation did not affect the initial generation of serum RV-specific IgG (Figure 7C and 7D). However, DSS treatment resulted in significantly lower levels of RV-specific fecal and serum IgA from 3 to 9 weeks after inoculation (Figure 7E–H). These results suggest that the state of basal innate immune activation at the time of RV inoculation may modulate the levels of RV-specific antibodies. Accordingly, manipulation of this parameter may be a way to modulate generation of these protective responses.
Figure 7.

Increasing basal immune activation impairs rotavirus (RV)–induced antibody generation. C57BL6 mice were treated with dextran sodium sulfate (DSS) as described in Materials and Methods and inoculated with RV. A, Fecal lipocalin-2 expression was analyzed with enzyme-linked immunosorbent assay. B, Feces specimens were collected daily, and RV antigen was assayed. C, E, and G, Serum and fecal supernatant was obtained, and serum RV immunoglobulin G (IgG; C), serum RV immunoglobulin A (IgA; E), and fecal RV IgA (G) production, as measured by RV immune reactivity at a single dilution of serum or fecal supernatant, was probed up to 9 weeks after inoculation. D, F, and H, Serum RV IgG (D), serum RV IgA (F), and fecal RV IgA (H) titers, as measured by the sample dilution at which OD450 equaled 0.2 over blank, at 9 weeks after inoculation. *P < .05.
DISCUSSION
The gut microbiota is increasingly appreciated as a modulator of numerous infectious and immunologic processes. In many circumstances, the gut microbiota serves to protect the host from infectious disease, such that use of antibiotics results in increased susceptibility to a number of bacterial infections. Moreover, generation of immune responses to some viruses is impaired in mice subjected to microbiota ablation [12, 13]. Thus, we originally hypothesized that elimination of microbiota might increase susceptibility to RV infection. In accordance with the observation that microbiota are essential for development of GALT [11], which generates the fecal IgA that promotes RV clearance [18], germ-free mice exhibited a delayed generation of fecal anti-RV IgA and concomitantly delayed RV clearance. Nonetheless, the overall consequence of microbiota ablation, particularly when achieved via the more clinically relevant approach of antibiotic treatment, seemed to benefit the host. Specifically, treatment with antibiotics resulted in a delay in RV infection and a reduction in total infectivity. Such reduction in infection was associated with, and likely resulted in, a substantial reduction in the incidence and duration of RV-induced diarrhea.
The mechanism by which ablation of microbiota retards RV infectivity is not clear but may be similar to the case for poliovirus, reovirus, and mouse mammary tumor virus, which were all recently reported to infect less efficiently in the absence of a microbiota [14, 23]. In such cases, bacteria-derived LPS was observed to bind the virus and facilitate its entry [14]. While our experiments have not, to date, demonstrated a direct role for bacterial ligands in promoting RV entry, antibiotics had a clear effect on viral loads with little effect on RV replication, which suggests that a similar paradigm might be operative. Another possibility is that absence of bacteria may be lessening the expression of RV receptors needed for viral entry. Indeed, it is known that microbiota promotes expression of certain Toll-like receptors [24, 25], which have been postulated to facilitate enteric virus entry [14, 23].
The reduction in RV infectivity upon antibiotic treatment likely resulted in reduced exposure of the immune system to RV antigens, which we predicted would reduce the antibody response to the virus. In contrast, antibiotics enhanced the antibody response, particularly resulting in one that was more durable. Such higher titers at later time points would likely offer more lasting and broader protection against subsequent infection by heterologous RV strains. The mechanism mediating this effect is not entirely clear but might reflect that so-called RV danger signals provide a greater stimulatory effect on the antigen presenting cell–lymphocyte interactions when such signals occur in the context of reduced basal/background signaling. In support of this possibility, microbiota ablation results in reduced fecal levels of the immune inflammatory markers such as lipocalin-2 (data not shown) and relm-β [26], where use of DSS to increase the state of immune activation before RV infection resulted in a less durable antibody response. In this context, we speculate that failure of RV vaccines to consistently elicit strong antibody responses in developing countries may reflect altered microbiota composition, possibly enriched with pathobionts that result in chronic immune activation upon RV vaccine administration. Another possibility is that microbiota ablation reduces regulatory T-cell functioning, allowing for heightened inflammatory T cell responses [27]. Deciphering such complex mechanisms remains an important research challenge.
Presuming the RV mouse model is translatable to humans, our study has several implications on antibiotic use in the context of RV infection and vaccination. First, given that many clinical diagnoses of infections are based on symptoms rather than laboratory-based detection of a specific pathogen, our results suggest that decisions to prescribe antibiotics need not fear that this would increase susceptibility to RV. Moreover, our results suggest that administration of RV vaccines need not be postponed in the event that an individual is presently taking antibiotics. Rather, our study suggests this might be an ideal time to administer RV and, perhaps, other orally administered vaccines. Furthermore, the low cost of antibiotics may justify their selective use in managing RV disease. Perhaps an active outbreak of RV infection might be managed by administration of antibiotics quickly followed by RV vaccination. This action might reduce the severity of arising infections and increase the vaccine efficacy. Alternatively, incorporating antibiotics into a broader RV vaccine campaign might increase seroconversion rates. Indeed, on the basis of our proposed mechanism by which antibiotics promote RV antibodies, we would envisage the effect might be greatest in areas with low seroconversion rates, such as Malawi, that may have higher levels of basal immune activation. Even a modest increase in vaccine efficacy might result in dramatic reduction in the societal disease burden due to herd immunity. In the event that our observations in mice prove to be relevant in humans, careful consideration of the detrimental consequences of antibiotic use, including increased susceptibility to bacterial infections [28] and promotion of antibiotic resistant bacteria [29], is warranted before large-scale deployment of antimicrobial agents to manage the RV disease burden. In light of these concerns, development of approaches to selectively manipulate the microbiota may provide a better means to safely enhance immune responses to RV vaccines. Such selective manipulations might take the form of more specifically acting antibiotics and/or administration of probiotics. Indeed, administration of a probiotic increased seroconversion rates in a cohort of RV-vaccinated Finnish infants [30]. Thus, while mechanistic understanding and optimization require additional experimentation, the approach of manipulating the microbiota may be a useful strategy to combat RV.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Caitlin Bohannon, Sarah Blutt, Daniel Rios, and Jason Oh, for technical advice; and Rama Amara, Timothy Denning, and Ifor Williams, for helpful discussion.
Financial support. This work was supported by the National Institutes of Health (grant DK083890).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Greenberg HB, Estes MK. Rotaviruses: from pathogenesis to vaccination. Gastroenterology. 2009;136:1939–51. doi: 10.1053/j.gastro.2009.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel MM, Glass R, Desai R, Tate JE, Parashar UD. Fulfilling the promise of rotavirus vaccines: how far have we come since licensure? Lancet Infect Dis. 2012;12:561–70. doi: 10.1016/S1473-3099(12)70029-4. [DOI] [PubMed] [Google Scholar]
- 3.Rotavirus. Available at: http://www.cdc.gov/rotavirus/about/symptoms.html. Accessed 17 December 2013.
- 4.Rotavirus surveillance—worldwide, 2009. Morbid Mortal Wkly Rep. 2011;60:514–6. [PubMed] [Google Scholar]
- 5.Babji S, Kang G. Rotavirus vaccination in developing countries. Curr Opin Virol. 2012;2:443–8. doi: 10.1016/j.coviro.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Holmgren J, Svennerholm AM. Vaccines against mucosal infections. Curr Opin Immunol. 2012;24:343–53. doi: 10.1016/j.coi.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Madhi SA, Cunliffe NA, Steele D, et al. Effect of human rotavirus vaccine on severe diarrhea in African infants. N Engl J Med. 2010;362:289–98. doi: 10.1056/NEJMoa0904797. [DOI] [PubMed] [Google Scholar]
- 8.Vesikari T, Matson DO, Dennehy P, et al. Safety and efficacy of a pentavalent human-bovine (WC3) reassortant rotavirus vaccine. N Engl J Med. 2006;354:23–33. doi: 10.1056/NEJMoa052664. [DOI] [PubMed] [Google Scholar]
- 9.Patel M, Shane AL, Parashar UD, Jiang B, Gentsch JR, Glass RI. Oral rotavirus vaccines: how well will they work where they are needed most? J Infect Dis. 2009;200(Suppl 1):S39–48. doi: 10.1086/605035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper PJ, Chico ME, Losonsky G, et al. Albendazole treatment of children with ascariasis enhances the vibriocidal antibody response to the live attenuated oral cholera vaccine CVD 103-HgR. J Infect Dis. 2000;182:1199–206. doi: 10.1086/315837. [DOI] [PubMed] [Google Scholar]
- 11.Sommer F, Backhed F. The gut microbiota—masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–38. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 12.Ichinohe T, Pang IK, Kumamoto Y, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A. 2011;108:5354–9. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abt MC, Osborne LC, Monticelli LA, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 2012;37:158–70. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuss SK, Best GT, Etheredge CA, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–52. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carvalho FA, Nalbantoglu I, Ortega-Fernandez S, et al. Interleukin-1beta (IL-1beta) promotes susceptibility of Toll-like receptor 5 (TLR5) deficient mice to colitis. Gut. 2012;61:373–84. doi: 10.1136/gut.2011.240556. [DOI] [PubMed] [Google Scholar]
- 16.Blutt SE, Warfield KL, O'Neal CM, Estes MK, Conner ME. Host, viral, and vaccine factors that determine protective efficacy induced by rotavirus and virus-like particles (VLPs) Vaccine. 2006;24:1170–9. doi: 10.1016/j.vaccine.2005.08.090. [DOI] [PubMed] [Google Scholar]
- 17.Fenaux M, Cuadras MA, Feng N, Jaimes M, Greenberg HB. Extraintestinal spread and replication of a homologous EC rotavirus strain and a heterologous rhesus rotavirus in BALB/c mice. J Virol. 2006;80:5219–32. doi: 10.1128/JVI.02664-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blutt SE, Miller AD, Salmon SL, Metzger DW, Conner ME. IgA is important for clearance and critical for protection from rotavirus infection. Mucosal Immunol. 2012;5:712–9. doi: 10.1038/mi.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vijay-Kumar M, Aitken JD, Kumar A, et al. Toll-like receptor 5-deficient mice have dysregulated intestinal gene expression and nonspecific resistance to Salmonella-induced typhoid-like disease. Infect Immun. 2008;76:1276–81. doi: 10.1128/IAI.01491-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–94. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 21.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng N, Franco MA, Greenberg HB. Murine model of rotavirus infection. Adv Exp Med Biol. 1997;412:233–40. doi: 10.1007/978-1-4899-1828-4_35. [DOI] [PubMed] [Google Scholar]
- 23.Kane M, Case LK, Kopaskie K, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334:245–9. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lundin A, Bok CM, Aronsson L, et al. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol. 2008;10:1093–103. doi: 10.1111/j.1462-5822.2007.01108.x. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Devkota S, Musch MW, et al. Regional mucosa-associated microbiota determine physiological expression of TLR2 and TLR4 in murine colon. PLoS One. 2010;5:e13607. doi: 10.1371/journal.pone.0013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang ML, Shin ME, Knight PA, et al. Regulation of RELM/FIZZ isoform expression by Cdx2 in response to innate and adaptive immune stimulation in the intestine. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1074–83. doi: 10.1152/ajpgi.00442.2004. [DOI] [PubMed] [Google Scholar]
- 27.Bollrath J, Powrie FM. Controlling the frontier: Regulatory T-cells and intestinal homeostasis. Semin Immunol. 2013;25:352–7. doi: 10.1016/j.smim.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 28.Dupont HL. Diagnosis and management of Clostridium difficile infection. Clin Gastroenterol Hepatol. 2013;11:1216–23. doi: 10.1016/j.cgh.2013.03.016. ; quiz e73. [DOI] [PubMed] [Google Scholar]
- 29.Lynch JB. Multidrug-resistant Tuberculosis. Med Clin North Am. 2013;97:553–79. doi: 10.1016/j.mcna.2013.03.012. ix-x. [DOI] [PubMed] [Google Scholar]
- 30.Isolauri E, Joensuu J, Suomalainen H, Luomala M, Vesikari T. Improved immunogenicity of oral D×RRV reassortant rotavirus vaccine by Lactobacillus casei GG. Vaccine. 1995;13:310–2. doi: 10.1016/0264-410x(95)93319-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.