

Abstract
Background
Wide genome screening through array comparative genomic hybridization made possible the recognition of the novel 19q13.11 deletion syndrome. There are very few cases reported with this deletion, but clinically this condition seems to be recognizable by pre and postnatal growth retardation, microcephaly, developmental delay/intellectual disabilities, speech disturbance, hypospadias (in males) and signs of ectodermal dysplasia and cutis aplasia over the posterior occiput.
Results
Using oligoarray CGH, a 4.6 Mb deletion in 19q13.11q13.12 was detected in a 23 year old female patient that presented clinical features previously associated with 19q13.11 deletion.
Conclusions
Our work reinforces the idea that a region encompassing four zinc finger genes is likely to be responsible for the syndrome, and that the difference in minor clinical manifestation depends on the genes present outside the minimal overlapping region proposed for this syndrome. We also review all cases described in the literature and discuss the correlation between haploinsufficiency of UBA2 gene and cutis aplasia present in the majority of the patients reported, and its importance as a clinical hallmark of 19q13.11 deletion syndrome, when associated with more common features like developmental delay, microcephaly, speech disturbance and hypospadias in males.
Keywords: 19q13.11 deletion, UBA2 gene, Cutis aplasia
Background
The development of array comparative genomic hybridization technique (array CGH) greatly improved the detection of cryptic unbalanced rearrangements in mental retardation patients and made possible the identification of novel microdeletion and microduplication syndromes [1].
In 1998, Kulharya et al., reported a cytogenetically visible 19q12q13.1 deletion, in a fetus with intrauterine growth retardation and decreased fetal activity. At the age of 3 years the child presented mental retardation, developmental delay, absence of speech, multiple minor anomalies and cutis aplasia [2]. Eleven years later, Malan et al., identified by array CGH a 19q13.11 microdeletion in three patients who share common clinical features with Kulharya et al.’s patient, and proposed the 19q13.11 microdeletion syndrome as a novel clinically recognizable syndrome [3]. In 2009, Schuurs-Hoeijmarkers et al. narrowed the critical region responsible for the new syndrome to a 750 kb segment within the 19q13.11 deletion [4]. Recently Forzano et al. further refined the critical region and Gana et al., proposed a minimal overlaping region (MOR) of 324 kb encompassing four zinc finger genes [5,6].
A total of 8 mental retardation patients and 1 aborted fetus, carrying the 19q13.11 deletion have been reported until now in addition to two more cases included in the Database of Chromosome Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) [2-7]. Despite some phenotypic variability, all these patients presented common features and the deletion of 19q13.11 is proposed as a new clinical recognizable syndrome [3].
Patients with this deletion are characterized by intrauterine and postnatal growth retardation, microcephaly, developmental delay/intellectual disabilities, speech disturbance, slender habitus, feeding difficulties, cutis aplasia over the posterior occiput, signs of ectodermal dysplasia, and genital malformation in males (hypospadias). In this study we report a case of a mentally retarded woman carrier of a 19q13.11 deletion and compare our findings with the ones previously reported in the literature [2-7].
Results
Patient karyotype was normal 46,XX. Array CGH analysis identified a 4.6 Mb deletion at the long arm of chromosome 19 (Figure 1A-C). The deletion breakpoints were ascertained between 33,203,635 and 38,108,990. Multiplex Ligation-dependent Probe Amplification (MLPA) analysis, using SALSA Probe MLPA – P347-A1 of the proband DNA confirmed the 19q13.11q13.12 de novo deletion (Figure 1D). Array CGH final results was arr [hg 19] 19q13.11q13.12 (33,203,635-38,108,990) ×1dn according to the International System for Human Cytogenetic Nomenclature (ISCN) 2013 [8]. The deleted region contains several coding genes, including CEBPA [OMIM ID: 116897], PEPD [OMIM ID: 613230], LSM14A [OMIM ID: 610677], UBA2 [OMIM ID: 613295], WTIP [OMIM ID: 614790], SCGB2B [OMIM ID:615063], ZNF302, ZNF181 [OMIM ID: 606741], ZNF599, ZNF30, SCN1 [OMIM ID: 202700], USF2 [OMIM ID: 600390], COX6B1 [OMIM ID: 124089] and HPHS1.
Figure 1.
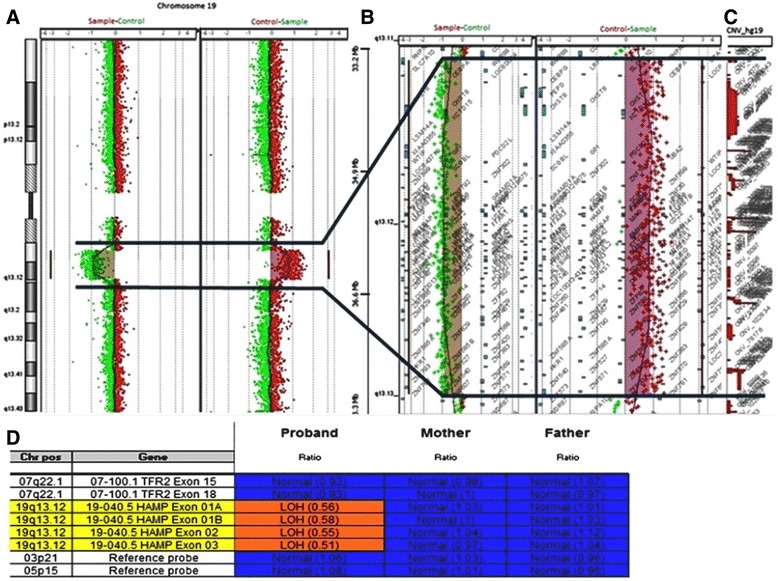
Molecular cytogenetic analysis. (A) Array CGH comparative profile for chromosome 19 disclosing the 19q13.12 deletion. (B) Zoom in of the deleted region detected by array CGH. (C) Zoom in of the CNV profile of the deleted region according to the Database of Genomic Variants (DGV). (D) MLPA analysis result of the proband and progenitors (SALSA Probe MLPA – P347-A1) confirming a de novo deletion of the probes corresponding to the deletion observed by array CGH.
Discussion
Since the Kulharya et al. report, of a 3 year-old patient, to the Lin et al. report, of an aborted fetus, a total of 8 cases had been reported, in the literature, as examples of the emerging 19q13.11 deletion syndrome [2-7].
In this study we identified another patient with a de novo 19q13.11q13.12 deletion, harboring the minimal overlapping region (MOR) pointed by Gana et al. as the critical region for the 19q13.11 deletion syndrome (Figure 2) [6]. Consistent with typical clinical features in literature [2-7], our case presented intrauterine and postnatal growth retardation, mental retardation, speech disturbance, microcephaly, slender habitus with little subcutaneous fat, facial dysmorphic features, signs of ectodermal dysplasia, cutis aplasia and feeding problems. Comparison of clinical features between our case and the previous reported, evidences that although many anomalies are common they are not all present in all patients (Table 1). The 4.6 Mb deleted region in our patient overlaps the MOR and, contains over 50 coding genes. The specific functions of ZNF302, ZNF181, ZNF399 and ZNF30, present in the MOR and pinpointed as important genes in this syndrome, have not been completely understood. These zinc finger proteins belong to the KRAB-ZNF family, known to be involved in cell differentiation, proliferation, cycle regulation and apoptosis, and in the embryonic development [9,10]. Moreover, the hypothesis that the haploinsuffiency of these genes could be the cause for mental retardation and speech disturbance is supported by the fact that zinc finger genes are involved in X-linked mental retardation in males, and that ZNF clusters are suspected to contribute to higher cognitive function in primates [11-14].
Figure 2.
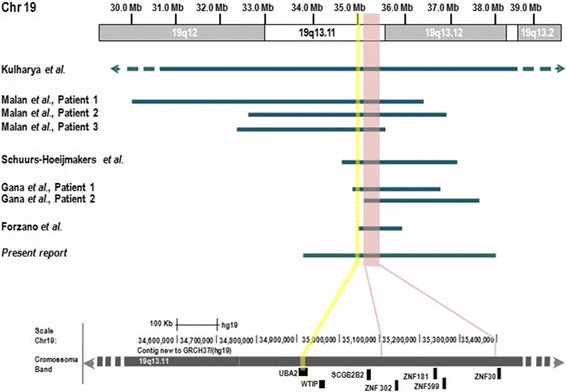
Schematic representation of the 13.11 deletions at the long arm of chromosome 19. Representation of eight cases with 19q13.11 deletion, evidencing the MOR region (chr19: 35,111,870-35,436,076) and the genes present. The UBA2 gene location (GRCh37:34,919,268-34,960,798) is highlighted in yellow.
Table 1.
Clinical features present in patients with 19q13.11 deletion
| Kulharya et al. | Malan et al. | Schuurs- Hoeijmakers et al. | Gana et al. | Forzano et al. | Present report | N/T | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 1 | Patient 2 | ||||||
| Approximated size of the deletion | 11 Mb | 6,2 Mb | 4,3 Mb | 3 Mb | 2,4 Mb | 1,7 Mb | 2,6 Mb | 1,4 Mb | 4,9 Mb | |
| Gender | Female | Male | Male | Male | Male | Male | Female | Female | Female | |
| Weight at birth | 1,295 Kg | 1,560 Kg | 1,930 Kg | 1,620 Kg | 1,590 Kg | 1,900 Kg | 1,580 Kg | 1,950 Kg | ||
| Developmental characteristics | ||||||||||
| Intrauterine growth retardation | + | + | + | + | + | + | + | + | + | 9/9 |
| Postnatal growth retardation | + | + | + | + | + | + | + | + | + | 9/9 |
| DD/ID | + | + | + | + | + | + | + | + | + | 9/9 |
| Speech disturbance | + | + | + | + | + | + | + | + | + | 9/9 |
| Feeding problems | + | + | + | + | + | + | + | + | 8/9 | |
| Slender habitus | + | + | + | + | + | + | + | + | + | 9/9 |
| Microcephaly | + | + | + | + | + | + | + | + | + | 9/9 |
| Failure to thrive | + | - | + | - | + | + | + | - | + | 6/9 |
| Facial anomalies | ||||||||||
| Long face | - | + | + | - | - | - | - | + | + | 4/9 |
| High forehead | - | + | + | - | - | - | - | - | + | 3/9 |
| Micrognathia/Retrognathia | + | + | + | - | + | + | + | + | + | 8/9 |
| Low set ears | + | - | + | - | + | - | - | + | + | 4/9 |
| V shaped nasal tip | - | + | + | - | - | - | - | - | - | 2/9 |
| Thin lips | - | + | + | + | + | + | + | - | + | 8/9 |
| Ectodermal dysplasia | ||||||||||
| Hair/eyebrows/eyelashes anomalies | - | + | + | + | + | + | + | + | + | 8/9 |
| Thin/dry skin | - | + | + | - | - | - | - | + | + | 4/9 |
| Cutis aplasia in midline scalp | + | + | + | + | + | + | - | + | + | 8/9 |
| Dysplastic nails | - | + | + | - | + | - | - | + | + | 5/9 |
| Cutaneous syndactyly | - | - | + | + | + | + | - | - | - | 4/9 |
| Extremity abnormalities | ||||||||||
| Clinodactyly | + | + | + | + | + | + | - | + | + | 8/9 |
| Overlapping of the toes | + | + | - | - | - | - | - | + | + | 4/9 |
| Long fingers | - | + | + | - | + | + | + | - | + | 6/9 |
| Genital abnormalities | ||||||||||
| Hypospadias | Not applicable | + | + | + | + | + | Not applicable | Not applicable | Not applicable | 5/5 |
| Organ abnormalities | ||||||||||
| Congenital heart disease | + | - | + | - | - | + | - | + | + | 5/9 |
| Livedo and cutaneous hypersensibility | - | + | - | - | - | - | - | - | + | 2/9 |
| Little subcutaneous fat | + | - | + | - | - | - | - | - | + | |
| Hypotonia | + | - | - | - | - | - | + | - | + | 3/9 |
| Dystonia | - | - | - | - | - | + | - | - | + | 2/9 |
| Airways infections | + | - | - | + | + | - | - | - | - | 3/9 |
(+) – feature present, (−) – absent feature, DD – development delay, ID – intellectual delay, N/T – number of patients/total of patients.
Neighboring the MOR there are also genes that may be implicated in the syndrome pathogenesis, either because they are also deleted or just because their regulation regions may be disrupted. Schuurs-Hoeijmarkers et al., suggested that the cause of mental retardation in 19q13.11 deletion syndrome could also be associated to haploinsuffiency of genes LSM14A and UBA2 [4]. The LSM14A is a Sm-like protein thought to have a role in the control of mRNA translation, and UBA2 is important in the ubiquitin pathway [4]. Other genes outside the MOR, whose deletions could be associated with major clinical features, are: CEBPA gene, as possible cause of lack of subcutaneous fat; COX6B1 gene, associated with severe infantile encephalomyopathy; and WT1 and WTIP genes that are involved in mammalian urogenital development [15,16].
Cutis aplasia is one of the major features presented by 19q13.11 deletion syndrome patients. However, Gana et al. Patient 2, does not present such features (Table 1). Comparing patients 19q13.11 deleted regions, it becomes evident that 34,9 – 35,1 Mb segment is deleted in all patients except in Gana et al. Patient 2. This segment harbors three genes: SCGB2B2, WTIP and UBA2, whose haploinsuffiency could be considered as a cause for cutis aplasia. This hypothesis can be ruled out for SCGB2B2 since this gene is not expressed in skin cells and is not likely for WTIP gene because its haploinsuffiency has been pointed as a cause for hypospadias [5]. Deletions of the UBA2 gene (codifier of a ubiquitin-like modifier), results into proteins with abnormal post-translational modifications, and in particular could result in prolidase deficiencies. It has been reported that prolidase deficiencies, may impairs proline recycle, causing recurrent cutaneous ulcers that are difficult to heal resulting in cutis aplasia. This could also account for the thin skin, observed in some patients, reinforcing the role for UBA2 gene in cutis aplasia [2].
Apart from the core phenotype, other clinical features seemed to be associated with genes outside the MOR: hypotonia has been reported and associated with UQCRFS1 and VIB genes; renal anomalies, associated with HPHS1 and USF2 genes; and cardiac defects associated with SCN1B gene [2-5]. Although these genes are deleted in several cases, patients do not present the same phenotype, leading to suppose that they can act like a predisposing factor that can be trigged under a certain genetic, epigenetic or environmental context.
Conclusions
In conclusion, 19q13.11 deletion syndrome is a continuous genetic disease where a critical region is responsible for the major clinical features. Other genes outside that region contribute to clinical features observed less frequently. Reviewing all the reported cases and ours we believed that cutis aplasia could be related to haploinsuffiency of UBA2 gene. The report of further patients with this deletion would reinforce this evidence.
Methods
Patient report
Our patient was born from non-consanguineous healthy parents, after 39 weeks of gestation and a complicated pregnancy due to intrauterine growth retardation. At birth she weight 1950 g (<5th centile), measured 42 cm (<5th centile) and her head circumference was 30 cm (<5th centile). After birth, she presented feeding difficulties, and it was necessary the presence of a gastrostomy button. Some dysmorphic features were presented, like microcephaly, long face, high forehead, low set ears, deep set eyes, V shaped nasal tip, hipoplasic alae nasi, thin lips, retrognathia and high arched palate. After teething, teeth abnormalities were presented. Signs of ectodermal dysplasia included sparse eyebrows and eyelashes, cutis aplasia in midline scalp, thin and dry skin and dysplastic nails (Figure 3). Extremity abnormalities were noticed, with clinodactyly, long finger and overlapping toes. Other anomalies included congenital heart disease, congenital dislocation of the hip, livedo and cutaneous hypersensibility. Her early years were compromised by feeding difficulties and failure to thrive. From the first (7 month old) to her last examination (23 years old) she always presented slender habitus, she was very thin with very little subcutaneous fat tissue. Throughout her growth dystonia, hiperlaxity of the joints, poor strength, walking disabilities, myopia, absence of verbal skills, and growth retardation were noticed.
Figure 3.
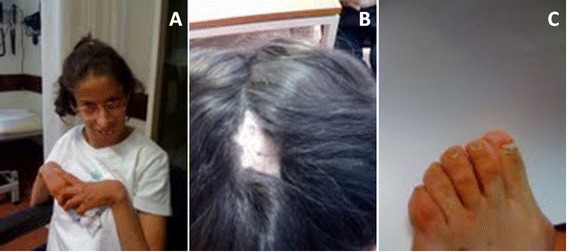
Dysmorphic features presented by the 23 year old patient. (A) Photograph of the patient with 23 years old. (B) Photograph evidencing cutis aplasia over the posterior occiput. (C) Photograph evidencing dysplastic nails.
Standard karyotyping
Chromosome analysis was performed on blood lymphocytes, using GTG high resolution banding technique, according to standard procedures [17].
Array CGH
DNA was extracted from the proband and parent’s peripheral blood, using the QIAmp DNA Mini kit (Qiagen, Valencia, CA, USA) according to manufacturer’s instructions. DNA concentration was determinate with NanoDrop ND1000 spectrophotometer and software (NanoDrop Technologies, Berlin, Germany). Array CGH analysis was performed using the Agilent kit 4×180K (Human Genome CGH Microarray, Agilent Technologies, Santa Clara, CA, USA), with a 17 Kb resolution, according to manufacturer’s protocol [18]. Genomic positions are referred to the Human Genome February 2009 assembly (hg19).
Multi ligation-dependent probe amplification
Multi ligation-dependent probe amplification was used to confirm oligoarray-CGH results for chromosome 19 in the proband and parents, with commercially available SALSA P347-A1 for microdeletions syndromes (MRC Holland, Amsterdam, Netherlands) and was performed according to manufacturer’s instructions. Amplification products were electrophoresed on an ABI PRISM 3100 Genetic Analyzer and the data obtained analyzed by excel spreadsheet (MRC Holland, Amsterdam, Netherlands) [19].
Written informed consent was obtained for publication and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- CGH
Comparative genomic hybridization
- DECIPHER
Database of Chromosome Imbalance and Phenotype in Humans using Ensembl Resources
- ISCN
International System for Human Cytogenetic Nomenclature
- MLPA
Multiplex Ligation-dependent Probe Amplification
- MOR
Minimal overlaping region
- OMIM ID
Online Mendelian Inheritance in Man Identification
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JBM conceived the study, designed it, carried out the molecular and conventional cytogenetic analysis and helped to draft the manuscript. AE drafted the manuscript. JS clinically evaluated the patient at her first’s years of life. LR clinically evaluated the patient at adult age. IMC participated in the study coordination and helped to draft the manuscript. All authors read and approved the manuscript.
Contributor Information
Joana B Melo, Email: mmelo@fmed.uc.pt.
Alexandra Estevinho, Email: alex.estevinho@gmail.com.
Jorge Saraiva, Email: j.saraiva@chuc.min-saude.pt.
Lina Ramos, Email: l.ramos@chuc.min-saude.pt.
Isabel M Carreira, Email: icarreira@fmed.uc.pt.
References
- 1.Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mut. 2010;31(12):1326–42. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kulharya AS, Michaelis RC, Norris KS, Taylor HA, Garcia-Heras J. Constitutional del (19) (q12q13.1) in a three-year-old girl with severe phenotypic abnormalities affecting multiple organ systems. Am J Med Genet. 1998;77:391–4. doi: 10.1002/(SICI)1096-8628(19980605)77:5<391::AID-AJMG7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 3.Malan V, Raoul O, Firth HV, Royer G, Turleau C, Bernheim A, et al. 19q13.11 deletion syndrome: a novel clinically recognizable genetic condition identified by array comparative genomic hybridisation. J Med Genet. 2009;46:635–40. doi: 10.1136/jmg.2008.062034. [DOI] [PubMed] [Google Scholar]
- 4.Schuurs-Hoeijmakers JHM, Vermeer S, van Bon BWM, Plund R, Marcelis C, de Brouwer APM, et al. Refining the critical region of the novel 19q13.11 microdeletion syndrome to 750 Kb. J Med Genet. 2009;46(6):421. doi: 10.1136/jmg.2009.066910. [DOI] [PubMed] [Google Scholar]
- 5.Gana S, Veggiotti P, Sciacca G, Fedeli C, Bersano A, Micieli G, et al. 19q13.11 cryptic deletion: description of two new cases and indication for a role of WTIP haploinsufficiency in hypospadias. Eur J Hum Genet. 2012;20:852–6. doi: 10.1038/ejhg.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forzano F, Napoli F, Uliana V, Malacarne M, Viaggi C, Bloise R, et al. 19q13.11 microdeletion syndrome: further refining the critical region. J Med Genet. 2012;55:429–32. doi: 10.1016/j.ejmg.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Lin SY, Lee CN, Chen TC, Tu MP, Lin CY, Chang TY, et al. A fetus with 19q13.11 microdeletion presenting with intrauterine growth restriction and multiple cystic kidney. Case Rep Perinat Med. 2012;1(1–2):69–74. [Google Scholar]
- 8.Shaffer LG, McGowan-Jordan J, Schimid M. Annual international system for human cytogenetic nomenclature. ISCN: Karger. 2013. [Google Scholar]
- 9.Urritia R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003;4:231–7. doi: 10.1186/gb-2003-4-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleefstra T, Yntema HG, Oudakker AR, Banning MJG, Kalscheuer VM, Chelly J, et al. Zinc finger 81 (ZNF81) mutations associated with X-linked mental retardation. J Med Genet. 2004;41(5):394–9. doi: 10.1136/jmg.2003.016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lugtenberg D, Yntema HG, Banning MJG, Oudakker AR, Firth HV, Willatt L, et al. ZNF674: a new krüppel-associated box–containing zinc-finger gene involved in nonsyndromic X-linked mental retardation. Am J Hum Genet. 2006;78(2):265–78. doi: 10.1086/500306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoichet SA, Hoffmann K, Menzel C, Trautmann U, Moser B, Hoeltzenbein M, et al. Mutations in the ZNF41 gene are associated with cognitive deficits: identification of a new candidate for X-linked mental retardation. Am J Hum Genet. 2003;73(6):1341–54. doi: 10.1086/380309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tadepally HD, Burger G, Aubry M. Evolution of C2H2-zinc finger genes and subfamilies in mammals: species-specific duplication and loss of clusters, genes and effector domains. BMC Evol Biol. 2008;8:1471–2148. doi: 10.1186/1471-2148-8-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang N-D, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, et al. Impaired energy homeostasis in C/EBP-alpha knockout mice. Science. 1995;269:1108–12. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- 15.Massa V, Fernandez-Vizzarra E, Alshahwan S, Bakhsh E, Goffrini P, Ferrero I, et al. Severe infantil encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am J Hum Genet. 2008;82:1281–9. doi: 10.1016/j.ajhg.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regev M, Kirk R, Mashevich M, Bistritzer Z, Reish O. Vertical transmission of a mutation in exon 1 of the WT1 gene: lessons for genetic counseling. Am J Med Genet Part A. 2008;146A:2332–6. doi: 10.1002/ajmg.a.32330. [DOI] [PubMed] [Google Scholar]
- 17.Rooney DE, Czepulkowski BH. Human cytogenetics: a practical approach. New York: Oxford University Press; 1992. [Google Scholar]
- 18.Matoso E, Melo JB, Ferreira SI, Jardim A, Castelo TM, Weise A, et al. Insertional translocation leading to a 4q13 duplication including the EPHA5 gene in two siblings with attention-deficit hyperactivity disorder. Am J Med Genet A. 2013;161A(8):1923–8. doi: 10.1002/ajmg.a.36032. [DOI] [PubMed] [Google Scholar]
- 19.Ferreira SI, Matoso E, Venâncio M, Saraiva J, Melo JB, Carreira IM. Critical region in 2q31.2q32.3 deletion syndrome: reporto f two phenotypically distinct patients, one with an additional in alagille syndrome region. Mol Cytogenet. 2012;5:25–30. doi: 10.1186/1755-8166-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]