

Abstract
Objective
HLA-B27 forms misfolded heavy chain dimers, which may predispose individuals to inflammatory arthritis by inducing endoplasmic reticulum (ER) stress and the unfolded protein response (UPR). We wanted to define the role of the UPR induced ER associated degradation (ERAD) pathway in the disposal of HLA-B27 dimeric conformers.
Methods
HeLa cell lines expressing only two copies of a carboxy terminally Sv5 tagged HLA-B27 were generated. The ER stress induced EDEM1 protein was over expressed by transfection and dimer levels monitored by immunoblotting. EDEM1, the UPR associated transcription factor XBP-1, the E3 ubiquitin ligase HRD1, the degradation associated derlin 1 and 2 proteins were inhibited by either short hairpin RNA or dominant negative mutants. The UPR associated ERAD of HLA-B27 was confirmed using ER stress inducing pharamacological agents in kinetic and pulse chase assays.
Results
We demonstrate that UPR induced machinery can target HLA-B27 dimers, and that dimer formation can be controlled by alterations to expression levels of components of the UPR induced ERAD pathway. HLA-B27 dimers and misfolded MHC class I monomeric molecules were detected bound to EDEM1, with overexpression of EDEM1 inhibiting HLA-B27 dimer formation. EDEM1 inhibition resulted in upregulation of HLA-B27 dimers, whilst UPR induced ERAD of dimers was prevented in the absence of EDEM1. HLA-B27 dimer formation was also enhanced in the absence of XBP-1, HRD1 and derlin1/2.
Conclusion
The UPR ERAD pathway as described here can dispose of HLA-B27 dimers and presents a potential novel therapeutic target for the modulation of HLA-B27 associated inflammatory disease.
Keywords: EDEM1, HRD1, MHC class I degradation, XBP1
HLA-B27 is strongly associated with a group of inflammatory arthritic conditions known as the spondyloarthropathies, with Ankylosing Spondyltis (AS) representing the prototypical presentation (1, 2). Up to 90–95% of patients with AS express HLA-B27 but despite this genetic association being known for almost four decades, the disease mechanism remains poorly understood. Genome-wide genetic screens have identified a number of other genes contributing to AS, but none have yet fully explained the disease process (3). One current theory linking HLA-B27 to AS involves the predisposition of HLA-B27 heavy chain to misfold and form non-native heavy chain dimeric structures (4). HLA-B27 dimers have been correlated with disease in the rat model for SpA (5). Dimers may interact aberrantly with immune receptors at the cell surface, or act within the environment of the endoplasmic reticulum (ER) to induce cellular stress responses, potentially leading to the onset of pro-inflammatory responses (6, 7).
MHC class I molecules present unique protein folding challenges to the ER quality control machinery and HLA-B27 is particularly prone to misfolding forming disulfided bonded dimers (8). Misfolding proteins within the ER can induce the Unfolded Protein Response (UPR), which is a cellular stress response initiating transcriptional changes whose function is to restore ER homeostasis (9, 10). There are three main effector molecules of the UPR, the ATF6 transcription factor and the two kinases IRE1 and PERK, which reside within the ER and all are maintained in an inactive state by the ER resident chaperone Immunoglobulin Binding Protein (BiP) (10–12). One of the most potent transcriptional changes induced by the UPR follows the oligomerisation and autophosphorylation of IRE1. Phosphorylated IRE1 can splice the cytosolically located X-box binding protein 1 (XBP-1) mRNA by excising a 26 nucleotide intronic sequence, generating the XBP-1 spliced(s) transcription factor (13). XBP-1s can activate chaperones to enhance both cellular folding capacity and/or proteins involved in degradation of misfolding substrates (14).
ER degradation-enhancing α-mannosidase-like protein 1 (EDEM1) was originally cloned as an ER stress induced gene (15), which is targeted by XBP-1s (14). EDEM1 overexpression studies have demonstrated a role in the degradation of misfolded proteins. EDEM1 is thought to handle misfolded proteins in concert with the ER chaperone calnexin and the UPR induced XBP-1 target E3 ubiquitin ligase HRD-1-SEL1 complex (16–18). Together with the E2-ubiquitin conjugating enzyme UBE2J1 they were shown to participate in the degradation of misfolded MHC class I heavy chains at steady state (19). Very recently, a role for EDEM1 in the MHC class I pathway has also been demonstrated (20), although its interaction with HLA-B27 was not investigated.
In this current study we demonstrate that EDEM1 participates in the degradation of HLA-B27 dimers under ER stress conditions. This degradation pathway requires XBP-1 and HRD1. Significantly, our data highlight the ER stress induced ERAD pathway as a potential route for therapeutic intervention in HLA-B27 associated diseases.
Materials and Methods
Cell lines and antibodies
Cells were maintained in DMEM, supplemented with 10% FBS (Globepharm), penicillin/streptomycin (D10 media) and maintained in a 5% CO2 37°C incubator. The Invitrogen Flp-In™ system was used to generate isogenic HeLa lines stably expressing HLA class I constructs. HeLa cells with two integrated copies of the pFRT/lacZeo plasmid were identified by Southern blot analysis. Clone H2Z, was β-galactosidase positive and Zeocin™ resistant. HLA class I constructs for expression were cloned into the pEF5/FRT/V5-D-TOPO® vector. The relevant FRT-containing expression plasmid was co-transfected together with the Flp recombinase expressing plasmid pOG44, into H2Z cells and selected using hygromycin B. Recombinants were identified by the lack of β-galactosidase expression and Zeocin™ sensitivity, thus generating stable isogenic lines containing two copies of HLA class I expression constructs driven by the human EF-1α promoter or CMV promotor.
The following antibodies were used: anti-V5 (pK) (Serotec), anti-FLAG, anti-EDEM1 and anti-rabbit monoclonal HRP (Sigma) and goat anti-mouse horseradish peroxidase (DAKO).
Transient transfections
HeLa and 293T cells were plated at 1 × 105 and 5 × 105 cells/well respectively and transfected with cDNA expression constructs and JetPrime PEI (PolyPlus Transfection) or Fugene respectively according to manufacturers’ recommended conditions.
Immunoprecipitation analysis
For EDEM1 immunoprecipitation experiments, HeLa cells expressing HLA-B*27:05 were transfected with a FLAG tagged EDEM1, incubated with 20mM NEM/PBS pH7.4 and lysed in 1% digitonin lysis buffer, immunoprecipitated with anti-FLAG antibody and washed in 1% digitonin buffer. For analysis of EDEM1 binding to MHC class I molecules, 293T cells co-transfected with EDEM1-FLAG and HLA class I molecules were treated in 20mM NEM, lysed in 1% NP40 lysis buffer and immunoprecipitated with anti-FLAG antibody.
Immunoblotting
Samples were resolved by non-reducing SDS-PAGE, transferred onto nitrocellulose (BA85, Schleicher and Scheull), blocked for 1 hr with 5% skimmed milk powder in PBS/0.1% Tween, followed by incubation with antibodies. Images were revealed by chemiluminescence using Supersignal Femto reagents (Pierce).
Knockdown analysis
For XBP-1 knockdown, transductions with shRNA-expressing lentiviral vector (pCSBX/pIG) targeting XBP-1 (shXBP-1) were as previously described (21). An equivalent control vector expressing an shRNA targeting luciferase (shLuc) was generated using the same methodology. HEK 293T cells were transfected with p8.91, pMDG, and the shRNA-expressing lentiviral vector genome using FuGENE-6. Samples showing high levels of viral transduction (>90% GFP+ by FACS) were processed and immunoblotted. For HRD1 and EDEM1 knockdowns, previously described 19mers matching HRD1 (19) or EDEM1 cDNAs (22) were incorporated into DNA oligos that were cloned into pSIREN-RetroQ (Clontech) shRNA expression vector and sequence verified. HEK 293Ts were transfected with plasmids encoding Moloney MLV gag-pol (pCMVi), VSV-G env (pMDG) and pSIREN-RetroQ-shHRD1 or shEDEM1 to make pseudotyped virus-like particles. Supernatants were harvested at 48hrs post-transfection.
Chemical inhibitor assays
HeLa cells expressing HLA-B*27:05 were incubated at 1 × 105 cells/well with varying concentrations of kifuensine (Calbiochem) or DMSO as controls and harvested 18 hrs later. ER induction and dimer half life assays; HeLa.B27 cells were incubated with varying concentrations of TUN, TPG and MG132 for ER stress studies or 50μM cycloheximide for half life analyses. Equal number of cells were harvested, NEM treated and lysed in 1% NP40 lysis buffer. Protein loading was normalised by cell number or MTT assay.
Monocyte-derived dendritic cells and quantitation
After full local ethical committee review, and informed consent, peripheral blood mononuclear cells were isolated by centrifugation of whole blood over Histopaque (Sigma, Poole, UK). Two hour plastic adherent monocytes (in RPMI, 10% FBS) were cultured with 50ng/ml interleukin 4 (IL-4) and 50ng/ml granulocyte macrophage-colony stimulating factor (GM-CSF) (R&D Systems, Abingdon, UK). DCs were allowed to differentiate for 5 days, before treatment with 100ng/ml lipopolysaccharide (LPS) (Sigma, Poole, UK). Cell lysates were analysed by SDS-PAGE and immunoblotted for MHC class I heavy chain with HC10, and for HRD1, and developed with Supersignal Femto chemiluminescent reagents. HRD1 and B27D values were generated from the Integrated Density in Image J. All values were then normalised to β-actin. For quantitation of HLA-B alleles lysates were analysed for HC10 reactivity and values generated as described.
Pulse chase analysis
Cells were serum starved for 15 mins and labeled with 35S cys/met 300μCi for 15 mins. Radiolabel was removed and cells resuspended in D10. For ER stress or pharmacological treatment, drugs were added at the point of chase. Equal number of cells were removed at each time point, treated with 20mM NEM pH7.4 on ice for 20 mins, lysed in 1% NP40 and immunoprecipitated for MHC class I. Prior to resolution on non-reducing SDS-PAGE, samples were treated with endoH for 1hr at 37°C.
Results
EDEM1 can associate with aberrant MHC class I conformers
To determine the role of EDEM1 in the processing of HLA-B27 dimers, we employed isogenic HeLa cells expressing two copies of either Sv5 C-terminally tagged HLA-B27 (HeLa-B27.Sv5) or HLA-B*35:01 (-B35) under the control of the E1Fα promoter, the latter as a control HLA-B allele which does not form disulfide linked dimers.
Flow cytometric analysis of HeLa.B27.Sv5 and -B35.Sv5 with ME1 and 4E antibodies respectively demonstrated efficient cell surface expression of these MHC class I molecules (Fig 1A, top and bottom left panels), whilst W632 staining demonstrated that all lines expressed endogenous MHC class I molecules. We assessed the amount of HLA-B proteins expressed by HeLa compared to HeLa.B27.Sv5 using immunoblotting with HC10. Quantitation revealed approximately twice as much HLA-B expression in HeLa.B27.Sv5 compared to wild type HeLa cells, in keeping with the additional two copies of HLA-B27 (Fig. 1B). We previously showed that HLA-B27 exhibits slow maturation kinetics (23) and pulse chase analysis of HeLa.B27.Sv5 confirmed the slow maturation kinetics compared to HLA-B35 as demonstrated by the acquisition of endoglycosidase H (endoH) resistant material within 2hrs (Fig. 1C, second and third panels). Pulse chase analysis of endogenous W632 reactive MHC class I molecules in wildtype HeLa (Fig. 1C top panel) and the analysis of HLA-B35, demonstrated that MHC molecules can be processed efficiently and rapidly by HeLa cells and that the slow kinetics of HLA-B27 are independent of cell line expression.
Figure 1.

HLA-B27 expression and maturation in HeLa cells; (A) HLA-B27 and –B35 are expressed efficiently at the cell surface. HeLa.B27.Sv5 and –B35.Sv5 were analysed by flow cytometry with antibodies ME1 (top left) and 4E (bottom left) respectively. HeLa.B27, -B35 and mock transfected control E84 express W632 reactive MHC class I molecules. (B) Quantitation of HLA-B alleles using HC10 antibody signal reveals approximately twice as much HLA-B expression in HeLa.B27.Sv5 compared to wild type HeLa cells. (C) HeLa cells process MHC class I molecules efficiently. Pulse chase analysis of endogenous MHC class I molecules using antibody W632 followed by endo H digestion reveals that MHC class I molecules mature within 1hr (top panel). Pulse chase of HLA-B27.Sv5 using the anti-Sv5 antibody pK reveals little endo H resistant material being acquired during 2 hrs of chase (middle panel). Control HLA-B35.Sv5 exhibit a rapid acquisition of endo H resistance in HeLa cells (bottom panel).
To determine the role of EDEM1 in the processing of HLA-B27 dimers, we both enhanced and inhibited EDEM1 expression. The HeLa-B27.Sv5 line expresses three distinct dimer populations (arrows 1–3) which we have previously determined corresponds to dimers in different redox states (24). Overexpression of EDEM1 resulted in a modest reduction in the levels of all HLA-B27 dimer populations (Fig. 2A, arrows 1–3). Depletion of EDEM1 expression (Fig. 2B, left panel) led to enhancement in all three dimer populations (Fig. 2B, right top panel). As both of these detectable differences in dimer levels were made at steady state, we then induced ER stress in the absence of EDEM1 using thapsigargin (TPG). Immunoblotting revealed that HLA-B27 dimers were more abundant in EDEM1 depleted cells (Fig. 2B, right bottom panel).
Figure 2.
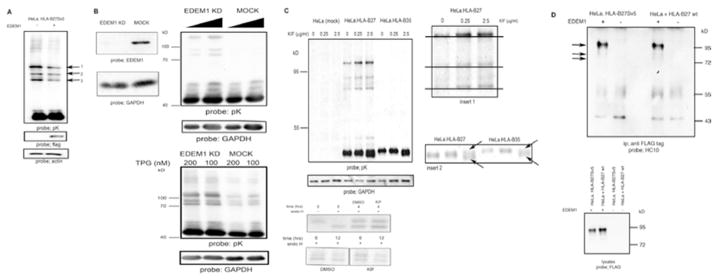
EDEM1 modulates HLA-B27 dimers; (A) Over expression of EDEM1 can enhance HLA-B27 dimer degradation. HeLa-B27 were transfected with EDEM1-FLAG, lysed and immunoblotted for MHC class I heavy chain. Arrows 1, 2 and 3 indicate HLA-B27 dimers. Lysates immunoblotted with anti-FLAG demonstrate EDEM1 expression. (B) shRNA inhibition of EDEM1 expression (left panel) in HeLa-B27 cells prevents HLA-B27 dimer ERAD when compared to mock transduction with a control scrambled shRNA (middle panel). HLA-B27 dimers are resistant to ER stress induced degradation following EDEM1 knockdown (right panel). (C) Increasing concentrations of kifuesine (KIF) lead to enhanced detection of dimers as determined by immunoblotting of HeLa-B27.Sv5. Immunoblotting for GAPDH reveals equal loading. Insert 1 demonstrates that with increasing KIF concentrations the high Mw dimers exhibit small changes in Mw. Insert 2 arrows indicate that at 2.5 μg/ml KIF, the monomer of both HLA-B27:05 and –B*35:01 can be resolved as a doublet (left panel) Kifuensine does not alter the maturation rate of HLA-B27. HeLa-B27.Sv5 were radiolabeled and chased in the presence of kifuensine or vehicle control DMSO (right panel). (D) HLA-B27 ER resident dimers preferentially associate with EDEM1 (left panel). HeLa-B27.Sv5 were transfected with EDEM1-FLAG, or HeLa cells were co-transfected with HLA-B27 and EDEM1-FLAG and cell lysates were immunoprecipitated with an anti-FLAG tag antibody and immunoblotted for class I heavy chain (left panel). Immunoblotting lysates with anti-FLAG from HeLa and HeLa-B27Sv5 cells transfected with EDEM1 demonstrates equal levels of EDEM1 expression (right panel).
To further verify that EDEM1 can participate in the degradation of aberrant HLA-B27 dimers we employed a pharmacological approach. HLA-B27 dimers were enhanced after treatment with kifuensine (KIF) which inhibits ERAD and the interaction of EDEM1 with the SEL1-HRD1 degradation complex (18, 25, 26) (Fig. 2C, top left panel). In addition, the dimer complexes displayed a small increase in Mw, characteristic of the retention of high mannose groups due to the inhibition of class I α-mannosidase activity (Fig. 2C, Insert 1 enlarged image of dimers). The monomers for both HLA-B27 and –B35 were also resolved as two closely migrating bands in the presence of KIF, which may represent a mannosylated heavy chain and/or intermediates of HLA heavy chains prior to ERAD (Fig. 2C, Insert 2 arrows, enlarged image of monomeric heavy chains). KIF treatment could potentially affect dimer formation by altering the maturation rate of HLA-B27. However, pulse chase analysis in the presence of KIF revealed no alteration in HLA-B27 maturation rates (Fig. 2C, bottom left panel).
To determine whether EDEM1 can directly associate with different forms of HLA-B27, EDEM1-FLAG cDNA was either transfected into HeLa-B27.Sv5 cells, or co-transfected with HLA-B*27:05 cDNA into HeLa cells. Following immunoprecipitation of EDEM1, immunoblotting for HLA-B27 with antibody HC10, demonstrated the preferential association of HLA-B27 dimers compared to monomeric HLA-B27 (Fig. 2D, top panel). These data suggest that EDEM1 can both associate with MHC class I dimeric heavy chains and modulate their levels.
Misfolded MHC class I heavy chains interact with EDEM1 in a glycan independent manner
To address whether EDEM1 can associate directly with misfolded monomeric heavy chains, we used a cell based binding assay. 293T cells were co-transfected with EDEM1-FLAG and a C-terminally Sv5 tagged wild type HLA-B*27:05 or with similarly tagged misfolding HLA-B27 molecules which have had the conserved structural cysteines of the α2 domain mutated to serines at position 101 (C101S) or 164 (C164S). We employed the structural HLA-B27.C101S mutant as the monomer misfolds but retains the ability to extensively dimerise and the C164S mutant exhibits enhanced levels of misfolded monomer with a reduced ability to dimerise (23). Therefore by using these mutants we could determine whether EDEM1 associates with dimeric and/or monomeric conformations. 24hrs post-transfection, 293T cells were harvested, treated in 20mM NEM and lysed in 1% NP40. Lysates were immunoprecipitated for EDEM1 using an anti-FLAG antibody and resolved by reducing SDS-PAGE. Immunoblotting for HLA-B27 with the anti-Sv5 pK antibody indicated that WT HLA-B*27:05 and the C101S and C164S mutants could be co-immunoprecipitated with EDEM1 (Fig. 3A). When resolved under non-reducing conditions, fewer monomeric wildtype HLA-B27 heavy chains interacted with EDEM1. It is likely that this pool would contain both correctly and misfolded monomers, whereas both C101S and C164S monomer heavy chain pools would all be classified as misfolded (Fig. 3B). Interestingly, C101S predominantly associated as a dimer with EDEM1, whilst C164S demonstrated an altered dimeric conformation, but could associate with EDEM1 both as a monomer and dimer (Fig. 3B).
Figure 3.

EDEM1 associates with dimeric and monomeric MHC class I conformers. (A) MHC class I molecules are detected only when coexpressed with EDEM1. 293T cells were cotransfected with EDEM1 and HLA-B27.Sv5 or along with two misfolding mutants B27.C101S and B27.C164S. Lysates were immunoprecipitated for anti-FLAG, resolved by reducing SDS-PAGE and immunoblotted for V5 using the pK antibody. (B) Both monomers and dimers can be detected in association with EDEM1. Similar immunoprecipitates were resolved by non-reducing SDS-PAGE and immunoblotted with pK antibody. Note that dimeric structures are preferentially detected in the presence of WT HLA-B27 and the C101S mutant (?). * denotes non-specific background band. (C) EDEM1 associates with HLA-B27 in a glycosylation independent manner. 293T cells were cotransfected with EDEM1, E225Q, D370N, E493Q or a triple mutant (TM) contaning all three mutations, along with HLA-B27 wt or a glycosylation site deficient mutant N86K. Lysates were immunoprecipitated with anti-FLAG, resolved by reducing SDS-PAGE and probed with pK. (D) EDEM1 was immunoprecipitated and endo H digested following co-transfection with HLA-B27 in 293T cells, followed by immunoblotting for MHC class I heavy chain.
To determine whether the EDEM1 mannosidase-like domain was important in HLA-B27 heavy chain recognition we employed a series of EDEM1 mutants whose acidic active site residues within the mannosidase-like domain have been mutated to neutral amino acids which abolish glycan dependent substrate binding; E225Q, D370N or E493Q. In addition we also employed a triple mutant of this domain (TM, E225Q/D370N/E493Q). To further address the requirement of glycan binding, we employed a glycosylation site deficient mutant of HLA-B27 (N86K). Using our cell based binding assay we co-expressed EDEM1 WT or the four EDEM1 mutants with HLA-B27 or the B27.N86K mutant. Cells were NEM treated, lysed in 1% NP40 and immunoprecipitated for EDEM1. Immunoblotting for MHC class I heavy chains demonstrated both WT and N86K HLA-B27 could be co-immunoprecipitated with all EDEM1 mutants (Fig. 3C).
HLA-B27 dimers can be ER resident and expressed at the cell surface. To determine which pool of dimers were associated with EDEM1, we subjected the EDEM1-associated HLA-B27 molecules to digestion with endoH. Immunoblotting revealed that following endoH digestion, the dimeric population associating with EDEM1 exhibited an apparent lower Mw, correlating with the removal of glycans from ER resident dimers (Fig. 3D, right panel). Furthermore, we could also detect monomeric HLA-B27 under these overexpression experimental conditions, which were also susceptible to endoH digestion (Fig. 3D, right panel).
Therefore the EDEM1-MHC class I interaction is not dependent on glycosylation of the substrate or the glycan binding site of EDEM1, with EDEM1 binding ER resident dimers and misfolding monomeric heavy chains.
HLA-B27 dimer ERAD requires the XBP-1 pathway, HRD1 and can use Derlin 1 and 2
As EDEM1 is a target gene for the potent UPR XBP-1s transcription factor (14, 27) we predicted that heavy chain dimer degradation would require an intact XBP-1 pathway. We transduced HeLa.B27.Sv5 with a lentivirus expressing a short hairpin (sh) RNA targeting XBP-1. Immunoblotting revealed that in the absence of XBP-1, HLA-B27 dimers are more prevalent (Fig. 4A, left panel and enlarged insert panel).
Figure 4.

XBP-1, HRD1 and Derlin1/2 participate in HLA-B27 dimer degradation. (A) Degradation of HLA-B27 is dependent on the XBP-1 pathway. shXBP1 inhibition demonstrates enhanced dimerisation of HLA-B27, with Insert showing the dimers following longer exposure (left panel). shRNA inhibition of HRD1 expression in HeLa-B27 cells prevents HLA-B27 dimer ERAD when compared to mock transduction with a control shRNA (shLUC). Immunoblotting lysates demonstrates that HRD1 has been down regulated (right panel). (B) HRD1 and EDEM1 depend on XBP-1 for ER stress induction. ShRNA inhibition of XBP-1 does not lead to induction of HRD1 and EDEM1 following ER stress induction with thapsigargin (TPG) compared to shLUC mock control cells. (C) HRD1 and HC10 reactive HLA-B27 dimer levels were quantified in both immature (I) and mature (M) dendritic cells generated from HLA-B27 patients. HRD1 levels can be elevated in dendritic cells from AS patients. Open bars represent normalised B27 dimer (D) levels and shaded bars represent normalised HRD1 levels. (D) Derlin-1 and –2 can participate in HLA-B27 dimer ERAD. Derlin-1.GFP and Derlin-2.GFP expression following retroviral infection of HeLa cells expressing HLA-B27 and HLA-B35 (left panel). HeLa-B27 cells were transduced with Derlin-1 and -2 GFP tagged proteins, which act as dominant negative inhibitors of the respective Derlin activities. Immunoblotting revealed that both Derlin-1 and –2 can modestly enhance HLA-B27 dimer formation, leading to the enhanced detection of dimers as highlighted by arrows 1, 2 and 3 (middle panel). Quantitation of the different dimer bands 1, 2 and 3 reveals a 2–3 fold increase in their detection when Derlin 1 and 2 activities are inhibited (right panel).
EDEM1 and HRD1 are targets for XBP-1s induction (14, 27) and EDEM1 can associate with the HRD1-SEL1 degradation complex (18), we therefore wanted to confirm the role of HRD1 in HLA-B27 dimer degradation. We hypothesized that inhibition of HRD1 expression should prevent ERAD of HLA-B27 dimers. HeLa-B27.Sv5 cells were infected with a retrovirus expressing an shRNA construct targeting HRD-1. Immunoblotting for HLA-B27 dimers revealed a large accumulation of dimers and larger aggregates (Fig. 4A, right panel). We then wanted to determine how inhibiting XBP-1 affected HRD1 and EDEM1 expression. XBP-1 inhibition led to a reduction in steady state levels of HRD1 but did not greatly affect EDEM1. We then subjected HeLa cells that were either mock transduced or had XBP-1 inhibited with the ER stress inducing agent TPG. HRD1 and EDEM1 could both be induced by increasing concentrations of TPG, however in the absence of XBP-1, neither of these proteins could be significantly induced (Fig. 4B). Therefore, inhibiting XBP-1 can prevent the induction of EDEM1 and HRD1 under stress conditions.
To determine the physiological significance of the above pathway for HLA-B27 dimer degradation, we analysed HLA-B27 dimer and HRD1 levels in cells from HLA-B27 positive AS patients. Monocyte derived dendritic cells from peripheral blood were stimulated with LPS and MHC class I dimer and HRD1 levels analysed by immunoblotting. Intriguingly, HRD1 levels were consistently higher in patient derived DC compared to HLA-B27 positive healthy controls (Fig. 4C) suggesting HRD1 may regulate HLA-B27 dimer levels in AS patient cells.
The derlin proteins have been postulated to be part of putative degradation pores and act as adaptors for ERAD substrates (28). Derlin 1 is involved in US11 mediated retrotranslocation of monomeric MHC class I molecules (29), whilst derlin 2 links EDEM1 with the ERAD p97 component. Furthermore derlins 1–3 can be induced by ER stress (30). As derlin 1 has a role in MHC class I ERAD and derlin 2 has been demonstrated to associate with EDEM1 we therefore wanted to determine whether HLA-B27 dimer degradation could be affected by derlin 1 and/or 2. We employed derlin 1 and 2 Green Fluorescent Protein (GFP) fusion proteins (Fig. 4D, left panel) which act as dominant negative proteins for their respective products (29). The inhibition of derlin 1 and 2 by their respective dominant negative proteins resulted in enhanced dimers (Fig. 4D, middle and right panel). Thus HLA-B27 dimers can be targeted for degradation via an XBP-1 dependent pathway and their degradation involves the E3 ubiquitin ligase HRD1 and the putative adaptor proteins derlins 1 and/or 2.
HLA-B27 heavy chain dimers are long lived
HLA-B27 dimers have been postulated to be pathogenic and participate in inflammatory arthritic disease (5). However, the duration of HLA-B27 dimers has been poorly analysed, therefore we initially wanted to determine the half life of HLA-B27 dimers. HLA-B27 HC10 reactive dimers have been reported to have a half life of approximately 2hrs (31), however using our controlled expression system of HLA-B27 we could determine the duration of all populations of dimers.
Using HeLa.B27 cells, we metabolically labelled the cells for 15 mins with 35S cys/met before chasing for 0, 2, 4, 6 and 8hrs and immunoprecipitating with the anti-Sv5 pK antibody. Pulse chase analysis revealed that dimers were long lived and evident up to 8 hrs (Fig. 5A). To further assess the half life of HLA-B27 dimers, we treated cells with the protein synthesis inhibitor cyclohexamide (50μM) and assessed dimer levels by immunoblotting. Again this analysis revealed that HLA-B27 dimers were extremely long lived with detectable levels remaining 18hrs after cyclohexamide treatment (Fig. 5B). Therefore, at steady state levels HLA-B27 dimers exhibit a prolonged lifetime and appear to be resistant to steady state degradation pathways.
Figure 5.
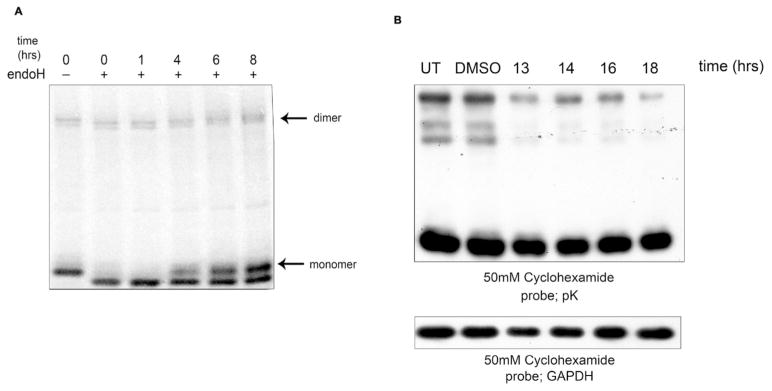
Half life and UPR induced ERAD of HLA-B27 heavy chain dimers. (A) HLA-B27 dimers have a prolonged duration. HeLa.B27Sv5 cells were metabolically labeled, chased and immunopecipitated with pK antibody. HLA-B27 dimers were apparent at 8hrs of chase. (B) HLA-B27 dimers have a half life in excess of 16hrs. HeLa.B27Sv5 cells were treated with 50μM cycloheximide and dimer levels detected by immunoblotting lysates resolved by non-reducing SDS-PAGE.
ERAD of HLA-B27 dimers is enhanced by pharmacological induction of the UPR
Our data indicate that induction of ER stress could aid in dimer removal and could potentially be employed as a therapeutic intervention strategy. If EDEM1 and/or other proteins upregulated during ER stress are involved in ERAD of HLA-B27 dimers, we hypothesised that induction of the UPR should enhance the removal of HLA-B27 dimers. To test this HeLa-B27.Sv5 cells were incubated with pharmacological agents known to induce the UPR, such as tunicamycin (TUN), TPG and MG132 (32). Immunoblotting revealed significantly fewer HLA-B27 dimers, suggesting that ER stress-induced ERAD was regulating dimer levels (Fig. 6A).
Figure 6.

(A–D); Incubation with UPR inducing drugs leads to loss of HLA-B27 dimers. (A) Treatment with tunicamycin (TUN), thapsigargin (TPG) and MG132 lead to loss of HLA-B27 dimers. (B) TUN and TPG lead to dimer disappearance after 8hrs of treatment (top left and right panels). ER stress drugs exhibit a biphasic effect of action. TUN and MG132 lead to the accumulation of dimers up to 8 hrs (left and right panels) TUN results in the appearance of glycan free (-CHO) forms of dimers (left). (C) Tunicamycin enhances the rate of dimer degradation. HLA-B27 cells were labeled with 35S cys/met and then chased in the presence (top panel) or absence (bottom panel) of tunicamycin (5μg/ml). Cells were chased for 0, 2, 4, 13, 14, 16 and 18hrs and immunoprecipitated for MHC class I heavy chain with the pK antibody. Dimers (D) and monomers (M) are highlighted, * denotes BiP. Note dimers are not apparent after 4hrs of chase (top panel). (D) The maturation rate of HLA-B27 as determined by pulse chase analysis is not grossly affected by ER stress inducing drugs (TPG and TUN, panels 1–3). Endogenous MHC class I molecules expressed by HeLa cells can mature within 1 hr (panel 4). HLA-B35 matures rapidly compared to HLA-B27 when expressed in HeLa cells (panel 5–6).
We then determined whether ER stress reduced the half life of dimers by performing kinetic and pulse chase analyses. A kinetic study of the effects of these drugs revealed initial enhanced dimerisation, as might be expected from their ability to inhibit normal folding processes, but this was followed by dimer disappearance (Fig. 6B–C and unpublished observations). For pulse chase analysis, cells were metabolically labelled and at the point of chase, cells were incubated with TUN (Fig. 6C, top panel). Following such treatment, dimers were indeed degraded far more rapidly disappearing within 8–12hrs post treatment (Fig. 6C bottom panel, compare top with middle panel). Previously, we demonstrated that changes within the 1–2hrs of maturation can affect the ability of MHC class I molecules to dimerise (23). It was possible that dimer levels could be modulated by the effects of these drugs on the maturation rates of the HLA-B27 monomer. However, pulse chase analysis in the presence of TPG and TUN did not reveal any alterations to the maturation phenotype following ER stress induction during the first 4hrs. However TUN appeared to enhanced the maturation rate markedly, whilst TPG had little if any effect (Fig. 6D, compare panels 1–3). Therefore, induction of ER stress, using a pharmacological approach can modulate levels of aberrant misfolded MHC class I molecules.
Discussion
We have defined a role for EDEM1 in the degradation pathway of HLA-B27 dimeric molecules. The XBP-1 induced ERAD of HLA-B27 dimers involves EDEM1, the E3 ubiquitin ligase HRD1 and the adaptor proteins derlin-1 and –2 (Fig. 4). EDEM1 shows specific affinity for HLA-B27 dimers as well as binding to misfolded monomers (Figs 2&3).
The induction of ERAD via the UPR has been demonstrated to lead to the degradation of misfolded proteins (33). UPR induced ERAD appears to target misfolded polypeptides in preference to folded monomers and this could in part be explained by the activity of EDEM1. How EDEM1 distinguishes such structures remains unresolved, but could involve interactions with calnexin and/or BiP. EDEM1 was originally proposed to act on misfolding substrates through its proposed mannosidase activity. However, our findings demonstrate that the mannosidase active site of EDEM1 is not necessary for substrate interactions (Fig. 3C). Our study suggests that the glycosylation status of this substrate does not influence its interaction with EDEM1 (Fig. 3C). These observations are consistent with those made for EDEM1 interactions with the model misfolding α-1-antitrypsin null Hong Kong protein (18).
Our study strongly suggests that HLA-B27 dimers are long lived. Relatively little is known regarding the half life and duration of such aberrant conformers. It has been proposed that HC10 reactive dimers have a half life of approximately 2hrs (31). However, in our system HLA-B27 expression is controlled and employs a C-terminally tagged epitope which enables us to perform an unbiased analysis of dimeric populations. It is possible that dimer duration and expression could be affected by additional factors. Recently, ER Aminopeptidase (ERAP)1, which trims MHC class I associated peptides (34) has also been found to be associated with AS (3). The peptide trimming activity could be altered by ERAP1 polymorphisms associated with AS or by increased expression levels (35), which could generate unstable peptides similar to those observed in ERAP1 knockout cells (36). An altered peptide subset could lead to MHC class I misfolding, increasing the propensity for HLA-B27 to dimerise and potentially overload the ERAD cell machinary. Recently, a poor supply of peptides was postulated to participate in the expression of cell surface HLA-B27 dimeric conformers (37). Therefore it is entirely possible that the peptide repertoire can participate in the expression of aberrant ER resident or plasma membrane associated HLA-B27 conformers.
Our findings have three potentially important implications. First, if misfolding and aggregation of HLA-B27 cause the associated inflammatory diseases then they could be targeted for therapeutic intervention in a manner similar to other protein conformational diseases. HRD1 activity has been implicated in such disorders (38) and our demonstration that HRD1 expression and possibly activity correlates with disease, suggest that this pathway may have an important role in how these conformers are handled in AS patients. Though we demonstrate that HRD1 and EDEM1 participate in the degradation of HLA-B27 dimers and that this pathway may be activated in AS patients, it is possible that differences exist in these pathways when comparing ex vivo DC expressing physiological levels of MHC class I molecules compared to transfected cell lines, and as such will require further investigation. Secondly, EDEM1 and engagement of the HRD1-SEL1 complex appear to be the rate limiting steps in the degradation of pathogenic disulfide bonded dimers of HLA-B27. It has previously been postulated that the resolution of disulfide bonds of HLA-B27 homodimers determined the rate of their degradation, however at steady state these molecules are far more long lived and stable than originally proposed. Thirdly, the degradation of HLA-B27 dimers offers the potential for therapeutic targetting by manipulating the ER stress response and induction of the ERAD pathway. Our study demonstrates that the ER stress pathway can be manipulated to modulate dimer expression. The use of ER stress inducing drugs highlights this “proof of principle” application with dimers becoming less apparent following the use of such ER stress inducing drugs (Fig. 6). Pulse chase analysis with the drugs employed at the point of chase demonstrate that the heavy chain can still mature slowly and that maturation is not altered significantly to account for the reduced detection of dimeric HLA-B27 populations (Fig. 6D). Thus the effects that we observe on dimer levels by these drugs is not due to their effects on folding. These drugs in theory should perturb folding and as such cause an accumulation of dimers. However, kinetic studies demonstrate a biphasic action of these drugs with early stages (0–8hrs) showing a brief accumulation of dimers (Fig. 6B) which then decline at later time points correlating with the induction of HRD1 (unpublished observations). Thus the initial effect of ER stress inducing drugs is to interfere with protein folding, with the accumulation of misfolding protein triggering ER stress pathways.
The spondyloarthropathies have been hypothesised to be disorders resulting from HLA-B27 misfolding, with dimerisation contributing to the disease process (4,39). Understanding the factors that contribute to the misfolding of HLA-B27 could have an impact on future therapeutic approaches. Our study suggests that modulating ER stress could offer a novel therapeutic intervention point. Drugs modulating ER stress responses are already under clinical development and testing for the treatment of multiple myelomas (40, 41). These could be tested and if effective repurposed for the treatement of HLA-B27 associated group of diseases and also other disorders associated with protein misfolding.
Acknowledgments
H.F is supported by an Arthritis Research (AR) UK project grant (17222). I.L is supported by ARUK studentship (17868) A.N.A is supported by an ARUK Fellowship (15293). E.C.C is supported by the Chief Scientist Office of the Scottish Government. D.N.H. is supported by US Public Health grant GM086874. G.J.T. is supported by a Wellcome Trust Senior Biomedical Fellowship. We thank Prof. H. Ploegh for the supply of reagents.
Abbreviations
- AS
Ankylosing Spondylitis
- BiP
Immunoglobulin Binding Protein
- ER
endoplasmic reticulum
- CST
casternospermine
- DC
dendritic cell
- DMJ
Deoxymannojirimycin
- DNJ
deoxynorjirimycin
- EDEM1
ER degradation-enhancing α-mannosidase-like protein 1
- ERAD
ER-associated degradation pathway
- ERAP
ER Aminopeptidase
- GFP
Green fluorescent protein
- HLA
Human Leukocyte Antigen
- HRD-3
hydroxyl-3-methylglutaryl reductase degradation
- KIF
kifuensine
- NEM
N-etheylmalemide
- SWN
swansonine
- TPG
tapsigargin
- TUN
tunicamycin
- UPR
unfolded protein response
- XBP1
X-box binding protein 1
Footnotes
CONFLICT OF INTEREST; The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
David B. Guiliano, Helen Fussell, Izabela Lenart, Nasim Yousef; performing and designing experiments and contributions to the manuscript
Ed Tsao, Adam Fletcher, Paul Kellam, Greg Towers; generation of shRNA targeting XBP-1 and HRD1 and confirmation of specificity
Darren Nesbeth, Keith Gould; generation of the HeLaFLP lines and contributions to the manuscript
Elaine C. Campbell, Sarah Lynch, Susana Santos, Amy Cameron; performing and designing experiments.
Daniel N. Hebert; cloning and tagging EDEM1 and contributing to the manuscript. Simon J. Powis, and Antony N. Antoniou; performing and designing experiments, directing the programme of research and writing the manuscript.
References
- 1.Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med. 1973;288(14):704–6. doi: 10.1056/NEJM197304052881403. [DOI] [PubMed] [Google Scholar]
- 2.Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DC, Sturrock RD. Ankylosing spondylitis and HL-A 27. Lancet. 1973;1(7809):904–7. doi: 10.1016/s0140-6736(73)91360-3. [DOI] [PubMed] [Google Scholar]
- 3.Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39(11):1329–37. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antoniou AN, Lenart I, Guiliano DB. Pathogenicity of Misfolded and Dimeric HLA-B27 Molecules. Int J Rheumatol. 2011;2011:486856. doi: 10.1155/2011/486856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tran TM, Satumtira N, Dorris ML, May E, Wang A, Furuta E, et al. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol. 2004;172(8):5110–9. doi: 10.4049/jimmunol.172.8.5110. [DOI] [PubMed] [Google Scholar]
- 6.Kollnberger S, Bird L, Sun MY, Retiere C, Braud VM, McMichael A, et al. Cell-surface expression and immune receptor recognition of HLA-B27 homodimers. Arthritis Rheum. 2002;46(11):2972–82. doi: 10.1002/art.10605. [DOI] [PubMed] [Google Scholar]
- 7.Layh-Schmitt G, Colbert RA. The interleukin-23/interleukin-17 axis in spondyloarthritis. Curr Opin Rheumatol. 2008;20(4):392–7. doi: 10.1097/BOR.0b013e328303204b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mear JP, Schreiber KL, Munz C, Zhu X, Stevanovic S, Rammensee HG, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol. 1999;163(12):6665–70. [PubMed] [Google Scholar]
- 9.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332(6163):462–4. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 10.Welihinda AA, Tirasophon W, Kaufman RJ. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr. 1999;7(4–6):293–300. [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66(2 Suppl 1):S102–9. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 12.Foti DM, Welihinda A, Kaufman RJ, Lee AS. Conservation and divergence of the yeast and mammalian unfolded protein response. Activation of specific mammalian endoplasmic reticulum stress element of the grp78/BiP promoter by yeast Hac1. J Biol Chem. 1999;274(43):30402–9. doi: 10.1074/jbc.274.43.30402. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 14.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, et al. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2(5):415–22. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science. 2003;299(5611):1397–400. doi: 10.1126/science.1079474. [DOI] [PubMed] [Google Scholar]
- 17.Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299(5611):1394–7. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 18.Cormier JH, Tamura T, Sunryd JC, Hebert DN. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34(5):627–33. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burr ML, Cano F, Svobodova S, Boyle LH, Boname JM, Lehner PJ. HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proc Natl Acad Sci U S A. 2011;108(5):2034–9. doi: 10.1073/pnas.1016229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burr ML, van den Boomen DJ, Bye H, Antrobus R, Wiertz EJ, Lehner PJ. MHC class I molecules are preferentially ubiquitinated on endoplasmic reticulum luminal residues during HRD1 ubiquitin E3 ligase-mediated dislocation. Proc Natl Acad Sci U S A. 2013;110(35):14290–5. doi: 10.1073/pnas.1303380110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, et al. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol. 2007;81(24):13578–86. doi: 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groisman B, Shenkman M, Ron E, Lederkremer GZ. Mannose trimming is required for delivery of a glycoprotein from EDEM1 to XTP3-B and to late endoplasmic reticulum-associated degradation steps. J Biol Chem. 2011;286(2):1292–300. doi: 10.1074/jbc.M110.154849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antoniou AN, Ford S, Taurog JD, Butcher GW, Powis SJ. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J Biol Chem. 2004;279(10):8895–902. doi: 10.1074/jbc.M311757200. [DOI] [PubMed] [Google Scholar]
- 24.Lenart I, Guiliano DB, Burn G, Campbell EC, Morley KD, Fussell H, et al. The MHC Class I Heavy Chain Structurally Conserved Cysteines 101 and 164 Participate in HLA-B27 Dimer Formation. Antioxid Redox Signal. 2011;16(1):33–43. doi: 10.1089/ars.2010.3693. [DOI] [PubMed] [Google Scholar]
- 25.Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol. 2002;158(2):247–57. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saeed M, Suzuki R, Watanabe N, Masaki T, Tomonaga M, Muhammad A, et al. Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J Biol Chem. 2011;286(43):37264–73. doi: 10.1074/jbc.M111.259085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto K, Suzuki N, Wada T, Okada T, Yoshida H, Kaufman RJ, et al. Human HRD1 promoter carries a functional unfolded protein response element to which XBP1 but not ATF6 directly binds. J Biochem. 2008;144(4):477–86. doi: 10.1093/jb/mvn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334(6059):1086–90. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429(6994):834–40. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 30.Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol. 2006;172(3):383–93. doi: 10.1083/jcb.200507057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dangoria NS, DeLay ML, Kingsbury DJ, Mear JP, Uchanska-Ziegler B, Ziegler A, et al. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem. 2002;277(26):23459–68. doi: 10.1074/jbc.M110336200. [DOI] [PubMed] [Google Scholar]
- 32.Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134(5):769–81. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2(7):379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 34.Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3(12):1169–76. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- 35.Campbell EC, Fettke F, Bhat S, Morley KD, Powis SJ. Expression of MHC class I dimers and ERAP1 in an ankylosing spondylitis patient cohort. Immunology. 2011;133(3):379–85. doi: 10.1111/j.1365-2567.2011.03453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammer GE, Gonzalez F, James E, Nolla H, Shastri N. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol. 2007;8(1):101–8. doi: 10.1038/ni1409. [DOI] [PubMed] [Google Scholar]
- 37.McHugh K, Rysnik O, Kollnberger S, Shaw J, Utriainen L, Al-Mossawi MH, et al. Expression of aberrant HLA-B27 molecules is dependent on B27 dosage and peptide supply. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2012-203080. [DOI] [PubMed] [Google Scholar]
- 38.Kaneko M, Koike H, Saito R, Kitamura Y, Okuma Y, Nomura Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci. 2011;30(11):3924–32. doi: 10.1523/JNEUROSCI.2422-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colbert RA. HLA-B27 misfolding: a solution to the spondyloarthropathy conundrum? [In Process Citation] Mol Med Today. 2000;6(6):224–30. doi: 10.1016/s1357-4310(00)01699-3. [DOI] [PubMed] [Google Scholar]
- 40.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100(17):9946–51. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]