

Abstract
The development of adenocarcinoma of the lung is believed to proceed from in situ disease (adenocarcinoma in situ, AIS) to minimally invasive disease with prominent lepidic growth (minimally invasive adenocarcinoma, MIA), then to fully invasive adenocarcinoma (AD), but direct evidence for this model has been lacking. Because some lung adenocarcinomas show prominent lepidic growth (AD-L), we designed a study to address the lineage relationship between the lepidic (noninvasive) component (L) and the adjacent nonlepidic growth component representing invasive disease within individual tumors. Lineage relationships were evaluated by next-generation DNA sequencing to define large genomic rearrangements in microdissected tissue specimens collected by laser capture. We found a strong lineage relationship between the majority of adjacent lepidic and invasive components, supporting a putative AIS–AD transition. Notably, many rearrangements were detected in the less aggressive lepidic component, although the invasive component exhibited an overall higher rate of genomic rearrangement. Furthermore, a significant number of genomic rearrangements were present in histologically normal lung adjacent to tumor, but not in host germline DNA, suggesting field defects restricted to zonal regions near a tumor. Our results offer a perspective on the genetic pathogenesis underlying adenocarcinoma development and its clinical management.
Introduction
The development of cancer is thought to be a multistep process characterized by sequential molecular changes. The molecular changes associated with such a multistep process are poorly understood in the evolution of lung adenocarcinoma. Recent histopathologic updates (1) have clarified the definition of bronchioloalveolar carcinoma (BAC) and invasive adenocarcinoma (AD). The new classification introduces the concept and definition of adenocarcinoma in situ (AIS), previously known as BAC, for small solitary adenocarcinomas (≤ 3 cm) with pure lepidic (noninvasive) growth, and minimally invasive adenocarcinoma (MIA) for small solitary AD (≤ 3 cm) with predominant lepidic growth and ≤ 5-mm invasion. ADs with predominant lepidic growth pattern greater than 3 cm or with invasion > 5 mm are classified as lepidic predominant AD (LPA) and shown to have better prognosis than other types of invasive AD. This definition implies that lepidic growth within an invasive tumor represents an in situ component and a progression from in situ disease to invasion within a given tumor. Further evidence for a hypothesized AIS–AD transition comes from epidemiologic data and the coexistence of some, or all, of these lesions within a single patient. Current genetic evidence demonstrating AIS as a precursor of AD is sparse, and based largely on the sharing of genetic anomalies such as k-ras and p53 mutations, loss of p16 and loss of heterozygosity for 3p (2). AIS and MIA define patients with almost 100% disease-specific survival with complete surgical resection, clearly differentiating them from patients with stage I AD, in which up to 30% of patients will recur and die from their disease (1, 3). Given the dramatic differences in clinical outcome across this spectrum of lung adenocarcinoma, it is clear that these newly defined pathologic intermediates represent clinically relevant entities, and present an opportunity to understand events in disease progression.
Molecular understanding of a potential AIS–MIA–AD transition, characterized by accumulation of genomic alteration/mutations in progression of the disease, has been hampered by a number of challenges. One is the difficulty in obtaining pathologically well-characterized lesions in sufficient quantity for study. Although a number of large-scale sequencing efforts have been applied to non–small cell lung cancer (NSCLC; refs. 4–7), none to date has focused on precursor lesions in the progression of lung adenocarcinoma. As a consequence, AIS and the development of lung adenocarcinoma remain poorly understood.
Whole-genome sequencing of several cancers has revealed that solid tumors harbor many somatic chromosomal rearrangements and thousands of single-nucleotide variations (SNV). Both of these types of alterations can be used to investigate the commonality of two phenotypically different parts of a tumor from the same individual (8, 9). The vast array of single-nucleotide polymorphisms (SNP) within the human genome and also the extensive numbers of background SNVs within the local environment of a tissue makes the derivation of tumor specific, somatic SNVs very challenging and can complicate lineage analysis. In contrast, sequencing data to date have demonstrated that the probability of detecting an identical chromosomal breakpoint in two unrelated tumors is extremely unlikely. Even for recurrent chromosomal rearrangements affecting specific genes, such as those between ERG and TMPRSS2 in >50% of patients with prostate cancer (10), or between EML4 and ALK in lung adenocarcinoma (11), very rarely or never will identical breakpoints be shared between different tumors or patients. Recognizing that the term in situ is recommended to be only used in the restricted setting of AIS as defined above, we hypothesized that the lepidic growth of LPA represents an in situ component of an invasive AD and would show common (lineage relationship) and diverse genomic alterations with the invasive component as a marker of progression. To find unique tumor-associated genomic alterations and track lineage relationships between adjacent invasive and lepidic components of AD, we therefore focused on chromosomal rearrangements, which can be readily obtained by a mate-pair (MP) library approach and next-generation DNA sequencing.
Materials and Methods
Case selection
Hematoxylin and eosin (H&E)–stained lung tumor frozen sections with a diagnosis of LPA, were reviewed by a pulmonary pathologist (M.C. Aubry). Fourteen cases, where tumor comprised at least 80% of the histologic section and the lepidic component varied between 40% and 80%, were selected for this study. All non-lepidic components of the adenocarcinomas were considered invasive. Invasive versus lepidic components needed to be easily distinguishable by routine light microscopy to allow for laser capture micro-dissection (LCM) of each component separately with no contamination. H&E-stained adjacent normal lung sections were also reviewed by the pulmonary pathologist to confirm the absence of tumor in these sections.
LCM frozen tissue specimens
Frozen lung tissue sections cut to 10-μm size and pure cell populations of lepidic and invasive components were isolated using the Arcturus PixCell II microscope and CapSure Macro LCM Caps (Arcturus; LCM 0211). Associated histologically nonneoplastic (aN) tissue was also collected by LCM from adjacent benign tissue blocks associated with each case. Whole-genome amplification was performed directly on LCM-captured cells using a single-step procedure (9). LCM cells were incubated for 10 minutes in 0.5 × REPLI-g D2 buffer (6.5 μL; Qiagen) and then in REPLI-g Stop Solution (3.5 μL). Cells were then mixed with REPLI-g Mini Kit Master Mix (40 μL) and incubated at 30° C for 16 hours. Four individual 50-μL whole-genome amplification (WGA) reactions were pooled for each sample. DNA was quantified by Quant-iT PicoGreen analysis (Invitrogen; P7581) and qualitative multiplex PCR was performed (Sigma-Aldrich; P0982). Germline DNA was also extracted from patient blood when a clinical sample was available in the Mayo lung tissue depository.
Next-generation sequencing
MP libraries were assembled from WGA DNA according to a previously published protocol (9) using the Illumina Mate Pair Library Preparation Kit v2 (Illumina, PE-112-2002). Briefly, WGA DNA (10 μg) was fragmented to 3 to 5 kb and DNA intramolecular circles assembled by ligation following biotin end labeling. Additional fragmentation to 350 to 650 bp was followed by immobilization of biotinylated terminal fragments on M-280 streptavidin beads (Dynal) and assembly of adapter-flanked Illumina indexed paired end libraries using Illumina adapters (Illumina). Two multiplexed libraries were loaded per lane of an Illumina flow cell and sequenced to 101 × 2 paired-end reads on an Illumina HiSeq. Base calling was performed using Illumina Pipeline v1.5.
Data analysis
Bioinformatics protocols to rapidly and efficiently process next-generation sequencing MP data using a 32-bit binary indexing of the Hg19 reference genome, to which consecutive 32-bit binary sequences from associated MP reads are aligned, have been previously published from our laboratory (12, 13). The algorithm maps both MP reads successively to the whole genome, selecting reads <15-kb apart allowing up to 10 mismatches, with the lowest cumulative mismatch count sent to the output. Discordant MPs mapping >15-kb apart or in different chromosomes were selected for further analysis and associated fragments were clustered together. Replication was calculated for each chromosome and replicate read-pairs were removed. Bridged coverage was calculated as the sum of the fragment lengths (distance between read1 and read2) of correctly mapping MP and paired-end reads, divided by the mappable chromosome size. Base coverage was calculated from the total number of mappable reads, multiplied by the read length and divided by the mappable chromosome size. Coverage was calculated per chromosome and the average was found using chromosomes 1 to 22.
Algorithmic filters to determine lineage relationships were set to reduce false positives (FP) and false negatives (FN). Namely, the lowest limit of MP numbers to call an event for lineage (also referred to as the number of associates) was raised to 7. This lower limit was set by analyzing data from normal samples as it was observed that the FP rate was practically zero with number of associates of seven and above and when a mask of breakpoints was used to eliminate common variants (9, 12, 13, 14) and discordant MPs that clustered because of experimental or algorithmic errors. Furthermore, the combined nucleotide distance to cluster associates to an event was set to 3,000, thereby eliminating closely related but not identical breakpoints from being called as shared. In addition, breakpoints near gaps of reference genome sequence were also eliminated. After initial exclusion of all events with less than three associates, the number of FP events eliminated during filtering ranged from 1,000 to 10,000 events per case, with a median of 5,387. The variation in this number was due in part to sequencing depth of each sample, which correlated well with the number of FP (R2 = 0.63). FN rate was estimated to be less than 15% (dictated by the incompleteness of the reference genome and by regions that are difficult to map). Using a probability statistic, we estimated that the probability of relatedness between two samples is less than 0.15n when the expected number of shared breakpoints is n, and no shared events are found.
Validation of genomic rearrangements
MP sequence reads were mapped to the human genome and primers spanning the fusion junctions were used in validation PCRs (25 μL, 50 ng template, 35 cycles) using the Easy-A High-Fidelity polymerase (#600404; Stratagene). A mixed population human Genomic DNA control (gC) was used (G304A; Promega). Validations on blood-extracted germline DNA were performed using identical conditions to the WGA DNA. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control PCRs was performed using primers, forward: ACAGTCCATGCCATCACTGC, and reverse: GCTTGACAAAGTGGTCGTTG.
Results
To investigate genetic similarities and differences between adjacent lepidic and invasive components, pure histologically distinct cell populations were isolated from fresh frozen lung cancer tissues using LCM. Fourteen cases of LPA with lepidic growth of varying from 40% to 80% on a given histologic frozen section were selected and LCM of the lepidic and invasive components was performed separately (Fig. 1A). WGA was performed directly from captured cells (Supplementary Fig. S1) for MP sequencing. An average of 84 million mappable reads was obtained per sample (Supplementary Table S1). Binary algorithms developed in our group and previously demonstrated to efficiently detect high-confidence large chromosomal abnormalities from MP sequencing data, including translocations, deletions, and insertions, were used in this study (9, 12–14). Figure 1B presents the number of large genomic rearrangements for each case divided into shared or unique events for an individual tumor mass. Numbers of detected genomic breakpoints varied greatly between the AD cases studied, ranging from 7 to 349 per case (Fig. 1B and Table 1). Only case 12 failed to reveal any shared identical genetic breakpoints between both the lepidic and invasive components. Specifically, 22 of the 28 components sequenced demonstrated at least 20% genetic breakpoints in common between lepidic and invasive components (Table 1). Twelve samples presented more than 50% commonality with their adjacent component. One case (Lu18) presented with the highest number of shared events (107) encompassing approximately 70% of the total breakpoints for that tissue. In addition to case 18, cases Lu11, 7, 14, 20, and 8 shared >40% of their detected breakpoints in both components (Table 1). Commonality within the lepidic and invasive components of cases Lu5 and Lu8 was less conclusive due to low numbers of total genomic breakpoints within those tissues. Interestingly, case Lu12 presented with a disproportionate number of genetic breakpoints observed solely in the lepidic component, none of which were detected as shared events within the invasive component (Fig. 1B). However, the majority of these events (>90%) stem from two extensive chromosomal catastrophes (15) on chromosomes 1 and 8 (Supplementary Table S2). Ignoring this case (Lu12), an average of 42 and 37 breakpoints was observed in the invasive and lepidic components, respectively. Independent agglomerative hierarchical clustering displayed a high degree of commonality within cases, which is displayed in the dendogram of Fig. 1C. In 13 of the 14 cases, the adjacent invasive and lepidic components are observed to cluster together. Only the components of case Lu12 failed to indicate commonality with another sample in the study (Fig. 1C).
Figure 1.
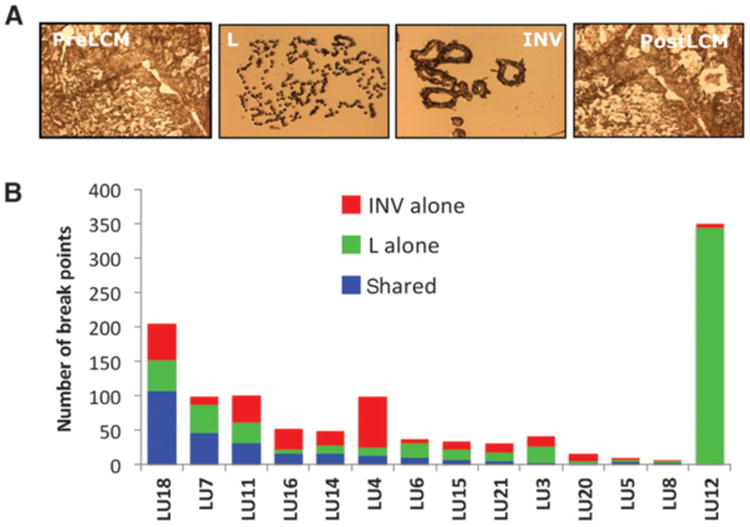
LCM and case mutational status. A, images of frozen sections before (PreLCM) and after (PostLCM) cell capture and the cell populations captured on the LCM caps for invasive (INV) and lepidic (L) components. B, numbers of large genomic rearrangements observed per case divided into lepidic and invasive shared (blue bars), invasive alone (red bars), and lepidic alone (green bars).
Table 1. Distribution of breakpoints between lepidic/invasive lesions and smoking status.
| Lepidic | Invasive | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Total # of eventsa | % Shared eventsb | Total # of eventa | % Shared eventsb | Total # of unique events/casec | Smoking statusd | |
| Lu12 | 344 | 0.0% | 5 | 0.0% | 349 | Former × 7 years, unknown packs/year |
| Lu18 | 151 | 66.5% | 161 | 70.9% | 205 | Never smoker |
| Lu11 | 61 | 44.3% | 70 | 50.8% | 100 | Never smoker |
| Lu7 | 87 | 79.3% | 58 | 52.9% | 99 | Former × 24 years, 15 packs/years |
| Lu4 | 22 | 33.3% | 45 | 68.2% | 98 | Never smoker |
| Lu16 | 25 | 15.1% | 86 | 52.0% | 52 | current 75–100 packs/year |
| Lu14 | 27 | 40.5% | 37 | 55.6% | 49 | Former × 40 years, 5–10 packs/year |
| Lu3 | 26 | 11.8% | 17 | 7.7% | 41 | Former × 45 years 50 packs/year |
| Lu6 | 22 | 38.9% | 18 | 31.8% | 36 | Never smoker |
| Lu15 | 30 | 62.5% | 16 | 33.3% | 33 | Former × 17 years, 35 packs/year |
| Lu21 | 17 | 27.8% | 18 | 29.4% | 30 | Former × 25 years 8 packs/year |
| Lu20 | 7 | 66.7% | 6 | 57.1% | 16 | Former × 11 years, 55 packs/year |
| Lu5 | 5 | 8.3% | 12 | 20.0% | 9 | Current 80 packs/year |
| Lu8 | 5 | 50.0% | 4 | 40.0% | 7 | Former × 19 years, 30 packs/year |
Total numbers of breakpoint events detected in each individual lepidic and invasive lesion.
The percentage shared with the adjacent lesions.
Total number of unique breakpoint events per case counting shared events as 1.
Current smoking status of patient together with the number of years since quitting and the number of packed smoked per year.
The total number of rearrangements and the number of shared rearrangements did not correlate with the smoking history (Table 1). For example, cases Lu18, L11, and Lu4 were among cases with the highest numbers of unique and common genomic breakpoints between the lepidic and invasive components, and were from never-smoker patients. Conversely, case Lu5, with just nine unique breakpoints, was a current smoker with an 80-pack year history.
Although no identical breakpoints were observed across all patients, a number of broader genomic loci were recurrently hit in the 14 cases studied. Fifty-six localized gene regions hit by rearrangement in at least two cases in this study are presented in Table 2. Several of these genes have major functions in cellular processes involved in cancer development, with four of the genes; ALK, NCOA2, WIF1, and EBF1, listed in the COSMIC census database of commonly mutated driver genes in cancer. The well reported ALK-EML4 fusion was observed in just one case (case Lu6), with an additional ALK intragenic rearrangement observed in case Lu3 (Supplementary Table S2). Other common breakpoints involving genes linked with cancer development included a mitotic spindle checkpoint gene (MAD1L1), cadherin 4 (CDH4), dihydropyrimidine dehydrogenase (DPYD), a proprotein convertase (PCSK2), and two protein tyrosine phosphatases, PTPRT and PTPRR. Other cosmic census-listed genes hit in just single cases included RET, CDNK2A, NF1, GPHN, ABL2, JAK1, PCM1, NSD1, and BCL2 (Supplementary Table S2).
Table 2. Recurrently hit genes.
| GENE NAMES | GENE LOCI | LU3 | LU4 | LU5 | LU6 | LU7 | LU8 | LU11 | LU12 | LU14 | LU15 | LU16 | LU18 | LU20 | LU21 | # Cases | # Samples | No. L | No. INV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HTR4 | 5q32d | 3 | 3 | 1 | 2 | ||||||||||||||
| CSMD1 | 8p23.2d | 3 | 4 | 3 | 1 | ||||||||||||||
| FAM19A2 | 12q14.1d | 3 | 5 | 2 | 3 | ||||||||||||||
| SRGAP1 | 12q14.2b | 3 | 5 | 2 | 3 | ||||||||||||||
| NUP107 | 12q15b | 3 | 4 | 2 | 2 | ||||||||||||||
| MACROD2 | 20p12.1d | 3 | 4 | 3 | 1 | ||||||||||||||
| COL24A1 | 1p22.3d | 2 | 3 | 2 | 1 | ||||||||||||||
| DPYD | 1p21.3b | 2 | 3 | 2 | 1 | ||||||||||||||
| COL11A1 | 1p21.1d | 2 | 2 | 2 | 0 | ||||||||||||||
| CELSR2, PSRC1 | 1p13.3b | 2 | 3 | 2 | 1 | ||||||||||||||
| ALK | 2p23.2a | 2 | 3 | 1 | 2 | ||||||||||||||
| CACNA2D3 | 3p21.1a | 2 | 2 | 1 | 1 | ||||||||||||||
| VPS8 | 3q27.2a | 2 | 2 | 0 | 2 | ||||||||||||||
| ZDHHC11 | 5p15.33 | 2 | 3 | 1 | 2 | ||||||||||||||
| LOC100132536 | 5p15.33 | 2 | 2 | 2 | 0 | ||||||||||||||
| BRD9, TRIP13 | 5p15.33 | 2 | 3 | 2 | 1 | ||||||||||||||
| LOC728613 | 5p15.33 | 2 | 4 | 2 | 2 | ||||||||||||||
| CTNND2 | 5p15.2d | 2 | 3 | 2 | 1 | ||||||||||||||
| CDH10 | 5p14.2a | 2 | 4 | 2 | 2 | ||||||||||||||
| SPEF2 | 5p13.2c | 2 | 2 | 0 | 2 | ||||||||||||||
| RAB3C | 5q11.2h | 2 | 2 | 1 | 1 | ||||||||||||||
| EBF1 | 5q33.3c | 2 | 2 | 0 | 2 | ||||||||||||||
| ODZ2 | 5q34f | 2 | 2 | 0 | 2 | ||||||||||||||
| ATXN1 | 6p22.3f | 2 | 2 | 0 | 2 | ||||||||||||||
| MAD1L1 | 7p22.3 | 2 | 2 | 1 | 1 | ||||||||||||||
| SDK1 | 7p22.2b | 2 | 3 | 1 | 2 | ||||||||||||||
| SP4 | 7p15.3e | 2 | 2 | 0 | 2 | ||||||||||||||
| IGF2BP3 | 7p15.3c | 2 | 3 | 2 | 1 | ||||||||||||||
| DPY19L2P1 | 7p14.2a | 2 | 3 | 1 | 2 | ||||||||||||||
| ELMO1 | 7p14.2a | 2 | 2 | 1 | 1 | ||||||||||||||
| C7orf10 | 7p14.1c | 2 | 3 | 1 | 2 | ||||||||||||||
| NCOA2 | 8q13.3a | 2 | 2 | 2 | 0 | ||||||||||||||
| COL22A1 | 8q24.23c | 2 | 2 | 2 | 0 | ||||||||||||||
| GABBR2 | 9q22.33b | 2 | 2 | 1 | 1 | ||||||||||||||
| PALM2 | 9q31.3a | 2 | 2 | 1 | 1 | ||||||||||||||
| CCNT1 | 12q13.11c | 2 | 3 | 2 | 1 | ||||||||||||||
| LIMA1 | 12q13.12b | 2 | 3 | 1 | 2 | ||||||||||||||
| WIBG, DGKA | 12q13.2c | 2 | 3 | 2 | 1 | ||||||||||||||
| MYL6, SMARCC2 | 12q13.2c | 2 | 3 | 2 | 1 | ||||||||||||||
| PPM1H | 12q14.1d | 2 | 4 | 2 | 2 | ||||||||||||||
| XPOT | 12q14.2b | 2 | 4 | 2 | 2 | ||||||||||||||
| GNS | 12q14.3a | 2 | 2 | 1 | 1 | ||||||||||||||
| LOC100289646 | 12q14.3a | 2 | 3 | 1 | 2 | ||||||||||||||
| WIF1 | 12q14.3a | 2 | 2 | 2 | 0 | ||||||||||||||
| MSRB3 | 12q14.3a | 2 | 4 | 2 | 2 | ||||||||||||||
| GRIP1 | 12q14.3c | 2 | 4 | 2 | 2 | ||||||||||||||
| CPSF6 | 12q15c | 2 | 4 | 2 | 2 | ||||||||||||||
| PTPRR | 12q15d | 2 | 4 | 2 | 2 | ||||||||||||||
| LOC100128674 | 12q21.1c | 2 | 4 | 2 | 2 | ||||||||||||||
| ATL1 | 14q22.1a | 2 | 2 | 1 | 1 | ||||||||||||||
| SAMD4A | 14q22.2b | 2 | 3 | 2 | 1 | ||||||||||||||
| MEIS2 | 15q14c | 2 | 3 | 1 | 2 | ||||||||||||||
| PCSK2 | 20p12.1a | 2 | 2 | 1 | 1 | ||||||||||||||
| PTPRT | 20q12d | 2 | 2 | 1 | 1 | ||||||||||||||
| CDH4 | 20p13e | 2 | 2 | 1 | 1 | ||||||||||||||
| TTC28 | 22q12.1c | 2 | 2 | 0 | 2 |
NOTE: Genes hit in more than one of the 14 cases are listed together with the gene locus and number of cases and samples. Cases hit are presented as lepidic alone (green), invasive alone (red), or shared (blue).
We examined the distribution of high confidence breakpoints for all 28 samples across the human genome (Fig. 2A). Although several regions stand out with high densities of breakpoints, often these originate from a single case. Specifically, the high-density region on chromosome 1 restricted to an approximately 35-Mb region (1p13.3b–1p31b), represents the extensive catastrophe observed solely in sample Lu12 lepidic (Fig. 2B). Similarly, the majority of events across chromosome 8 also originates from Lu12 lepidic, but in this instance is dispersed across the entire chromosome (Fig. 2B). Chromosome 12 was the most frequently rearranged within the 14 cases and the majority of events were shared between lepidic and invasive components, with a frequency of approximately 0.13 breakpoints per Mb DNA per case (Supplementary Fig. S2). Chromosomes 5, 7, 16, and 20 were similarly affected with even frequencies between the two adjacent components, but at reduced frequencies of around 0.04 breakpoints per Mb DNA per case. The most common breakpoint region was located on the q-arm of chromosome 12 (q13.11 to q21.2), with breakpoints present in 10 of the 14 cases (Fig. 2C). This region was also a site of common chromosomal catastrophe with three cases (Lu7, Lu11, and Lu18) seeing large numbers of breakpoints in this region and an additional three cases (Lu3, Lu14, and Lu16) with lesser multiple localized breakpoints (Fig. 2C). Other common breakpoint regions included 20q13.3, which hit multiple times in cases Lu15, Lu11, and Lu21 (Fig. 2D), and the p-arm of chromosome 7, which hit in 8 cases, 5 of which (Lu7, Lu16, Lu18, Lu4, and Lu21) presented significant chromosomal catastrophe (Fig. 2E). Other unique sites of catastrophe were located in the p-arm of chromosome 5 for Lu18 lepidic/invasive, 1q31–1q41 in Lu14 invasive, 11p15 of Lu18 lepidic/invasive, around the mid q-arm of chromosome 16 in case Lu4 lepidic/invasive, and chromosome 5 q-arm in Lu4 invasive (Supplementary Fig. S3). The majority of these catastrophe events were shared in both lepidic and invasive components of the AD tumors. Significantly, the cases with the most shared events between lepidic and invasive (Lu18, Lu7, Lu11, Lu16, Lu14, Lu4, Lu6, and Lu15) each presented with shared catastrophes between the lepidic and invasive components (Supplementary Fig. S3). Cases Lu5 and Lu8 presented no regions of chromosomal catastrophe, consistent with the small numbers of breakpoints determined in these cases. To further study the location of common breakpoints and regions of chromosomal catastrophe, we also contrasted the breakpoints from all samples with those of reported chromosomal fragile sites (16). No significant overlap was seen between the breakpoints predicted in these 14 cases and the known common chromosomal fragile sites (Fig. 2A–E).
Figure 2.
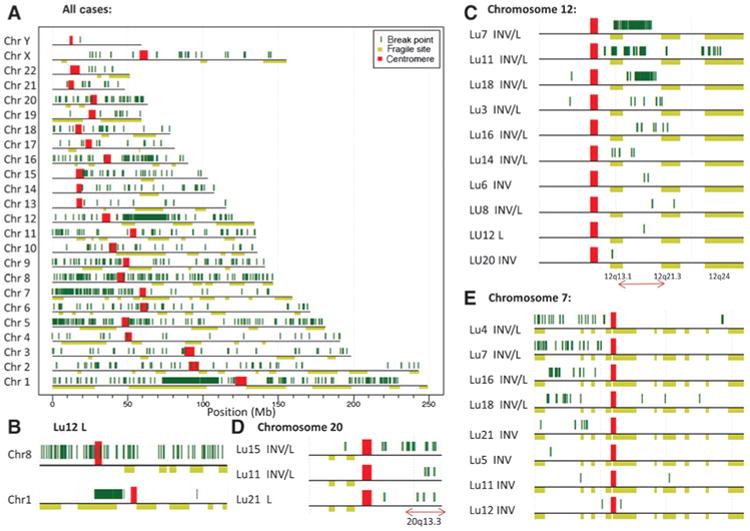
Mapping of breakpoints to the human genome. A, all breakpoints (dark green vertical lines) for the 28 samples mapping to each chromosome. Chromosomes are presented p to q arms (left to right) with the centromeres marked in red. Horizontal light green bars below each chromosome mark predicted chromosomal fragile sites described in ref. 16. B, breakpoints hitting chromosomes 1 and 8 for Lu12 lepidic. C–E, cases with breakpoints in common regions of chromosomes 12, 20, and 7 (respectively). INV, invasive; L, lepidic.
PCR validations were performed focusing on a selection of predicted rearrangements present in both the lepidic and invasive components of selected cases (Fig. 3A and Supplementary Fig. S4A). Events faithfully validated in tissues from both components in all cases studied, yielding identical breakpoints in associated tissues after Sanger sequencing of PCR bands. However, multiple genomic rearrangements were also present in adjacent histologically normal (aN) tissue on validation studies. Germline variation in cases where blood DNA was available only accounted for three small intrachromosomal deletions (Fig. 3B). An identical PCR band in both the lepidic and invasive components, which is absent from aN tissue, was seen in at least one validated event in 7 of the 14 cases studied (Lu7, Lu11, Lu4, Lu14, Lu21, Lu3, and Lu20; Fig. 3A). For two additional cases, LU16 and LU18, although no event was restricted to just the lepidic and invasive components, very weak bands were associated in aN tissues. Ten of the 13 cases in which PCR validations were performed in aN presented with significant banding in aN tissues; however, reduced levels of PCR banding in some aN tissues compared with the tumors were often observed. This was confirmed by quantitative PCR and by comparative even banding for GAPDH PCR controls (Supplementary Figs. S4B–S4D).
Figure 3.
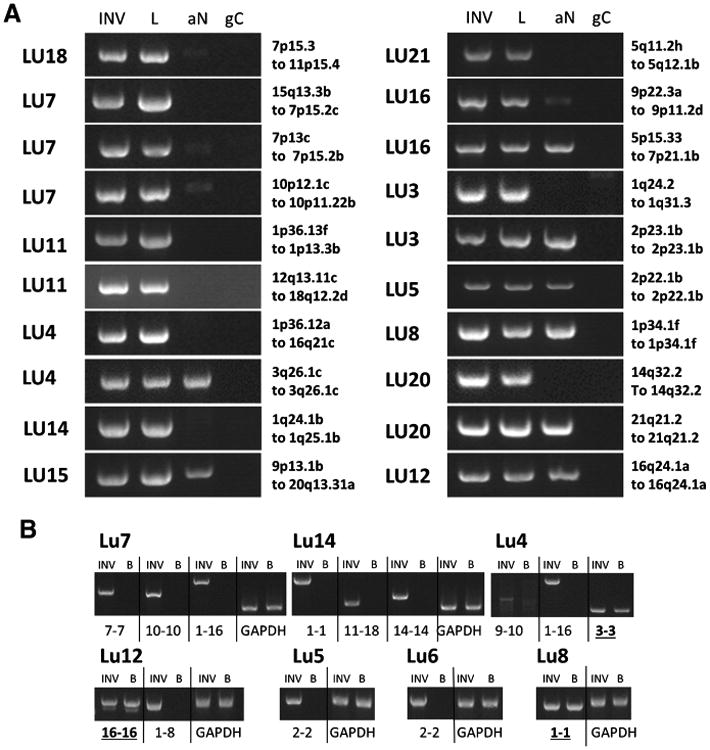
PCR validation of large genomic rearrangements. A, PCR products from primers flanking predicted genomic breakpoints for the invasive (INV) and lepidic (L) components, together with the associated normal (aN) and genomic control (gC). B, PCR validations on germline DNA for the indicated cases where blood was available and consented for that patient, compared with the associated lepidic tumor DNA. Case number and genetic loci of breakpoints or chromosomes involved are indicated; more precise genomic positions are presented in Supplementary Table S2. PCR products were run on 1.5% agarose gels together with 1 kb ladder (Ladder). Rearrangements present in blood are underlined and bold.
To investigate this observation further, sections were collected deeper into the nonneoplastic tissue blocks and multiple discrete areas were captured from the four edges of the tissue sections (Fig. 4A). Figure 4B presents the results for Lu15 for a deletion, d(9-9) and a translocation, t(9-20), as well as a GAPDH control. These deeper sections into the surrounding normal lung lost the validation banding. A similar study was also performed for case Lu16, but in addition to four deeper sections from the same face as the original aN sample (aN1-4), the block was also sectioned from the opposite face (aN5-8; Fig. 4A). In this case, both the d(1-1) deletion and t(5-7) translocation events were observed in restricted zones of both faces (Fig. 4C). Two further cases demonstrated identical banding in all aN tissues upon further sectioning (Fig. 4D and E). In all cases, the control GAPDH PCR bands were even for all tissues.
Figure 4.
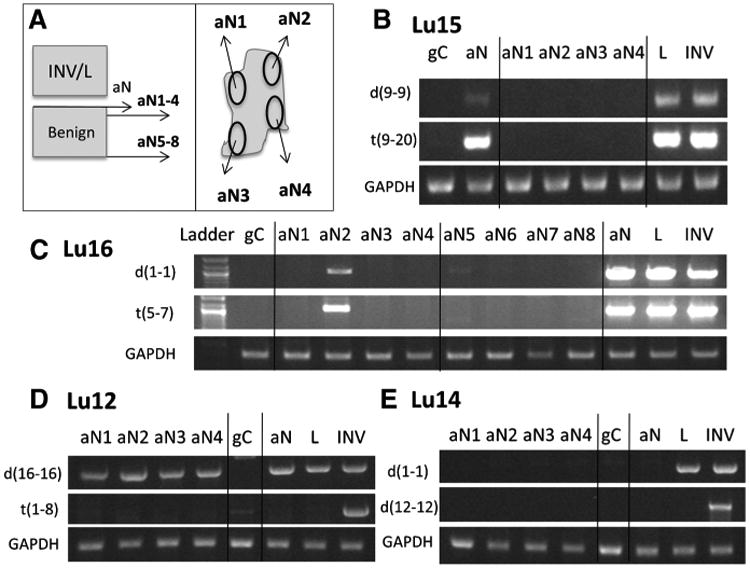
PCR validation of associated normal tissue. A, schematic of additional sectioning of associated benign tissue blocks. PCR banding of four aN regions (aN1-4) sectioned further into same block as original aN sample for case Lu15 (B), case Lu16 (C), case Lu12 (D), and case Lu14 (E). The original normal tissue collected at the same time as the lepidic/ invasive tissues is represented as aN. Additional sectioning of four regions from the opposing face of the benign block (aN5-8) also applied for case Lu16 (C). INV, invasive; L, lepidic.
Discussion
To our knowledge, this is the first study presenting molecular evidence supporting the concept of clonal relatedness and progression in lung adenocarcinoma. All our cases, with one exception, showed shared identical and unique genomic breakpoints, with 6 of the 14 cases harboring more than 40% genomic breakpoint commonality in both the lepidic and invasive components (Table 1). A single shared rearrangement in two lesions within a tissue, not present in the germline of that patient, is indicative of clonal relatedness/lineage. Additional numbers of unique breaks within each tissue present additional information on the distance and timing of divergence from a common lineage. AIS components contained an unexpectedly high number of genomic rearrangements, considering their hypothesized origin as precursors of more aggressive invasive disease. Excluding the Lu12 case, the average number of rearrangement events for the lepidic and invasive components was 37 and 42, respectively, suggesting that these events occur early in tumorigenesis. The large numbers of shared breakpoints in the surrounding histologically normal tissues also support this theory, with many rearrangements potentially emerging before the emergence of the AIS histology.
Our observation of genomic alterations in normal lung tissue surrounding AD raises interesting questions in regard to field effect theories. Indeed, the majority of these genomic alterations were found only in the surrounding normal tissue and not blood, ruling out germline alterations. Molecular changes such as EGF receptor (EGFR) mutation have also been previously reported in normal lung adjacent to lung adenocarcinomas (17). To explain the aN validations as contamination of captured histologically normal tissue with neoplastic cells, equivalent levels of PCR banding would not be expected between the aN and lepidic/invasive lesions (Fig. 3 and Supplementary Fig. S4). Furthermore, the validation studies showed these alterations to be restricted to focal areas of the normal tissue rather than being wide spread (Fig. 4). These observations could be explained by a field effect in which carcinogenetic changes have occurred even in histologically normal lung as a precursor to the development of cancer. The finding of more numerous genomic alterations in the adjacent normal of smokers (current and former), compared with never smokers, would support this theory.
Models of cancer progression have been widely presented in literature; however, due to technical restrictions of isolating neoplastic subpopulations within a single tumor, the majority has focused on progression from primary tumor to metastatic disease. Fearon and Vogelstein proposed the classic “clonal evolution” model, back in 1990, which simply describes an accumulation of mutations as a tumor progresses (18). Thus, a subpopulation of a primary tumor acquires additional driver mutations, leading to the outgrowth of the final metastatic form. However, this model generally presents the metastasis as the end stage of tumor progression and does not consider the continuous mutagenic state of all evolutionarily related tumor components previous to the metastatic form. The “parallel evolution” model incorporates the concept of constant change in all components of a tumor (19, 20, 21). Thus, just as a precursor form of a tumor is able to amass driver mutations, which result in the outgrowth of a more aggressive form of the tumor, other cells in that precursor population are also independently able to amass further unique mutations. These models of metastatic progression are similarly descriptive of the heterogenic state within a single tumor population as described in this current study. Through the evidence of commonality between the lepidic and invasive components, we attempted to relate our cases to models of AD progression. The clonal evolution model (Fig. 5) predicting the direct outgrowth of AD from AIS, was supported best in cases Lu4, Lu7, Lu11, Lu14, Lu16, and Lu18, those predicted most clonal in the independent hierarchical clustering (Supplementary Fig. 4). However, of these 6 cases, the lepidic components of Lu7, Lu11, and Lu18 are observed to have acquired a significant number of unique rearrangements, which are not in common with the adjacent invasive component, fitting the parallel evolution model, where both AIS and AD continue to progress independently from a common genetic precursor (Fig. 5). Cases Lu3 and Lu20, presented with 41 and 17 breakpoints, respectively (Fig. 1B), with only two shared rearrangements between their lepidic and invasive components. However, even from these limited numbers of shared breakpoints, PCR validation showed one event each to be specific to just the invasive and lepidic components, and fitted the parallel evolution model. Conversely, a more independent evolution from a common background (Fig. 5) is predicted in LU12. The model presented in Fig. 5 additionally incorporates this concept of mutations in the adjacent normal, in which mutations shared in the lepidic and invasive components could also be present in histologically normal tissue surrounding the tumor.
Figure 5.
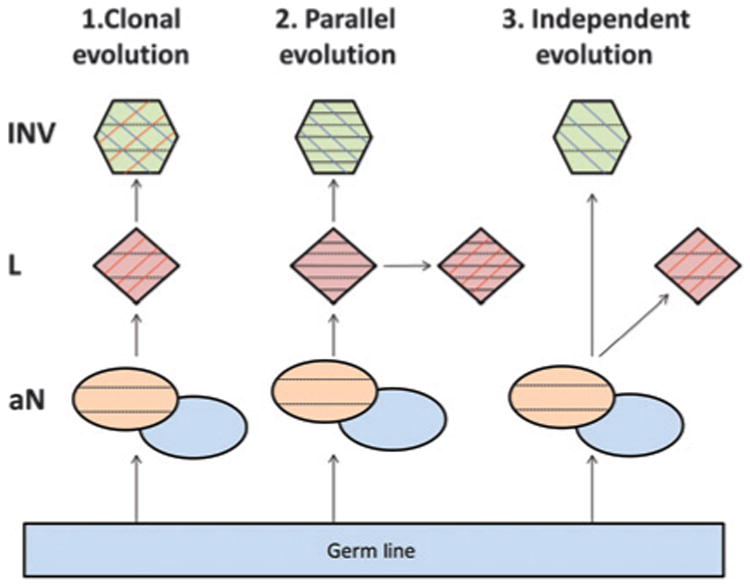
Model of lineage between adjacent lepidic and invasive adenocarcinomacomponents.Demonstrates the three models of lineage between adjacent tissues. 1, the clonal evolution model describes a direct progression from the in situ lepidic tumor components (red) to the invasive tumors (green). 2, the parallel evolution model introduces the further concept that the transition took place some time in the past and both the lepidic and invasive components are continuing to progress and accumulate further unique mutations. 3, the independent evolution model predicts the lepidic and invasive components to grow independently from a common background. Each model also incorporates the concept of the associated normal (aN) in the local environment from where the tumors emerged. This aN is split into two colors to indicate the germline (gray blue) and the abnormal (orange), which has accumulated background mutations above the germline but would be common to any tumor arising from that environment. Each component additionally contains black horizontal, red vertical, and/or blue vertical lines to represent mutations occurring in aN, lepidic, or invasive, respectively.
It is important to clarify that sensitivity of lineage detection is somewhat different from the sensitivity of complete breakpoint detection, although influenced by it. Sensitivity of lineage detection in two samples is dominated by recurrent high-coverage breakpoints, whereas sensitivity of complete breakpoint detection is influenced by tumor heterogeneity and contamination by normal cells. Recurrent breakpoints that define lineage are early events that are more likely to be present in a high percentage of tumor cells and therefore have higher sequencing coverage and are easier to detect. The use of LCM in our experiments likely increases the sensitivity of breakpoint detection for lineage. Furthermore, in these cases where foci are determined to be in-lineage via presence of many shared breakpoints and/or proven by SANGER sequencing, FN do not play a role. Therefore, the positive predictive value of lineage detection for 11 of 14 cases is nearly 100%. However, in LU12, in which the two tumors were rendered independent because no shared breakpoints were found by the algorithm, the potential FN rate is an issue and dependent on algorithmic filters. To find a compromise between FN and FP rates, we set the lower limit of breakpoint-supporting MPs to seven. Sequence coverage for this case was more than 25× and many breakpoints were detected. With the conservative estimate of a 15% FN rate (dictated by the incompleteness of the reference genome and by regions that are difficult to map), we estimated that the probability of relatedness between these two samples is less than 5.7e—9 when the expected number of shared breakpoints is 10 or more, and none were found. The two remaining cases, LU8 and LU5, had very few breakpoints detected and experimental validation of the few detected events did not resolve lineage; therefore, their lineage remained inconclusive.
Lu12 lepidic contained the highest number of breakpoints due to extensive chromosomal catastrophe on chromosomes 1 and 8 (Fig. 2B). Chromosomal catastrophe or chromothripsis, defined as clustered chromosomal rearrangements occurring in localized and confined genomic regions (15), was observed in a number of samples in this study. Interestingly, several regions of catastrophe were common between multiple cases. Specifically, the region 12q13.11-q21.2 displayed extensive catastrophe in three cases, and to a lesser extent in an additional three cases (Fig. 2D). Additional common breakpoint regions included the p-arm of chromosome 7 in five cases (Fig. 2E) and the telomeric end of chromosome 20 in three cases (Fig. 2C). Other unique sites of catastrophe were observed in individual cases on chromosomes 1, 5, 8, 11, and 16 (Supplementary Fig. S3). In the literature, region 12q13.3 has been previously reported as a site of common breakpoints in two lung adenocarcinoma cell lines (22), and amplifications/deletions in the region 12q13.3-q14.1 reported in lung adenocarcinoma tissues through SNP array (23). Allelic loss at region 7p14-15 has additionally been associated with breast cancer (24) and loss of the 7p-arm with acute lymphoblastic leukemia (25). A genome-wide association study in the lung also reported a linkage between 20q13.2 and lung disease severity (26). Interestingly, the majority of observed regions of common chromosomal catastrophe were present in both the lepidic and invasive components, suggesting that these are early events in tumorigenesis. For reasons of determining lineage between adjacent lesions, the presence of a shared chromosomal catastrophe was a strong indicator of a common origin, compared with single breakpoints. In line with this hypothesis, the 8 cases with the predicted most clonal lepidic and invasive component (Fig. 1B) also presented with commonality in chromosomal catastrophes (Supplementary Fig. S3). Just 7 of the 28 samples studied presented with no distinct evidence of chromosomal catastrophe (Supplementary Fig. S3). As expected, these were also the cases with least numbers of detected breakpoints, including both the lepidic and invasive components of Lu5, Lu8, and Lu20, and the invasive component of Lu12.
The additional alignment of common chromosomal fragile sites with genomic breakpoints observed in this study surprisingly demonstrated minimal concordance (Fig. 2A). Chromosomal fragile sites are defined as specific points on chromosomes that tend to form gaps or constriction, and are therefore more likely to break when the cell is exposed to replication stress (16). Considering the sensitivity of these sites to breakage, it has been hypothesized that these regions could also be hot spots for genomic recombination in cancer. Currently, >150 fragile sites are described in the human genome; however, very few of these sites (<10%) have been accurately molecularly characterized. The precise boundaries of the majority of these sites are poorly defined, with literature describing the positions as chromosomal loci, which often span hundreds of Mb of sequence (15). Nevertheless, even with these overrepresented fragile site regions imaged in Fig. 2A, the majority of breakpoints are not restricted to these regions. We conclude from these data that the chromosomal rearrangements observed in these 14 cases of lung adenocarcinoma seem to be minimally influenced by fragile site structures.
A number of recurrent genes are impacted by rearrangements in multiple cases in this study, some of which have been previously implicated in tumorigenesis. ALK rearrangements have been extensively reported in lung adeno-carcinoma as both tumor-driving events and therapeutic targets (27). In this study, a classic EML4-ALK fusion is present in both the lepidic and invasive components of case Lu6 and an intragenic rearrangement within the ALK gene between exons 3 and 4 is predicted in Lu3 invasive. The nuclear receptor cofactor, NCOA2, a transcription factor that plays important roles in various aspects of cell growth, development, and homeostasis by controlling expression of specific genes, was hit in two cases (Table 2). This gene has been previously linked with the prognosis of NSCLC (27), and rearrangements involving PAX and HEY genes have also been previously described (28). The tumor-suppressor gene; WNT inhibitory factor 1 (WIF1), was also rearranged in two cases. The DNA methylation status of WIF1 and other WNT antagonist genes have been linked with responses to EGFR-targeting therapies in NSCLC (29). Breakpoints in the mitotic spindle-assembly checkpoint protein gene, MAD1L1, was also observed in Lu4 invasive and Lu7 lepidic, which has been associated with a susceptibility to lung cancer (30). Other major genes included RET hit in both lepidic/invasive components of Lu4, a BCL2-IL18 fusion just in Lu14 lepidic, GPHN in Lu16 invasive, and CDNK2A in Lu15 invasive. Because of the limited number of cases involved and a lack of point-mutation data, an in-depth evaluation of driver genes was outside the scope of this study.
The heterogeneity of lung cancer tells us repeatedly that the natural history of tumors and the roads to progression vary among cases and ultimately all the models are true in certain cases. The next stage of cancer research is to be able to predict which road a tumor is likely to take. To generate data to even attempt to ask these questions, similar techniques to those undertaken in this article are required for genomic interrogation of progressive forms of a tumor from premalignant state to distal metastases. A knowledge that the lepidic growth patterns can progress to invasive disease allows us to design future studies to determine biomarkers to predict the potential for AIS progression. Although the current study was only designed to address the initial question of lineage relationship between adjacent lepidic and invasive histologies present in a single tumor, future studies with increased sample numbers will aim to discover insights into patterns between patient tumors. To address these issues, future cases will be additionally stratified according to the degree of lepidic growth patterns and patient outcome data, in an attempt to determine genomic modifications that drive these differing histologies.
The current findings have a number of implications for both the molecular pathogenesis and the clinical management of lung adenocarcinoma. First, our results suggest that a subset of lung adenocarcinoma truly do progress through an AIS–AD sequence, in which an accumulation of genomic alterations leads to progression of invasive disease. As suggested by clinical studies demonstrating improved disease-free and overall survival for treatment of lesions containing components of AIS, it may be that this represents a distinct clinical entity that can be treated less aggressively by either sublobar resection or even periods of watchful waiting with close imaging follow-up before any treatment. Second, the finding of somatic alterations in histologically normal lung surrounding a tumor mass provides further evidence for the concept of a field defect giving rise to malignant lesions. This might facilitate a combination of imaging and biomarker-based evaluation being incorporated into screening and surveillance for lung cancer that currently does not exist.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by the Mayo Clinic Center for Individualized Medicine (CIM) Biomarker Discovery (BMD) Program and grants from Uniting Against Lung Cancer (DAW).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Authors' Contributions: Conception and design: S.J. Murphy, D.A. Wigle, T. Peikert, P. Yang, M.C. Aubry, G. Vasmatzis
Development of methodology: S.J. Murphy, D.A. Wigle, J.F. Lima, S. Terra, M.C. Aubry, G. Vasmatzis
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S.J. Murphy, D.A. Wigle, F.R. Harris, G. Halling, M.K. Asiedu, S. Terra, P. Yang, M.C. Aubry, G. Vasmatzis
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S.J. Murphy, D.A. Wigle, F.R. Harris, S.H Johnson, G. Halling, C.T. Seto, S. Terra, P. Yang, M.C. Aubry, G. Vasmatzis
Writing, review, and/or revision of the manuscript: S.J. Murphy, D.A. Wigle, S.H Johnson, M.K. Asiedu, F. Kosari, T. Peikert, P. Yang, M.C. Aubry, G. Vasmatzis
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): F.R. Harris, S.H Johnson, G. Halling, M.K. Asiedu, S. Terra
Study supervision: D.A. Wigle, M.C. Aubry, G. Vasmatzis
References
- 1.Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger K, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6:244–85. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLoS ONE. 2012;7:e36530. doi: 10.1371/journal.pone.0036530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Schil PE, Asamura H, Rusch WV, Mitsudomi T, Tsuboi M, Brambilla E, et al. Surgical implications of the new IASLC/ATS/ERS adenocarcinoma classification. Eur Respir J. 2012;39:478–86. doi: 10.1183/09031936.00027511. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y, Yoon KA, Joo J, Lee D, Bae K, Han JY, et al. Prognostic implications of genetic variants in advanced non-small cell lung cancer: a genome-wide association study. Carcinogenesis. 2010;34:307–14. doi: 10.1093/carcin/bgs356. [DOI] [PubMed] [Google Scholar]
- 5.Tanney A, Oliver GR, Farztdinov V, Kennedy RD, Mulligan JM, Fulton CE, et al. Generation of a non-small cell lung cancer transcriptome microarray. BMC Med Genomics. 2008;30:1–20. doi: 10.1186/1755-8794-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–34. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navin NE, Hicks J. Tracing the tumor lineage. Mol Oncol. 2010;4:267–83. doi: 10.1016/j.molonc.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy SJ, Cheville JC, Zarei S, Johnson SH, Sikkink RA, Kosari F, et al. Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer. DNA Res. 2012;19:395–406. doi: 10.1093/dnares/dss021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.St John J, Powell K, Conley-LaComb MK, Chinni SR. TMPRSS2-ERG Fusion Gene Expression in Prostate Tumor Cells and Its Clinical and Biological Significance in Prostate Cancer Progression. J Cancer Sci Ther. 2012;4:94–101. doi: 10.4172/1948-5956.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 12.Feldman AL, Dogan A, Smith DI, Law ME, Ansell SM, Johnson SH, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117:915–19. doi: 10.1182/blood-2010-08-303305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasmatzis G, Johnson SH, Knudson RA, Ketterling RP, Braggio E, Fonseca R, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peritheral T-cell lymphomas. Blood. 2012;11:2280–9. doi: 10.1182/blood-2012-03-419937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovtun IV, Cheville JC, Murphy SJ, Johnson SH, Zarei S, Kosari F, et al. Lineage relationship of Gleason patterns in Gleason score 7 prostate cancer. Cancer Res. 2013;73:3275–84. doi: 10.1158/0008-5472.CAN-12-2803. [DOI] [PubMed] [Google Scholar]
- 15.Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell. 2012;148:29–32. doi: 10.1016/j.cell.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz M, Zlotorynski E, Kerem B. The molecular basis of common and rare fragile sites. Cancer Lett. 2005;232:13–26. doi: 10.1016/j.canlet.2005.07.039. [DOI] [PubMed] [Google Scholar]
- 17.Tang X, Varella-Garcia M, Xavier AC, Massarelli E, Ozburn N, Moran C, et al. Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas. Ca Prev Res. 2008;1:192–200. doi: 10.1158/1940-6207.CAPR-08-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 19.Gray JW. Evidence emerges for early metastasis and parallel evolution of primary and metastatic tumors. Cancer Cell. 2003;4:4–6. doi: 10.1016/s1535-6108(03)00167-3. [DOI] [PubMed] [Google Scholar]
- 20.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi K, Kohno T, Matsumoto S, Nakanishi Y, Arai Y, Yamamoto S, et al. Clonal and parallel evolution of primary lung cancers and their metastases revealed by molecular dissection of cancer cells. Clin Cancer Res. 2007;13:111–20. doi: 10.1158/1078-0432.CCR-06-0659. [DOI] [PubMed] [Google Scholar]
- 22.Peng KJ, Wang JH, Su WT, Wang XC, Yang FT, Nie WH. Characterization of two human lung adenocarcinoma cell lines by reciprocal chromosome painting. Dongwuxue Yanjiu. 2010;31:113–21. doi: 10.3724/SP.J.1141.2010.02113. [DOI] [PubMed] [Google Scholar]
- 23.Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–70. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 24.Kurose K, Iida A, Araki T, Sakamoto G, Kasumi F, Nakamura Y, et al. Frequent allelic loss at 7p14–15 associated with aggressive histologic types of breast cancer. Jpn J Cancer Res. 1998;89:533–8. doi: 10.1111/j.1349-7006.1998.tb03294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heerema NA, Nachman JB, Sather HN, La MK, Hutchinson R, Lange BJ, et al. Deletion of 7p or monosomy 7 in pediatric acute lymphoblastic leukemia is an adverse prognostic factor: a report from the Children's Cancer Group. Leukemia. 2004;18:939–47. doi: 10.1038/sj.leu.2403327. [DOI] [PubMed] [Google Scholar]
- 26.Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L, et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13. 2 Nat Genet. 2011;43:539–46. doi: 10.1038/ng.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;27:473–7. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 28.Sumegi J, Streblow R, Frayer RW, Dal Cin P, Rosenberg A, Meloni-Ehrig A, et al. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer. 2012;49:224–36. doi: 10.1002/gcc.20731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu J, Wang Y, Duan J, Bai H, Wang Z, Wei L, et al. DNA Methylation status of Wnt antagonist SFRP5 can predict the response to the EGFR-tyrosine kinase inhibitor therapy in non-small cell lung cancer. J Exp Clin Cancer Res. 2012;31:80. doi: 10.1186/1756-9966-31-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo Y, Zhang X, Yang M, Miao X, Shi Y, Yao J, et al. Functional evaluation of missense variations in the human MAD1L1 and MAD2L1 genes and their impact on susceptibility to lung cancer. J Med Genet. 2010;47:616–22. doi: 10.1136/jmg.2009.074252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.