

Abstract
We report the discovery and SAR of two novel series of imidazopyrimidinones and dihydroimidazopyrimidinones as metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulators (PAMs). Exploration of several structural features in the western and eastern part of the imidazopyrimidinone core and combinations thereof, revealed compound 4a as a mGlu5 PAM with good in vitro potency and efficacy, acceptable drug metabolism and pharmacokinetic (DMPK) properties and in vivo efficacy in an amphetamine-based model of psychosis. However, the presence of CNS-mediated adverse effects in preclinical species precluded any further in vivo evaluation.
Keywords: Positive allosteric modulator (PAM), Metabotropic glutamate receptor 5 (mGlu5), Imidazopyrimidinone, Dihydroimidazopyrimidinone, Schizophrenia
Schizophrenia is a devastating psychiatric illness that afflicts approximately 1% of the world population. Current therapies, based on the direct modulation of the monoaminergic system (dopamine and serotonin), are associated with severe adverse effects and do not provide sufficient relief across all symptom domains of the disease.1,2 Therefore, new treatments, possibly acting via different neurotransmitter systems are under investigation. Among the different existing alternatives, the modulation of the glutamatergic system has emerged as an obvious candidate.3,4 Glutamate (l-glutamic acid) is the major excitatory neurotransmitter in the mammalian central nervous system (CNS), where it mediates its effects through three ionotropic receptors (d,l-α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), N-methyl-d-aspartate (NMDA) and kainate) and eight metabotropic glutamate receptors (mGlu1 to mGlu8).5 Among the different mGlus, the mGlu5 receptor is structurally and pharmacologically associated with the NMDA receptor playing prominent roles in synaptic plasticity, cognition, learning and memory process1,6 and it has been reported that the activation of mGlu5 potentiates NMDA receptor function.7 Provided the advantages associated to PAMs over orthosteric agonists, that is, increased subtype selectivity, improved chemical tractability, better modulatory control of receptor function and low propensity for receptor desensitization; the potentiation of mGlu5 via activation at allosteric sites could be a preferred pharmacological mechanism to indirectly correct the hypothesized NMDA receptor hypofunction in schizophrenia.2,8 Consequently, the development of PAMs of mGlu5 has emerged as a potentially viable way to treat positive symptoms9,10 and also improve cognitive deficits11,12 in schizophrenia.
After nearly a decade of mGlu5 PAMs research, a variety of structurally different chemotypes have been reported.13 Among them, we have recently described the discovery of phenoxy-containing mGlu5 PAMs derived from dihydrothiazolopyridone14 and naphthyridinone15 bicyclic cores. As a progression of this work, and in order to further optimize the in vitro and in vivo potencies of dihydrothiazolopyridone 1, we envisioned the replacement of the thiazolyl ring system by other aromatic five-membered rings (Fig. 1).
Figure 1.

Evolution strategy of dihydrothiazolopyridone 1 to other 5,6-bicyclic systems.
Our initial approach involved the direct replacement by an oxazolyl ring affording dihydrooxazolopyridones 2. Encouraged by a comparable in vitro mGlu5 PAM activity to dihydrothiazolopyridone 1 series, the most potent analogues were tested in the reversal of amphetamine-induced hyperlocomotion (AIH) in rats, a predictive model of antipsychotic activity.10a,16 Unfortunately, none of the analogues tested showed robust in vivo activity after oral dosing. This lack of relevant in vivo activity could be explained by the limited absorption observed for these derivatives and thus, our subsequent exploration focused on the identification of other chemotypes with different properties (i.e., electronics, basicity, lipophilicity, etc.) that could eventually lead to an improvement in the in vivo efficacy. In this Letter we report our efforts towards the synthesis and optimization of imidazopyrimidinones 3 and dihydroimidazopyrimidinones 4 as novel series of mGlu5 PAMs.
Synthesis of these 5,6-bicyclic systems was achieved following two parallel approaches that allowed the introduction of diverse substituents in the eastern and western part of the targeted molecules, as shown in Schemes 1 and 2. Our first strategy was focused on the preparation of the key compound 3a, with the aim to survey the eastern diversity in the last step (Scheme 1). Condensation of chloropyrimidine 5 with 1,3-dichloroacetone under acidic conditions, following a procedure already reported in the literature afforded intermediate 6.17 Introduction of phenoxy group in the western part of the molecule using potassium carbonate as base was followed by the selective hydrolysis of the thiomethyl group yielding intermediate 7. De-chlorination was performed under hydrogenation conditions affording analogue 3a. This key compound was then derivatized to the desired N-aryl imidazopyrimidinones 3e and 3h–3n, by reaction either with p-fluoroiodobenzene (3e) or various heteroaryl halides (3h–3n) using a copper iodide–diamine ligand protocol to perform a modified Ullman-type lactam cross-coupling.
Scheme 1.

Reagents and conditions: (a) 1,3-dichloroacetone, AcOH, 110 °C, 16 h, 44%; (b) PhOH, K2CO3, ACN, 50 °C, 18 h, 53%; (c) LiOH, H2O/THF, 50 °C, 4 h, 98%; (d) Pd(OH)2/C, H2 atm, Et3N, AcOEt/MeOH, 50 °C, 45 psi, 16 h, in a Parr reactor, 92%; (e) R1X, CuI, N,N′-dimethylethylenediamine, K2CO3, toluene, 120 °C, 16 h, 22–73%.
Scheme 2.

Reagents and conditions: (a) for R1 = Alkyl: R1X, BuN4OH, DMF, rt, 2 h, 57–77% and for R1 = Aryl or heteroaryl: R1B(OH)2, Cu(OAc)2, TMEDA, MeOH/H2O, rt, 16 h, 48–65%; (b) 1,3-dichloroacetone, DMF, 150 °C, 30 min, microwave irradiation, 15–75%; (c) R2OH, K2CO3, ACN, 90 °C, 18 h, 58–95%; (d) I2, NaIO4, H2SO4, AcOH/water, 80 °C, 4 h, 84%; (e) MeB(OH)2, Pd(PPh3)4, K2CO3, 1,4-dioxane/DMF, 150 °C, 45 min, microwave irradiation, 72%.
Unfortunately, functionalization in the eastern amide moiety with alkyl substituents or other aryl halides following the same Ullman-type conditions was unsuccessful and for this purpose, the approach outlined in Scheme 2 was used. This second strategy allowed the introduction of several alkyl and aryl moieties and also to survey the substitution in the western part of the molecule. Furthermore, this approach could be additionally applied to the introduction of small groups in the 7 and 8-positions of the imidazopyrimidinone core (Scheme 2). Thus, for the functionalization with alkyl and aryl groups in the eastern part of the molecule we started from commercially available cytosine derivatives 9. The reaction of 9 with a variety of alkyl halides using tetrabutylammonium hydroxide as base allowed the selective alkylation on the lactam. On the other hand, for the functionalization with aryl moieties a Chan-Lam-type copper-catalyzed coupling reaction between cytosine derivatives 9 and the corresponding aryl boronic acids as depicted in Scheme 2 was followed.18 Then, condensation with 1,3-dichloroacetone followed by introduction of the corresponding phenoxy or pyridyloxy group in the western part of the molecule, via reaction with the corresponding phenols or hydroxypyridines using potassium carbonate as base, allowed the preparation of analogues 3b–3g, 3o–3s and 3u. The 8-methyl analogue 3v was prepared by Suzuki-type coupling reaction between methylboronic acid and intermediate 13, synthesized following a similar synthetic route as described before but starting from the corresponding substituted iodocytosine 12 as shown in Scheme 2.
Hydrogenation of the double bond in analogues 3 was performed using palladium hydroxide on carbon or Raney nickel as catalysts at high temperatures (Scheme 3). All attempts to reduce the double bond in compounds with substitution in the 7 or 8-positions different than hydrogen were unsuccessful. Finally, functionalization in the imidazole ring was achieved by halogenation of the corresponding derivatives 3 and 4 with N-chlorosuccinimide (NCS) or N-bromosuccinimide (NBS). Subsequent Suzuki-type coupling reaction of 14 with methylboronic acid yielded analogue 4l as depicted in Scheme 3.
Scheme 3.

Reagents and conditions: (a) Pd(OH)2/C, H2 atm, MeOH, 50 °C, 50 psi, 16 h, in a Parr reactor or Raney-Ni, 80 °C, full hydrogen mode, DMF/MeOH in an H-Cube reactor, 5–78%; (b) NBS, benzoyl peroxide, DCE, rt, 16 h, 35%; (c) MeB(OH)2, Pd(PPh3)4, K2CO3, 1,4-dioxane/DMF, 150 °C, 45 min, microwave irradiation, 56%; (d) NCS, DMF, 150 °C, 15–20 min, microwave irradiation, 27–55%.
The compounds synthesized were profiled in a human mGlu5 low receptor expression cell line using a ‘triple add’ calcium mobilization assay, allowing the detection of agonism as well as positive and negative allosteric modulation simultaneously.19 The most relevant SAR trends for the imidazopyrimidinones 3 are shown in Table 1. Initial exploration was focused on the introduction of substituents in the amide moiety with the aim to further improve the marginal activity observed for the unsubstituted analogue 3a (EC50 >30 µM, 19% Glu Max). Following a similar strategy to the dihydrothiazolopyridone series (1),14 we first targeted the introduction of alkyl substituents. Thus, N-methylation in compound 3b, despite having a positive effect in efficacy (67% Glu Max) did not improve the in vitro potency (EC50 >10 µM), which was still significantly decreased compared with analogue 1. According to previous work,14 the functionalization with an alkyl chain of increased size such as cyclopropylmethyl (3c) interestingly induced a significant improvement in the in vitro mGlu5 PAM activity, resulting in a potency in the same range as that of the dihydrothiazolopyridone prototype 1, although with reduced efficacy. Comparable potency again but significantly improved efficacy (93% Glu Max) was achieved by the introduction of a bulkier aromatic benzyl group in 3d.
Table 1.
Structures and activities of substituted imidazopyrimidinones 3
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | R | mGlu5 pEC50a (±SEM) |
mGlu5 EC50 (nM) |
% Glu maxb (±SEM) |
HLMc (%) |
RLMc (%) |
| 3a | H |  |
H | H | <4.52d | >30,000 | 19 | n.t.e | n.t.e |
| 3b |  |
H | H | <5d | <10,000 | 67 ± 4.2 | n.t.e | n.t.e | |
| 3c |  |
H | H | 5.95 ± 0.03 | 1120 | 47 ± 3.7 | n.t.e | n.t.e | |
| 3d |  |
H | H | 5.80 ± 0.06 | 1590 | 93 ± 5.0 | n.t.e | n.t.e | |
| 3e |  |
 |
H | H | 6.51 ± 0.15 | 310 | 83 ± 9.1 | 14 | 27 |
| 3f | 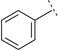 |
H | H | 6.29 ± 0.02 | 514 | 69 ± 0.9 | 47 | 71 | |
| 3g |  |
 |
H | H | 6.64 ± 0.05 | 230 | 93 ± 11.7 | n.t.e | n.t.e |
| 3h |  |
 |
H | H | 6.23 ± 0.05 | 587 | 87 ± 10.2 | n.t.e | n.t.e |
| 3i | 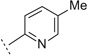 |
 |
H | H | 5.69 ± 0.14 | 2000 | 84 ± 15.4 | n.t.e | n.t.e |
| 3j |  |
H | H | 5.45 ± 0.11 | 3580 | 64 ± 10.3 | n.t.e | n.t.e | |
| 3k |  |
 |
H | H | 5.29 ± 0.07 | 5100 | 64 ± 9.0 | n.t.e | n.t.e |
| 3l | 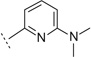 |
 |
H | H | 6.55 ± 0.08 | 284 | 87 ± 13.6 | 66 | 90 |
| 3m |  |
 |
H | H | 6.43 ± 0.05 | 375 | 77 ± 8.0 | 8 | 50 |
| 3n |  |
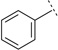 |
H | H | 6.36 ± 0.07 | 435 | 77 ± 11.9 | 17 | 49 |
| 3o |  |
 |
H | H | 6.64 ± 0.05 | 230 | 91 ± 11.1 | 13 | 28 |
| 3p |  |
 |
H | H | 6.43 ± 0.06 | 375 | 85 ± 11.6 | 11 | 29 |
| 3q |  |
H | H | 6.46 ± 0.11 | 344 | 30 ± 2.8 | n.t.e | n.t.e | |
| 3r |  |
H | H | 5.50 ± 0.09 | 3170 | 88 ± 12.4 | n.t.e | n.t.e | |
| 3s | 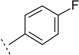 |
H | H | <5d | <10,000 | 65 ± 5.5 | n.t.e | n.t.e | |
| 3t |  |
Cl | H | 6.66 ± 0.08f | 218f | 5 ± 0.0.5f | 6 | 73 | |
| 3u | 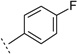 |
H | 7-Me | 5.68 ± 0.08 | 2070 | 60 ± 4.5 | n.t.e | n.t.e | |
| 3v | 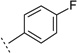 |
H | 8-Me | 6.00 ± 0.06f | 1000f | 16 ± 3.8f | 36 | 71 | |
Calcium mobilization assay using HEK293 cells stably expressing rat mGlu5 receptors; values are the average of three or more independent determinations.
Expressed as amplitude of response using 30 µM test compound (percentage of maximal response vs 100 µM glutamate).
HLM and RLM data refer to % of compound metabolized after incubation of tested compound with human and rat microsomes, respectively, for 15 min at 1 µM concentration.
Data obtained from a single experiment not replicated.
Not tested.
Antagonists/NAMs, data represents pIC50/IC50 from EC80 window and % Glu represents Emin not Emax.
We next evaluated the effects of the introduction of different substituted aryl and heteroaryl rings on the lactam. Based on our previous results,14 SAR investigation started with the introduction of fluorine-containing phenyl rings (3e–3g) resulting in a remarkable improvement in the mGlu5 PAM activity. Among this small set, the para- and the 3,4-substitutions (3e and 3g) proved to be more beneficial than the meta- (3f) in terms of potency and efficacy. Additionally, the meta-substitution was also found detrimental for the metabolic stability in rat liver microsomes (RLM). In a second step, we studied the effects of the replacement of the phenyl ring by more polar and weakly basic pyridines prioritizing the 2-substituted pyridine analogues over the 3 or 4 regioisomers (3h–3n).14 Thus, while only a slight decreased PAM activity was shown with compound 3h, the corresponding 2-pyridyl analogue of 3e; the introduction of a methyl substituent in 3-, 4- and 5-position of the 2-substituted pyridine ring (analogues 3i–3k), revealed a substantial loss of potency and efficacy. The recovery of both properties was achieved with a more basic analogue (3l) although unfortunately, this was accompanied by a high metabolic turnover in human liver microsomes (HLM) and RLM. Similarly, despite the combination of fluorine and methyl in the disubstituted pyridyl congeners 3m and 3n resulted in good in vitro PAM activity and efficacy, the improvement in metabolic stability in RLM was only moderate.
Encouraged by the promising profile of 3e, we next decided to evaluate the influence of the western aromatic ring in our molecules keeping constant for that purpose the para-fluorophenyl substitution on the amide moiety. Thus, the introduction of a fluorine atom in meta- and para-positions of the phenoxy ring resulted in similar PAM potencies and efficacies (EC50 = 230 nM, 91% Glu Max for 3o and EC50 = 375 nM, 85% Glu Max for 3p) to the parent analogue 3e. Similarly, the introduction of a methyl group also retained the in vitro activity although a remarkable decrease in efficacy was observed (3q, 30% Glu Max). In contrast, replacement of the western phenyl by a pyridine was found to be clearly detrimental for mGlu5 activity (compounds 3r and 3s).
Additionally, we also investigated the influence of small groups in different positions of the imidazopyrimidinone core, keeping again the para-fluorophenyl substitution on the lactam (compounds 3t–3v). This brief SAR survey led to an unexpected ‘molecular switch’ that changed the mode of the pharmacology. Thus, the introduction of a chlorine atom in the 3-position (3t) or a methyl substituent in the 8-position (3v) provided weak to moderate mGlu5 antagonists or negative allosteric modulators (NAMs) with IC50s of 218 and 1000 nM, respectively, and with additionally reduced metabolic stability in RLM. On the contrary, the 7-methyl analogue 3u still behaved as mGlu5 PAM although with a greatly reduced potency (EC50 = 2070 nM, 60% Glu Max) compared to parent congener 3e.
In parallel to our exploration of the imidazopyrimidinone series 3, we also synthesized a number of the more flexible dihydroimidazopyrimidinones 4 by hydrogenation of a set of analogues 3 presenting identified preferred modifications. SAR of this series is summarized in Table 2. Alkyl analogues in the lactam were found unstable under basic conditions20 and thus only aryl and heteroaryl substitutions were explored. Interestingly, this small library proved more productive than the previous one, providing several analogues with mGlu5 PAM activities below 250 nM. The specific comparison among pairs of analogues presenting fluorine decorations in the eastern aromatic aryl confirmed this trend; with a noticeable ~3.5 fold increase in potency for the para-fluorophenyl derivative 4a versus 3e and less prominent 1.8–2.4 increase for the meta- and the 3,4-difluoro substituted analogues (4b and 4c vs 3f and 3g, respectively). The same beneficial effect was also observed for the pyridyl-containing pairs (4d–4f vs 3h,i,m) with mGlu5 potency increases ranging from 1.6 to 4.1 fold, however counterparts 4g and 3n showed comparable activity. On the other hand, the increased flexibility in the central core did not result in any outstanding effect on microsomal stability.
Table 2.
Structures and activities of substituted dihydroimidazopyrimidinones 4
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | mGlu5 pEC50a (±SEM) |
mGlu5 EC50 (nM) |
% Glu maxb (±SEM) |
HLMc (%) |
RLMc (%) |
| 4a |  |
 |
H | 7.07 ± 0.02 | 86 | 70 ± 0.7 | 18 | 19 |
| 4b |  |
H | 6.66 ± 0.03 | 218 | 82 ± 9.2 | 20 | 78 | |
| 4c |  |
 |
H | 6.88 ± 0.06 | 130 | 91 ± 12.6 | 10 | 37 |
| 4d |  |
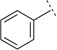 |
H | 6.44 ± 0.08 | 359 | 66 ± 2.1 | 1 | 14 |
| 4e |  |
 |
H | 6.31 ± 0.02 | 485 | 74 ± 9.6 | 35 | 98 |
| 4f |  |
 |
H | 6.74 ± 0.02 | 183 | 91 ± 13.0 | 31 | 93 |
| 4g |  |
 |
H | 6.43 ± 0.08 | 371 | 87 ± 16.1 | n.t.d | n.t.d |
| 4h |  |
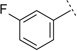 |
H | 7.16 ± 0.01 | 69 | 72 ± 0.7 | 0 | 29 |
| 4i |  |
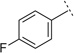 |
H | 6.70 ± 0.04 | 197 | 88 ± 12.2 | 3 | 11 |
| 4j | 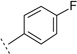 |
H | 6.55 ± 0.07 | 285 | 47 ± 3.3 | n.t.d | n.t.d | |
| 4k |  |
 |
Cl | 6.88 ± 0.05e | 133e | 14 ± 4.8e | n.t.d | n.t.d |
| 4l |  |
 |
Me | 6.28 ± 0.09e | 529e | 6 ± 1.4e | 21 | 46 |
Calcium mobilization assay using HEK293 cells stably expressing rat mGlu5 receptors; values are the average of three or more independent determinations.
Expressed as amplitude of response using 30 µM test compound (percentage of maximal response vs 100 µM glutamate).
HLM and RLM data refer to % of compound metabolized after incubation of tested compound with human and rat microsomes respectively, for 15 min at 1 µM concentration.
Not tested.
Antagonists/NAMs, data represents pIC50/IC50 from EC80 window and % Glu represents Emin not Emax.
Analogously, the introduction of a fluorine atom in the meta- (4h) and para-position (4i) of the western aryl, greatly increased mGlu5 potency up to ~3.3 fold versus series 3 corresponding counterparts, maintaining also a high efficacy. Gratifyingly, this exploration yielded analogue 4h, the most potent mGlu5 PAM across the two series described herein (EC50 = 69 nM, 72% Glu Max). However, the introduction of a methyl group in the meta-position (4j) contributed to a significant loss in efficacy in comparison with the other dihydroimidazopyrimidinones 4, confirming the tendency previously observed for series 3 (47% Glu Max for 4j vs 30% Glu Max for 3q).
Similarly to chemotype 3, the introduction of substituents in the 3-position of the dihydroimidazopyrimidinone core was responsible again for a change in the mode of pharmacology and resulted in compounds that behaved as antagonists/NAMs. Thus, the introduction of a chlorine atom in 4k or a methyl group in 4l, afforded mGlu5 antagonists/NAMs of moderate potency (133 and 529 nM, respectively). These data demonstrated again that with very subtle structural changes, a reasonably potent PAM can be transformed into an antagonist/NAM of comparable potency, confirming the findings previously reported for other chemotypes.15,21
After this initial exploration, analogues 3e and 4a were selected as representative examples from both chemotypes for additional characterization (Table 3). Thus, besides increased flexibility, 4a shows also a slightly reduced cLogP (0.5 log units). Both analogues were found adequately stable in HLM and RLM with no significant differences in the extent of metabolism. Additionally, in a cocktail assay using HLM and known substrates, 4a did not show any relevant inhibition of the major human cytochrome P450 (CYP) enzymes (2C9, 2D6, 3A4, 1A2) (IC50s >20 µM), while 3e was found to display moderate CYP inhibitory activity on 1A2 (IC50 = 9.3 µM, others IC50 >20 µM). Finally, 4a showed a slightly higher unbound fraction in both human and rat plasma compared with 3e.
Table 3.
Comparative profile of compounds 3e and 4a
| Compd | mGlu5 EC50a (nM) |
% Glu maxb (±SEM) |
c log Pc | mGlu3 EC50 (nM) |
HLMd (%) |
RLMd (%) |
hPPBe (%) |
rPPBe (%) |
|---|---|---|---|---|---|---|---|---|
| 3e | 310 | 83 ± 9.1 | 3.95 | 630 | 14 | 27 | 96.4 | 93.1 |
| 4a | 86 | 70 ± 0.7 | 3.42 | 290 | 18 | 19 | 90.3 | 88.1 |
Expressed as amplitude of response using 30 µM test compound (percentage of maximal response vs 100 µM glutamate).
Calculated with Biobyte software.
HLM and RLM data refer to % of compound metabolized after incubation of tested compound with human and rat microsomes respectively, for 15 min at 1 µM concentration.
Plasma protein binding, human and rat.
Subsequent selectivity profiling against the other mGlu receptors (mGlu1–4,6–8) revealed that both compounds were also active as full mGlu3 NAMs, (IC50 = 630 nM for 3e and 290 nM for 4a), resulting in moderate functional selectivities for mGlu5 of ~2 fold for 3e and ~3 fold for 4a respectively (other mGlu EC50 >10 µM). An analogous dual profile had already been observed for similarly substituted N-aryl analogues within our naphthyridinone series.15 Although the biological relevance of the mGlu3 receptor is still not well understood, inhibition of mGlu3 has been hypothesized to have potential therapeutic utility in the treatment of neurological and psychiatric disorders.22 Based on its mGlu5 PAM potency and efficacy, higher selectivity versus mGlu3, reduced cLogP, superior CYP profile and higher unbound fraction in plasma, 4a was selected for further in vivo pharmacokinetic (PK) and pharmacological evaluation.
Compound 4a was formulated at 1 mg/mL in 20% hydroxypropyl-β-cyclodextrin (HP-β-CD) and evaluated in in vivo PK studies in rats (1 mg/kg IV, 10 mg/kg PO).23 Compound 4a showed a moderate clearance (CLp = 23.3 mL/min/kg) and a terminal half-life (t1/2) of 0.93 h. Evaluation of drug concentrations in portal vein, plasma and brain (0.5, 1.0, 2.0, 4.0 and 7.0 h) revealed a moderate first-pass effect (Ehep = 0.43) with relatively low volume of distribution (Vss = 1.4 L/kg) and a good systemic exposure (plasmaAUC = 7.2 µM-h). In addition, 4a showed also a high brain exposure (brainAUC = 12.7 µM-h) with a very good distribution to the brain (brainAUC/plasmaAUC = 1.8), which together with a moderate unbound fraction in brain (rat fu brain = 6.2%) contributed to reach high absolute brain levels (Cmax = 6.05 µM). Encouraged by its overall profile, 4a was evaluated for its ability to revert AIH in rats, an established model of antipsychotic activity.10a,16 As seen in Figure 2, 4a showed a robust dose-dependent reversal of amphetamine effects with a lowest active oral dose of 3 mg/kg, and a maximal effect of ~50% at the highest doses tested of 56.6 and 100 mg/kg. At these doses the average terminal unbound brain concentrations (382 and 363 nM, respectively) are well above the in vitro mGlu5 PAM EC50.24
Figure 2.

Dose-dependent effect and calculated terminal unbound brain of 4a on the reversal of amphetamine-induced hyperlocomotion in rats.
In parallel to these explorations, potential mGlu5 related CNS adverse-effect liabilities driven by excessive glutamate fold potentiation25 or allosteric agonism26,27 were reported, suggesting that PAMs with lower functional cooperativity with glutamate and devoid of allosteric agonism may be preferred for an adequate therapeutic index.28 Concurrent with these findings and despite the fact that no significant adverse effects had previously been observed in PK and pharmacological experiments in rats up to 100 mg/kg p.o., investigation of 4a in the modified Irwin neurological test battery in rats at a high dose (120 mg/kg) and in in vivo PK studies in dogs (0.5 mg/mL i.v.) revealed clear signs of CNS-mediated side effects (pro-convulsive behavior and dizziness, respectively) which excluded any further in vivo evaluation. Although further in vitro characterization of 4a confirmed the lack of mGlu5 agonistic activity up to the highest concentration tested of 30 µM, fold shift experiments highlighted a high degree of cooperativity with glutamate (12.5-fold leftward shift in the glutamate concentration response curve at 10 µM) that could eventually explain the observed adverse effects, although a potential contribution of mGlu5/mGlu3 synergistic effects cannot be completely ruled out.
In summary, further optimization of our initial dihydrothiazolopyridone lead 1 resulted in two novel series of imidazopyrimidinones and dihydroimidazopyrimidinones as mGlu5 PAMs. Parallel SAR investigation of both series and further DMPK profiling of representative prototypes revealed dihydroimidazopyrimidinone 4a as a highly potent and efficacious mGlu5 PAM (EC50 = 86 nM, 70% Glu Max) and moderately potent mGlu3 NAM (IC50 = 290 nM, 3% Glu Max). 4a possessed an adequate PK profile in rats and showed robust dose-dependent effects in a preclinical model predictive of antipsychotic efficacy. However, the presence of CNS-mediated adverse effects in preclinical species precluded any further in vivo evaluation. Keeping in mind that more studies are necessary to address ongoing questions concerning the therapeutic index of mGlu5, further efforts within these and related series are in progress and will be reported in due course.
Acknowledgments
Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) research was supported by grants from Janssen Pharmaceutical Companies of Johnson & Johnson and in part by the NIH (NS031373 and MH062646).
References and notes
- 1.Conn PJ, Lindsley CW, Jones CK. Trends Pharmacol. Sci. 2009;30:25. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meltzer HY. Biol. Psychiatry. 1999;46:1321. [PubMed] [Google Scholar]
- 3.Kantrowitz J, Javitt DC. Curr. Opin. Psychiatry. 2012;25:96. doi: 10.1097/YCO.0b013e32835035b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moghaddam B, Javitt DC. Neuropsychopharmacology. 2012;37:4. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 6.(a) Anwyl R. Neuropharmacology. 2009;56:735. doi: 10.1016/j.neuropharm.2009.01.002. [DOI] [PubMed] [Google Scholar]; (b) Neisewander JL, Baker DA, Fuchs RA, Tran-Nguyen LT, Palmer A, Marshall JF. J. Neurosci. 2000;20:798. doi: 10.1523/JNEUROSCI.20-02-00798.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rosenbrock H, Kramer G, Hobson S, Koros E, Grundl M, Grauert M, et al. Eur. J. Pharmacol. 2010;639:40. doi: 10.1016/j.ejphar.2010.02.057. [DOI] [PubMed] [Google Scholar]
- 7.(a) Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Neurosci. 2000;20:7871. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Doherty AJ, Palmer MJ, Bortolotto ZA, Hargreaves A, Kingston AE, Ornstein PL, Schoepp DD, Lodge D, Collingridge GL. Br. J. Pharmacol. 2000;131:239. doi: 10.1038/sj.bjp.0703574. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ. J. Neurosci. 2001;21:5925. doi: 10.1523/JNEUROSCI.21-16-05925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser H, Rowe B, Sur C, Duggan ME, Pettibone DJ, Conn PJ, Williams DL., Jr J. Pharmacol. Exp. Ther. 2004;309:568. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- 8.Lindsley CW, Shipe WD, Wolkenberg SE, Theberge CR, Williams DL, Jr, Sur C, Kinney GG. Curr. Top. Med. Chem. 2006;8:771. doi: 10.2174/156802606777057599. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kinney GG, Burno M, Campbell UC, Hernandez LM, Rodriguez D, Bristow LJ, Conn PJ. J. Pharmacol. Exp. Ther. 2003;306:116. doi: 10.1124/jpet.103.048702. [DOI] [PubMed] [Google Scholar]; (b) Homayoun H, Stefani MR, Adams BW, Tamagan GD, Moghaddam B. Neuropsychopharmacology. 2004;29:1259. doi: 10.1038/sj.npp.1300417. [DOI] [PubMed] [Google Scholar]; (c) Lecourtier L, Homayoun H, Tamagnan G, Moghaddam B. Biol. Psychiatry. 2007;62:739. doi: 10.1016/j.biopsych.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Darrah JM, Stefani MR, Moghaddam B. Behav. Pharmacol. 2008;19:225. doi: 10.1097/FBP.0b013e3282feb0ac. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chavez-Noriega LE, Marino MJ, Schaffhauser H, Campbell UC, Conn PJ. Curr. Neuropharmacol. 2005;3:9. [Google Scholar]; (f) Chan MH, Chiu PH, Sou JH, Chen HH. Psychopharmacology (Berl.) 2008;198:141. doi: 10.1007/s00213-008-1103-1. [DOI] [PubMed] [Google Scholar]; (g) Schlumberger C, Pietraszek M, Gravius A, Danysz W. Pharmacol., Biochem. Behav. 2010;95:23. doi: 10.1016/j.pbb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, Smith S, Jacobson MA, Sur C, Duggan ME, Pettibone DJ, Conn PJ, Williams DL., Jr J. Pharmacol. Exp. Ther. 2005;313:199. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]; (b) Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, Smith D, Olsen M, Kouranova E, Lai M, Pruthi F, Pulicicchio C, Day M, Gilbert A, Pausch MH, Brandon NJ, Beyer CE, Comery TA, Logue S, Rosenzweig-Lipson S, Marquis KL. J. Pharmacol. Exp. Ther. 2008;327:827. doi: 10.1124/jpet.108.136580. [DOI] [PubMed] [Google Scholar]
- 11.(a) Gass JT, Olive MF. Biol. Psychiatry. 2009;65:717. doi: 10.1016/j.biopsych.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Olive MF. Eur. J. Pharmacol. 2010;639:47. doi: 10.1016/j.ejphar.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Uslaner JM, Parmentier-Batteur S, Flick RB, Surles NO, Lam JS, McNaughton CH, Jacobson MA, Hutson PH. Neuropharmacology. 2009;57:531. doi: 10.1016/j.neuropharm.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 12.Ayala JE, Chen Y, Banko JL, Sheffler DJ, Williams R, Telk AN, Noreen L, Watson NL, Xiang Z, Zhang Y, Jones PJ, Lindsley CW, Olive MF, Conn PJ. Neuropsychopharmacology. 2009;34:2057. doi: 10.1038/npp.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsley CW, Stauffer SR. Pharm. Pat. Anal. 2013;2:93. doi: 10.4155/ppa.12.82. [DOI] [PubMed] [Google Scholar]
- 14.Bartolome-Nebreda JM, Conde-Ceide S, Delgado F, Iturrino L, Pastor J, Pena MA, Trabanco AA, Tresadern G, Wassvik CM, Stauffer SR, Jadhav S, Gogi K, Vinson PN, Noetzel MJ, Days E, Weaver CD, Lindsley CW, Niswender CM, Jones CK, Conn PJ, Rombouts F, Lavreysen H, Macdonald GJ, Mackie C, Steckler TJ. Med. Chem. 2013;56:7243. doi: 10.1021/jm400650w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turlington M, Malosh C, Jacobs J, Manka JT, Noetzel MJ, Vinson PN, Jadhav S, Herman EJ, Lavreysen H, Mackie C, Bartolome-Nebreda JM, Conde-Ceide S, Martin-Martin ML, Tong HM, Lopez S, Macdonald GJ, Steckler T, Daniels JS, Weaver C, Niswender CM, Jones C, Conn JP, Lindsley CW, Stauffer SR. J. Med. Chem. 2014;57:5620. doi: 10.1021/jm500259z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindsley CW, Wisnoski DD, Leister WH, O’Brien JA, Lemaire W, Williams DL, Jr, Burno M, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. J. Med. Chem. 2004;47:5825. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 17.Siegel S, Wilmen A, Röhrig S, Svenstrup N, Gnoth MJ, Heitmeier S, Rester U, Zubov D, Strayle J, Sperzel M. WO 2008/113469 A2. Int. Pat. Appl. 2008
- 18.Yue Y, Zheng Z-G, Wu B, Xia C-Q, Yu X-Q. Eur. J. Org. Chem. 2005:5154. [Google Scholar]
- 19.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days E, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Mol. Pharmacol. 2010;78:1105. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buschauer A, Sattler HJ, Schunack W. Chem. Ber. 1984;117:2597. [Google Scholar]
- 21.Wood MR, Hopkins CR, Brogan JT, Conn JP, Lindsley CW. Biochemistry. 2011;50:2403. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Harrison PJ, Lyon L, Sartorius LJ, Burnet PWJ, Lane TA. J. Psychopharmacol. 2008;22:308. doi: 10.1177/0269881108089818. [DOI] [PubMed] [Google Scholar]; (b) Caraci F, Molinaro G, Battaglia G, Giuffrida ML, Riozzi B, Traficante A, Bruno V, Cannella M, Merlo S, Wang X, Heinz BA, Nisenbaum ES, Britton TC, Drago F, Sortino MA, Copani A, Nicoletti F. Mol. Pharmacol. 2011;79:618. doi: 10.1124/mol.110.067488. [DOI] [PubMed] [Google Scholar]
- 23.Intrinsic solubility of compound 4a in fasted simulated intestinal fluid was low (FaSSIF = 6 µg/mL at pH = 7.2).
- 24.Based upon rat brain homogenate binding fraction unbound (6.2%) and total brain levels ([brain]90min = 6.2 ± 2.9 and 5.9 ± 1.9 µM, respectively).
- 25.Parmentier-Batteur S, Hutson PH, Menzel K, Uslaner JM, Mattson BA, O’Brien JA, Magliaro BC, Forest T, Stump CA, Tynebor RM, Anthony NJ, Tucker TJ, Zhang XF, Gomez R, Huszar SL, Lambeng N, Faure H, Le Poul E, Poli S, Rosahl TW, Rocher JP, Hargreaves R, Williams TM. Neuropharmacology. 2013;62:1453. [Google Scholar]
- 26.Rook JM, Noetzel MJ, Pouliot WA, Bridges TM, Vinson PN, Cho HP, Zhou Y, Gogliotti RD, Manka JT, Gregory KJ, Stauffer SR, Dudek FE, Xiang Z, Niswender CM, Daniels JS, Jones CK, Lindsley CW, Conn PJ. Biol. Psychiatry. 2013;73:501. doi: 10.1016/j.biopsych.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bridges TM, Rook JM, Noetzel MJ, Morrison RD, Zhou Y, Gogliotti RD, Vinson PN, Xiang Z, Jones CK, Niswender CM, Lindsley CW, Stauffer SR, Conn PJ, Daniels JS. Drug. Metab. Dispos. 2013;41:1703. doi: 10.1124/dmd.113.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turlington M, Noetzel MJ, Chun A, Zhou Y, Gogliotti RD, Nguyen ED, Gregory KJ, Vinson PN, Rook JM, Gogi KK, Xiang Z, Bridges TM, Daniels JS, Jones C, Niswender CM, Meiler J, Conn PJ, Lindsley CW, Stauffer SR. J. Med. Chem. 2013;56:7976. doi: 10.1021/jm401028t. [DOI] [PMC free article] [PubMed] [Google Scholar]