

Abstract
Hybridoma cell lines producing natural autoantibodies (NAA), generated from A.SW mice with progressive experimental autoimmune encephalomyelitis (P-EAE), have been shown to cause demyelination and renal pathology when injected into naïve mice. To investigate the relative contribution of these antibodies to disease pathogenesis, B-1 cells, the major producers of NAA, were depleted by hypotonic shock. Depletion of B-1 cells during the effector phase of EAE significantly decreased the severity of demyelination and overall pathology in the brain. There was also a decreased incidence of P-EAE and a decrease in clinical score. Depletion during the induction phase of the disease resulted in an increase in the incidence of P-EAE and in the clinical score. Overall, B-1 cells were found to modulate EAE pathogenesis.
Keywords: Autoantibodies, Autoimmunity, CNS-demyelinating disease, CNS disease pathology, Multiple sclerosis
Introduction
Antibody producing B cells are divided into B-1 and B-2 cells [1]. B-2 cells circulate from the bone marrow to lymphoid organs and undergo T cell dependent isotype rearrangements, to produce different immunoglobulin (Ig) subclasses, and somatic hypermutation and affinity maturation, to produce antibodies with increased specificity. In contrast, B-1 cells are T cell independent and produce natural autoantibodies (NAA) of the IgM isotype. B-1 cells originate from fetal liver and to a lesser extent bone marrow precursor cells. They are phenotypically IgMhiIgDloCD23− B220lo and are subdivided based on expression of CD5 (Ly1) into B-1a (CD5+) and B-1b (CD5−) [2–5]. B-1 cells also have a strong self-renewal capacity [6], are found predominantly in the peritoneal cavity of mice [7] and are the major producer of NAA.
The term NAA was initially introduced to refer to autoreactive antibodies present in normal healthy subjects [8]. NAA are primarily of the IgM isotype and are produced by B-1 cells. NAA are polyreactive and exhibit cross-reactivity with both self and nonself antigens. These antibodies are encoded by Ig genes that have not undergone somatic hypermutation or affinity maturation and therefore have few or no mutations and N insertions in their variable regions [9–11]. NAA have been detected in sera of healthy humans and animals, in patients with autoimmune diseases and monoclonal gammopathies, and among first degree healthy relatives of patients with auto-immune diseases [8]. The precise biologic roles of NAA have not yet been fully elucidated. However, they have been suggested to serve a primary protective role in the innate immune response against microorganisms.
A pathogenic role for NAA in autoimmunity has been proposed based on reports that high titers of NAA in the serum of patients with primary anti-phospholipid syndrome (PAPS) and systemic lupus erythematosus (SLE) were shown to correlate with disease activity. In addition, NAA were detected on the surface of erythrocytes from patients with PAPS and in the kidneys of patients with SLE. Passive transfer of the NAA to naive mice was sufficient to induce the clinical and serological manifestations of the autoimmune diseases (reviewed in [8]). Higher frequencies of B-1 cells have been reported in patients with autoimmune diseases, such as Sjögren’s disease [12] and rheumatoid arthritis [13], and animal models of autoimmune disease, i.e. lupus in NZB mice [14]. However, the role of B-1 cells in the development of autoimmune diseases is controversial since there are claims both for a pathogenic role of B-1 cells in autoimmune hemolytic anemia [15], and cases against a pathogenic role in SLE [16] and graft versus host disease [17].
Multiple sclerosis (MS) has been suggested to be an autoimmune disease of the central nervous system (CNS). Patients with MS have higher levels of NAA in cerebrospinal fluid (CSF) compared to both healthy controls and patients with other neurological diseases [18]. Myelin reactive antibodies are typically presumed to be involved in oligodendrocyte loss and demyelination in MS and its animal model, experimental autoimmune encephalomyelitis (EAE). However, these antibodies are typically of the IgG isotype [19,20]. Recently, a few oligodendrocyte-reactive NAA were shown to promote remyelination in the Theiler’s murine encephalomyelitis virus-induced model for MS [9,10,21–23]. Therefore, NAA could potentially participate in both destruction and repair of myelin in MS and its animal models. B-1 cells have also been found at higher frequencies in the CSF and peripheral blood of patients with MS [24,25]. The frequency of B-1 cells has been demonstrated to correlate with disease activity, circulating levels of autoantibodies against myelin basic protein and the number of gadolinium-enhancing magnetic resonance imaging (MRI) lesions in patients with relapsing-remitting (RR) MS [26]. Patients with progressive MS were found to have a significantly higher percentage of B-1 cells than patients with RR disease [27]. A recent study reported an increase in CD5− B cells (B-2 or B-1b) in patients with MS, but similar numbers of CD5+ B-1a cells among patients with clinically definite MS, patients with a clinically isolated syndrome (CIS) suggestive of MS and control patients with noninflammatory neurological diseases [28]. Although these results are contradictory to previous studies with regard to differences in the frequency of CD5+ B cells between patients with MS and controls, they do not eliminate the possibility of a role for NAA in MS, since NAA are also produced by CD5− B-1b cells and by CD5 knockout mice [29–31].
A.SW mice sensitized with amino acids 92–106 from myelin oligodendrocyte glycoprotein (MOG) develop a progressive form of EAE, with antibody deposition associated with demyelinated lesions and serum MOG92–106 antibody titers that correlated with disease progression [32]. To investigate the contribution of MOG92–106 antibodies to disease, we generated hybridoma cell lines from an A.SW mouse with progressive EAE (P-EAE) [33]. Two hybridoma cell lines producing MOG92–106 antibodies were found to be of the IgM isotype, encoded by Ig genes in their germline configuration [34] and polyreactive. These monoclonal antibodies recognized not only the initiating antigen, but also antigens associated with systemic autoimmune disease such as double-stranded DNA (dsDNA) and histone H1a, as well as proteins isolated from organs outside the CNS. The MOG92 – 106 reactive antibodies were also cytotoxic in vitro, suggesting a mechanism by which they could contribute to systemic disease. Intracerebral (i.c.) injection of the MOG92–106 antibody producing hybridoma cells into naïve mice resulted in antibody deposition and demyelination in the CNS. In addition, intraperitoneal (i.p.) injection of the MOG92–106 antibody producing hybridoma cells into naïve mice resulted in antibody deposition in the kidney [33]. Thus, the MOG92–106 reactive NAA recognize a variety of antigens including several ubiquitously expressed antigens and transfer of the antibodies recapitulates disease manifestations observed in P-EAE. Since MOG92–106 antibodies established from mice with P-EAE are NAA, we hypothesized that NAA play a pathogenic role in EAE, and enhance disease progression. NAA are known to be produced by B-1 cells that are found predominantly in the peritoneal cavity. Thus, we tested whether depletion of B-1 cells could modulate the clinical course of EAE in A.SW mice.
In this study, we examined the involvement of B-1 cells, and the NAA they produce, in MOG92–106 induced EAE by depletion of peritoneal cells, which are the main source of B-1 cells, but not of other immune cells. B-1 cells were depleted using hypotonic shock as described by Murakami et al. [15]. Depletion of B-1 cells was confirmed by flow cytometry. Depletion of B-1 cells during the effector phase of EAE significantly decreased the severity of demyelination and overall pathology in the brain. There was also a decreased incidence of P-EAE and a decrease in clinical score. Depletion during the induction phase of the disease resulted in an increase in the incidence of P-EAE and in the clinical score. In addition, B-1 cell depletion did not alter the titer of IgM antibodies reactive against MOG92–106 and/or dsDNA. These results suggest that B-1 cells can modulate MOG92–106 induced EAE.
Materials and methods
Animals and EAE induction
SJL/J and A.SW mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Five week old mice were sensitized subcutaneously at the base of the tail with 200 nmol of MOG92–106 peptide (DEGGYTCFFRDHSYQ) (Peptide Core Facility, University of Utah, Salt Lake City, UT, USA) [35,36] in complete Freund’s adjuvant (CFA) containing 2 mg/ml Mycobacterium tuberculosis H37 Ra (Difco Laboratories, Detroit, MI, USA). Mice were weighed and observed for clinical signs for three months. Classical signs of EAE were assessed according to the following criteria: 0, no clinical disease; 1, loss of tail tonicity; 2, mild hind leg paresis; 3, moderate hind leg paralysis; 4, complete paraplegia; and 5, quadriplegia, moribund state or death [37]. Ataxic signs of EAE were assessed according to the following criteria: 0, no clinical disease; 1, mice turning their heads or bodies to one side with or without a waddling gait; 2, mice lean significantly to one side and fall while walking; 3, mice continuously roll by twisting their bodies or rotate laterally in a circle; 4, mice cannot stand but lie on their sides with or without rolling; and 5, moribund state or death [32,38].
Histology
Mice were euthanized using isoflurane, exsanguinated and perfused with phosphate-buffered saline (PBS) followed by a buffered 4% paraformaldehyde solution and brains and spinal cords were removed from the mice. Brains were divided into five coronal slabs and spinal cords into 10–12 transverse slabs. Tissues were then embedded in paraffin. Four-micrometer thick tissue sections were stained with Luxol fast blue for myelin visualization or with hematoxylin and eosin. Brain and spinal cord sections were scored for meningitis, demyelination and perivascular cuffing as described previously [32]. Briefly, brain sections were scored for meningitis (0, no meningitis; 1, mild cellular infiltrates; 2, moderate cellular infiltrates; 3, severe cellular infiltrates), perivascular cuffing (0, no cuffing; 1, 1–10 lesions; 2, 11–20 lesions; 3, 21–30 lesions; 4, 31–40 lesions; 5, more than 40 lesions) and demyelination (0, no demyelination; 1, mild demyelination; 2, moderate demyelination; 3, severe demyelination). The scores were then combined for a maximum score of 11 per mouse brain. Spinal cord sections were divided into four quadrants consisting of the anterior funiculus, the posterior funiculus and each lateral funiculus. Each quadrant containing meningitis, demyelination or perivascular cuffing was given a score of 1 in that pathologic class. The total number of positive quadrants for each pathologic class was determined, then divided by the total number of quadrants present on the slide and multiplied by 100 to give the percent involvement for each pathologic class. The overall pathology score was determined by counting the number of quadrants containing any lesions.
B-1 cell depletion
B-1 cell depletion was performed using an adaptation of the protocol reported by Murakami et al. [15], in which i.p. injection of double distilled water (ddH2O) results in the selective depletion of B-1 cells in the peritoneal cavity, but not in the spleen. Mice were injected i.p. with 0.5 ml of ddH2O every week from three weeks of age until sacrifice or from the onset of clinical signs until sacrifice. As a negative control, groups of mice received injections of PBS, an isotonic solution. To determine the efficiency of depletion of B-1 cells, cells were isolated from the peritoneal cavity and flow cytometric analysis performed.
Peritoneal cells were obtained by injecting 8 ml ice cold PBS into the peritoneal cavity of mice, gently massaging the cavity and collecting lavage fluid containing peritoneal cells using a 22-gauge needle. Single cell suspensions containing 1 × 106 cells in a volume of 100 μl of staining buffer [PBS containing 2% cosmic calf serum (CCS) (Hyclone, Logan, UT, USA) and 0.1% sodium azide (Sigma-Aldrich, St Louis, MO, USA)] were incubated with anti-mouse CD16/CD32 (BD Pharmingen, San Diego, CA, USA) for 5 min at 4°C to block Fc receptor binding. The cell suspensions were then incubated with fluorochrome-conjugated monoclonal antibodies against B220/CD45R (RA3-6B2; Caltag, Burlingame, CA, USA), CD5 (53–7.3; BD Biosciences, San Diego, CA, USA), CD3 (CT-CD3; Caltag), CD4 (GK1.5; BD Biosciences) and CD8a (CT-CD8a; Caltag) for 30 min at 4°C. The cell suspensions were then washed three times with buffer and analyzed by flow cytometry. For each combination of antibodies, 10,000 viable lymphocytes were analyzed by FACScan (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) using CELL Quest Software (Becton Dickinson).
Serum anti-MOG92 –106 and anti-dsDNA IgM ELISAs
Enzyme-linked immunosorbent assays (ELISAs) were performed to measure the amount of serum MOG and dsDNA antibodies as described previously [32]. Briefly, 96-well flat-bottom Nunc-Immuno Maxi-Sorp™ plates (Nalge Nunc International, Rochester, NY, USA) were coated overnight with 10 μg/ml MOG92–106 peptide or dsDNA (Sigma-Aldrich) at 4°C in a humidified chamber. After blocking with 10% CCS and 0.2% Tween 20, serial dilutions of sera were added to the plates and incubated for 90 min at room temperature. After washing, peroxidase-conjugated anti-mouse IgM (Stressgen Biotechnologies, Victoria, BC, Canada) or anti-IgG (Caltag) was added for 90 min. Immunoreactive complexes were detected with o-phenylenediamine dihydrochloride (Sigma-Aldrich), and were read at 492 nm in a Titertek Multiskan Plus MK II spectrophotometer (Flow Laboratories, McLean, VA, USA).
Lymphoproliferation assay
Lymphoproliferation assays were performed as described previously [39], using mononuclear cells (MNCs) from MOG-sensitized mice injected with PBS (control) or ddH2O (depleted). Spleens and inguinal lymph nodes were removed and pooled, and MNCs were isolated using Histopaque®-1083. A volume of 200 μl containing 2 × 105 cells in RPMI 1640 (Mediatech, Herndon, VA, USA), supplemented with 1% glutamine (Mediatech), 1% antibiotics (Mediatech), 50 μM 2-mercaptoethanol (Sigma-Aldrich) and 10% CCS, was added to each well. MNCs were stimulated with MOG92 – 106 peptide or myelin proteolipid protein (PLP139–151) peptide (HSLGKWLGHPDKF) (Peptide Core Facility, University of Utah) at a concentration of 50 μg/ml. The cells were cultured for 4 days, pulsed with 1 μCi of 3H-thymidine (PerkinElmer Life Sciences, Boston, MA, USA) per well, and cultured for another 18–24 h. Cultures were harvested onto filters using a multiwell cell harvester (Molecular Devices, Sunnyvale, CA, USA) and 3H incorporation was determined using standard liquid scintillation techniques. All cultures were performed in triplicate and results expressed as stimulation index [experimental counts per minute (cpm)/control cpm].
Results
Efficiency of B-1 cell depletion by hypotonic shock
Previously, Murakami et al. [15] reported that i.p. injection of 1 ml ddH2O resulted in a dramatic reduction of B cells, T cells and macrophages in the peritoneal cavity, but not in the spleen of mice. Although the initial killing was non-specific, the long-lasting depletion was specific to B-1 cells because B-1 cells are the only cells that depend on self-renewal within the peritoneal cavity for replenishment. In our experiments, we injected 0.5 ml ddH2O, because the strains of mice we use for our studies do not grow to the same size/weight as the strains used by Murakami et al. [15]. The frequency of B-1 cells (CD5+, B220lo) in the peritoneal cavity was 7.8% in A.SW mice treated with ddH2O (Figure 1a) compared with 23.2% in control A.SW mice treated with PBS (Figure 1b). The efficiency of depletion in the peritoneal cavity was approximately 70%, which is similar to the depletion reported by Murakami et al. [15] in BALB/c mice (75%), but less than that reported in NZB and NZB/NZW F1 mice (87%). Water injection decreased the number of B-1 cells, and increased the number of conventional B cells in the peritoneal cavity. Water treatment had no significant effect on the number of B-1 or B-2 cells in the spleen as compared to the control PBS treated group (data not shown).
Figure 1.
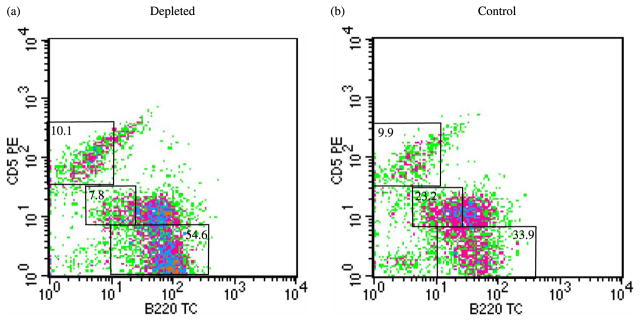
Depletion of CD5+ B-1 cells by repeated i.p. injection of ddH2O in A.SW mice sensitized with MOG92–106. Flow cytometric analysis of cells from peritoneal lavage using antibodies against B220 and CD5 was used to assess the depletion of CD5+ B-1 cells. Two months after sensitization with MOG92–106, A.SW mice, into which 0.5 ml of ddH2O had been injected weekly from three weeks of age (depleted), had a significant reduction in the frequency of CD5+ B-1 cells (a) compared to mice that had been injected with PBS (control) (b). The percentage of T cells (B220−, CD5+), B cells (B220+, CD5−) and B-1 cells (B220lo, CD5+) gated is indicated. The data is representative of mice sensitized with MOG92–106 and treated with ddH2O (depleted) or PBS (control) in three independent experiments.
B-1 cell depletion after disease onset (effector phase) decreases the extent of demyelination
Since we established NAA producing hybridomas from mice in the effector phase of P-EAE, we first speculated that B-1 cells play a role during the effector phase. To examine the effect of depletion of B-1 cells on the development of EAE, we evaluated the incidence, the time of onset and the severity of clinical signs of disease. Mice were sensitized with MOG92–106 at 5 weeks of age and monitored for weight change and ataxic signs of EAE. After the onset of clinical signs, groups of mice received ddH2O (depleted) or PBS (control) injections. No significant effects on disease course were observed between the groups (Figure 2a). Interestingly, control mice had a higher incidence of P-EAE and more severe clinical signs than the depleted group, although these differences did not reach statistical significance (Table I). Neuropathologically, MOG92–106 sensitized A.SW mice depleted after the onset of clinical signs of disease had significantly less demyelination (P < 0.05, t-test) and overall pathology (P < 0.05, t-test) in their brain compared to control mice (Figure 2b). In the spinal cord, no significant difference in the extent of meningitis (P = 0.41, t-test), perivascular cuffing (P = 0.40, t-test), demyelination (P = 0.36, t-test) and overall pathology (P = 0.83, t-test) was observed between depleted versus control mice (Figure 2c).
Figure 2.

B-1 cell depletion after the onset of clinical signs of EAE (effector phase) did not significantly alter the disease course, but decreased the severity of demyelination in A.SW mice sensitized with MOG92–106. A.SW mice were sensitized with MOG92–106 at five weeks of age and received i.p. injections of ddH2O (depleted) or PBS (control) weekly after the onset of clinical signs. Mice were weighed and observed for clinical signs for three months. (a) A.SW mice in which B-1 cells had been depleted by i.p. injection of ddH2O after the onset of clinical signs had a disease course similar to that observed in control mice injected with PBS. (b) A.SW mice treated with ddH2O (depleted) after the onset of clinical signs of disease had significantly less demyelination and overall neuropathology in their brain compared to mice treated with PBS (control). However, no significant differences in the extent of meningitis or perivascular cuffing were observed in the brain whether the mice were treated with ddH2O (depleted) or PBS (control). (c) Similarly, no significant difference in the extent of meningitis, perivascular cuffing, demyelination and overall neuropathology was observed in the spinal cord of mice treated with ddH2O (depleted) or PBS (control) after onset of clinical signs. *P < 0.05, t-test. Data shown are mean + standard error of the mean (SEM) for P-EAE and RR-EAE combined from three independent experiments.
Table I.
Clinical disease in mice treated with ddH2O (depleted) or PBS (control) during the effector phase of EAE.
| P-EAE/ mice | RR-EAE/ mice | Disease onset* | Clinical score* | |
|---|---|---|---|---|
| Depleted | 3/11 | 8/11 | 49.7 ± 7.4 | 3.1 ± 0.4 |
| Control | 5/11 | 6/11 | 50.1 ± 7.7 | 4.0 ± 0.3 |
ddH2O, double-distilled water; PBS, phosphate-buffered saline; P-EAE, progressive experimental autoimmune encephalomyelitis; RR-EAE, relapsing-remitting experimental autoimmune encephalomyelitis;
Mean ± SEM for P-EAE and RR-EAE combined.
Mice depleted during the induction phase have a higher incidence of P-EAE than control mice
We hypothesized that B-1 cells could also play a role in the induction phase of EAE. To examine the effects of B-1 cell depletion during the induction phase of EAE, mice received ddH2O (depleted) or PBS (control) injections i.p. every week from three weeks of age and were sensitized with MOG92–106 at five weeks of age. In contrast to the results observed in depleted and control mice treated after the onset of clinical signs (effector phase), A.SW mice depleted prior to sensitization with MOG92–106 (induction phase) had a higher incidence of progressive disease (9/16) with more severe clinical signs compared to control mice. However, there was no difference in disease onset (Table II). During the first two months of the observation period, depleted mice displayed higher mean clinical scores than control mice. However, in the last month of the observation period, depleted mice and control mice had similar mean clinical scores (Figure 3a). This was largely due to mice with high clinical scores having died of P-EAE.
Table II.
Clinical disease in mice treated with ddH2O (depleted) or PBS (control) during the induction phase of EAE.
| Disease incidence | P-EAE/ mice | RR-EAE/ mice | Disease onset* | Clinical score* | |
|---|---|---|---|---|---|
| Depleted | 16/22 | 9/16 | 7/16 | 35.1 ± 4.0 | 4.0 ± 0.4 |
| Control | 18/21 | 4/18 | 14/18 | 36.7 ± 4.7 | 2.8 ± 0.3 |
ddH2O, double-distilled water; PBS, phosphate-buffered saline; P-EAE, progressive experimental autoimmune encephalomyelitis; RR-EAE, relapsing-remitting experimental autoimmune encephalomyelitis;
Mean ± SEM for P-EAE and RR-EAE combined.
Figure 3.

B-1 cell depletion during the induction phase did not significantly alter the disease course or neuropathology of EAE in A.SW mice sensitized with MOG92–106. A.SW mice received i.p. injections of ddH2O (depleted) or PBS (control) weekly from three weeks of age and were sensitized with MOG92–106 at five weeks of age. Mice were weighed and observed for clinical signs for three months. (a) The PBS (control) injected group developed less severe clinical signs than the ddH2O (depleted) injected group. (b) No significant differences in the extent of meningitis, perivascular cuffing, demyelination or overall neuropathology in the brain were observed among the groups. (c) No significant differences in the extent of meningitis, perivascular cuffing, demyelination or overall neuropathology in the spinal cord were observed among the groups. Data shown are mean + SEM for P-EAE and RR-EAE combined from three independent experiments.
We observed more demyelination in mice that developed P-EAE than in mice with RR-EAE, regardless of treatment (Figure 4). Demyelination was most frequently observed in the cerebellum and spinal cord. A.SW mice with P-EAE developed large demyelinating lesions in the cerebellum (Figure 4a) and spinal cord (Figure 4c). In contrast, A.SW mice with RR-EAE developed small perivascular demyelinating lesions in the brain (Figure 4b) and severe inflammation with mild demyelination in the spinal cord (Figure 4d). Overall, when P-EAE and RR-EAE were combined within the depleted and control groups, similar pathology scores were observed in the brains of mice from the two groups (Figure 3b). No significant differences were observed in any brain pathology scores, demyelination, meningitis, perivascular cuffing and overall, between the two groups. Similarly, in the spinal cord, no significant differences were observed in pathology scores between depleted mice versus control mice, when P-EAE and RR-EAE were combined (Figure 3c).
Figure 4.

Neuropathology corresponded to the clinical course of EAE in A.SW mice sensitized with MOG92–106 and treated with ddH2O to deplete B-1 cells or PBS as a control. (a,c) A.SW mice with P-EAE developed large demyelinating lesions in the cerebellum (a) and spinal cord (c). Images shown are from a representative mouse that had been treated with ddH2O (depleted). (b,d) A.SW mice with RR-EAE developed small perivascular demyelinating lesions (arrows) in the brain (b) and severe inflammation with mild demyelination (arrowheads) in the spinal cord (d). Images shown are from a representative mouse that had been treated with PBS (control). Myelin was visualized using Luxol fast blue staining (a, b: × 23; c, d: × 230).
Water injection does not alter EAE in B-1 cell deficient SJL/J mice
Although the i.p. injection procedure was the same between depleted and control groups in the experiments described above, ddH2O injection might elicit other responses in mice, such as damage to the peritoneal cavity itself and stress, which potentially influence the immune response [40]. To determine whether the i.p. injection itself influences the disease course, we decided to use SJL/J mice, which have very few peritoneal CD5+ B-1 cells [41], to examine whether differences observed between depleted and control mice could be attributed to factors other than B-1 cells. SJL/J mice received ddH2O (depleted) or PBS (control) injections i.p. every week from three weeks of age and were sensitized with MOG92–106 at five weeks of age. SJL/J mice sensitized with MOG92–106 had a similar disease course, (Figure 5a), incidence (100%), time of onset (56.6 ± 8.5 and 45.1 ± 3.8) and severity of disease (2.8 ± 0.4 and 2.3 ± 0.2) whether they were depleted or control mice, respectively. Histological analysis of the CNS did not reveal any difference in the extent of meningitis (P = 0.75, t-test), perivascular cuffing (P = 0.92, t-test), demyelination (P = 0.74, t-test) and overall neuropathology (P = 0.82, t-test) in the brain whether the mice were depleted or control mice (Figure 5b). Similarly, no difference in the extent of meningitis (P = 0.66, t-test), perivascular cuffing (P = 0.47, t-test), demyelination (P = 0.91, t-test) and overall neuropathology (P = 0.74, t-test) was observed in the spinal cord of depleted or control mice (Figure 5c).
Figure 5.

B-1 cell depletion did not significantly alter the disease course or neuropathology of MOG92–106 induced EAE in mice with inherently low levels of B-1 cells, SJL/J mice. SJL/J mice received i.p. injections of ddH2O (depleted) and PBS (control) weekly from three weeks of age and were sensitized with MOG92–106 at five weeks of age. Mice were weighed and observed for clinical signs for three months. No significant differences in disease course (a) or neuropathology in the brain (b) and spinal cord (c) were observed between the depleted mice and control mice. Data shown are mean + SEM.
B-1 cell depletion does not alter MOG92 –106 specific immune responses or dsDNA antibody responses
Previously, we reported that two MOG92–106 anti-bodies produced by two hybridoma cell lines were NAA of the IgM isotype, which reacted not only with the initiating antigen, but also protein isolated from organs outside the CNS and antigens associated with systemic autoimmune disease [33], such as dsDNA and histone H1a [41]. If these NAA contribute significantly to the amount of IgM antibodies reactive with MOG92–106 and dsDNA, depletion of the major producer of these antibodies, B-1 cells, should drastically reduce serum titer of MOG92–106 and dsDNA antibodies of the IgM isotype. While the efficiency of B-1 cell depletion in the peritoneal cavity of A.SW mice was approximately 70%, this did not reduce the titer of MOG92–106 or dsDNA reactive IgM or total IgM in the sera compared to control mice (Figure 6a). These results suggest that B-1 cells are only partially responsible for the generation of MOG92 – 106 or dsDNA reactive IgM in mice sensitized with MOG92–106.
Figure 6.

Depletion of B-1 cells had no effect on MOG92–106 specific antibody production or proliferation. A.SW mice received i.p. injections of ddH2O (depleted) and PBS (control) weekly from three weeks of age and were sensitized with MOG92–106 at five weeks of age. (a) Total IgM, anti-dsDNA IgM and anti-MOG92–106 IgM and IgG antibody responses in the serum were measured by ELISA. No significant differences in antibody response were detected between depleted mice and control mice. (b) MNCs were isolated from spleens and stimulated with MOG92–106 or PLP139–151 for five days. No significant differences in responses to MOG92–106 were detected in depleted mice and control mice. All cultures were performed in triplicate. Results are representative of two independent experiments. Data shown are mean + SEM.
Lymphoproliferative responses to MOG92–106 were determined using MNCs isolated from spleen and inguinal lymph nodes. MOG92–106 specific proliferation was low in A.SW mice that developed clinical signs of EAE. Among the mice with clinical signs, no significant difference in lymphoproliferation was observed between depleted mice and control mice (Figure 6b).
Discussion
In this study, we examined the involvement of NAA in MOG92–106 induced EAE by depletion of peritoneal cells, which are the main source of B-1 cells, but not of other immune cells. B-1 cells were depleted using hypotonic shock as described by Murakami et al. [15]. This treatment has been shown to significantly decrease the number of peritoneal cells within minutes, and to be an effective method for depletion of B-1 cells from lamina propria of the gut, whereas, the number and relative proportion of the other lymphocyte subsets do not change in the spleen and bone marrow [15]. If B-1 cells producing NAA play a role in disease progression in MOG92–106 sensitized A.SW mice, elimination of B-1 cells should alter the disease course and neuropathology. This could be due to either the MOG92–106 NAA itself being functional (myelinotoxic or remyelinating antibody) or to other roles of B-1 cells such as antigen presentation to T cells. Thus, in addition to investigating disease modulation, we examined whether clinical signs correlated with MOG92 – 106 antibody titers or MOG92–106 specific immune responses. It is possible that MOG92–106 reactive NAA might be beneficial, and contribute to remyelination. If this were the case, elimination of NAA would exacerbate disease. We found that the effect of depletion of B-1 cells depended on whether the depletion was performed during the induction or the effector phase of EAE, though the effects were minimal. It is possible that B-1 cells and the NAA they produce could have different roles during the induction phase versus the effector phase.
SJL/J mice have very few peritoneal B-1 cells [41]. Our previous results suggest that MOG92–106 specific T cells, not antibody, play a major pathogenic role in SJL/J mice, which develop RR-EAE after sensitization with MOG92–106 [32]. Thus, modulation of disease course should not be observed in SJL/J mice with or without depletion, making SJL/J a good control for the effects of hypotonic shock on EAE. As expected, no difference in the clinical or pathological outcome of MOG92–106 induced EAE was observed between depleted or control SJL/J mice.
Several possibilities could explain why depletion of B-1 cells during the effector phase resulted in such a modest alteration in clinical and pathological outcomes in A.SW mice with MOG92–106 induced EAE. The simplest explanation is that they play a minor role in the pathogenesis of MOG92–106 induced EAE. However, the mild effect could also be due to the incomplete elimination of B-1 cells, or to production of pathogenic antibodies by conventional B-2 B cells. B-1 cells are found predominantly in the peritoneal cavity, but are also present in the pleural cavity and to a lesser extent in peripheral blood and lymphoid organs [7]. Therefore, B-1 cells outside the peritoneal cavity could be contributing to the pathogenesis in EAE. It is also possible that both beneficial (remyelinating) and pathogenic (demyelinating) NAA exist, and that it is the balance between these antibodies that determines their role in autoimmunity. Our method of depleting B-1 cells, likely depleted cells producing both types of NAA, and therefore did not alter the balance in the remaining B-1 cell population. Studies are currently ongoing to further delineate the contribution of B-1 cells in EAE by performing the reverse experiments of increasing the numbers of B-1 cells present in the peritoneum at various times in the disease course rather than depleting them.
Our results for the effect of B-1 cell depletion on autoimmunity differ, to some extent, from those reported by Murakami et al. [15] in NZB and NZB/NZW F1 mice. Depletion of B-1 cells by hypotonic shock in NZB mice was shown to dramatically decrease the incidence and severity of autoimmune hemolytic anemia, and the titer of anti-erythrocyte IgM and IgG in the sera. In NZB/NZW F1 mice, B-1 cell depletion was demonstrated to decrease the titer of total IgM and anti-DNA IgM and IgG in the sera, as well as the severity of renal pathology. In contrast, no significant effects on anti-DNA IgM titer were observed in our study. This could be due to slight differences in the depletion protocol, since the maximum dose of ddH2O tolerated by the mice in our study was 0.5 ml, while Murakami et al. [15] were injecting two to four times this volume in their mice. However, we achieved a similar level of CD5+ B-1 cell depletion, so this is not likely to explain the observed differences. It is possible that the contribution of B-1 cells and NAA differs between autoimmune diseases. Reap et al. [16,17] demonstrated that B-2 cells, not B-1 cells, were the source of autoantibodies in the mouse models of both SLE and graft-versus-host disease. It is also possible that the difference is due to the increased frequency (42% in NZB and NZB/NZW F1 mice compared to 23% in A.SW), and therefore a more dramatic depletion, of B-1 cells in the peritoneal cavity of naïve NZB and NZB/NZW F1 mice [15], compared to A.SW mice.
In our experiments in which A.SW mice were injected with 0.5 ml of ddH2O (depleted) or PBS (control) weekly from three weeks of age, we found that depleted mice showed more severe disease than the control group. Control A.SW mice had a similar time of onset of EAE compared to depleted mice. In contrast, more of the control mice had a RR disease course with a lower maximum clinical score compared to depleted mice (Table II).
Previously, we reported that A.SW mice sensitized with MOG92–106 develop P-EAE, with antibody deposition associated with demyelinated lesions in the CNS and serum MOG92–106 antibody responses that associated with disease progression [32]. We also demonstrated that antibodies produced by hybridoma cell lines generated from an A.SW mouse with P-EAE were NAA, capable of recognizing not only the initiating antigen, but also antigens associated with systemic autoimmune disease such as dsDNA, as well as protein isolated from organs outside the CNS. I.c. injection of the MOG92–106 antibody producing hybridomas into naïve mice resulted in antibody deposition and demyelination in the CNS [33]. This led us to hypothesize that NAA were involved in the disease progression and demyelination in A.SW mice with MOG92–106 induced P-EAE. B-1 cell depletion did show effects on the type of disease course (P-EAE versus RR-EAE), the severity of maximum clinical signs and the extent of demyelination, but the overall effects were relatively minor. Therefore, B-1 cells play a minor role in pathogenesis in MOG92–106 induced EAE. This suggests that B-2 cells play a more significant role in MOG induced EAE. This is supported by the study of EAE induction in transgenic mice engineered to produce increased levels of MOG autoantibodies [42]. These transgenic mice had a dramatic reduction in the number of CD5+ B-1 cells in their peritoneal cavity compared to wild type mice. Despite the reduced frequency of peritoneal B-1 cells, MOG antibody producing transgenic mice developed EAE with an earlier time of onset, and more severe clinical signs and CNS pathology compared to wild type mice [42]. Therefore, in this study, an increase in B-2 cells producing MOG antibody overcame the effect of decreased NAA production by B-1 cells. We are currently investigating this possibility.
Acknowledgments
The authors would like to thank Jane E. Libbey, MS, and Nikki J. Kirkman, BS, for many helpful discussions and Faris Hasanovic, J. Wes Peterson, Bryan A. Moore, Victor Gappmeier and Daniel G. Smith for excellent technical assistance. We are grateful to Ms Kathleen Borick for preparation of the manuscript. This work was supported by NIH grant 5R01NS040350.
References
- 1.Askenase PW, Szczepanik M, Itakura A, Kiener C, Campos RA. Extravascular T-cell recruitment requires initiation begun by Vα14+ NKT cells and B-1 B cells. Trends Immunol. 2004;25:441–449. doi: 10.1016/j.it.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Hardy RR, Hayakawa K. Development and physiology of Ly-1 B and its human homolog, Leu-1 B. Immunol Rev. 1986;93:53–79. doi: 10.1111/j.1600-065x.1986.tb01502.x. [DOI] [PubMed] [Google Scholar]
- 3.Herzenberg LA, Stall AM, Lalor PA, Sidman C, Moore WA, Parks DR, Herzenberg LA. The Ly-1 B cell lineage. Immunol Rev. 1986;93:81–102. doi: 10.1111/j.1600-065x.1986.tb01503.x. [DOI] [PubMed] [Google Scholar]
- 4.Stall AM, Adams S, Herzenberg LA, Kantor AB. Characteristics and development of the murine B-1b (Ly-1 B sister) cell population. Ann N Y Acad Sci. 1992;651:33–43. doi: 10.1111/j.1749-6632.1992.tb24591.x. [DOI] [PubMed] [Google Scholar]
- 5.Hardy RR. B-1 B cell development. J Immunol. 2006;177:2749–2754. doi: 10.4049/jimmunol.177.5.2749. [DOI] [PubMed] [Google Scholar]
- 6.Hayakawa K, Hardy RR, Stall AM, Herzenberg LA, Herzenberg LA. Immunoglobulin-bearing B cells reconstitute and maintain the murine Ly-1 B cell lineage. Eur J Immunol. 1986;16:1313–1316. doi: 10.1002/eji.1830161021. [DOI] [PubMed] [Google Scholar]
- 7.Marcos MAR, Huetz F, Pereira P, Andreu J-L, Martinez AC, Coutinho A. Further evidence for coelomic-associated B lymphocytes. Eur J Immunol. 1989;19:2031–2035. doi: 10.1002/eji.1830191110. [DOI] [PubMed] [Google Scholar]
- 8.George J, Gilburd B, Shoenfeld Y. The emerging concept of pathogenic natural autoantibodies. Hum Antibodies. 1997;8:70–75. [PubMed] [Google Scholar]
- 9.Miller DJ, Rodriguez M. A monoclonal autoantibody that promotes central nervous system remyelination in a model of multiple sclerosis is a natural autoantibody encoded by germline immunoglobulin genes. J Immunol. 1995;154:2460–2469. [PubMed] [Google Scholar]
- 10.Asakura K, Miller DJ, Pogulis RJ, Pease LR, Rodriguez M. Oligodendrocyte-reactive O1, O4, and HNK-1 monoclonal antibodies are encoded by germline immunoglobulin genes. Brain Res Mol Brain Res. 1995;34:283–293. doi: 10.1016/0169-328x(95)00190-4. [DOI] [PubMed] [Google Scholar]
- 11.Kirschning E, Jensen K, Dübel S, Rutter G, Hohenberg H, Will H. Primary structure of the antigen-binding domains of a human oligodendrocyte-reactive IgM monoclonal antibody derived from a patient with multiple sclerosis. J Neuroimmunol. 1999;99:122–130. doi: 10.1016/s0165-5728(99)00118-6. [DOI] [PubMed] [Google Scholar]
- 12.Shirai T, Okada T, Hirose S. Genetic regulation of CD5+ B cells in autoimmune disease and in chronic lymphocytic leukemia. Ann N Y Acad Sci. 1992;651:509–526. doi: 10.1111/j.1749-6632.1992.tb24658.x. [DOI] [PubMed] [Google Scholar]
- 13.Plater-Zyberk C, Maini RN, Lam K, Kennedy TD, Janossy G. A rheumatoid arthritis B cell subset expresses a phenotype similar to that in chronic lymphocytic leukemia. Arthritis Rheum. 1985;28:971–976. doi: 10.1002/art.1780280903. [DOI] [PubMed] [Google Scholar]
- 14.Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med. 1983;157:202–218. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murakami M, Yoshioka H, Shirai T, Tsubata T, Honjo T. Prevention of autoimmune symptoms in autoimmune-prone mice by elimination of B-1 cells. Int Immunol. 1995;7:877–882. doi: 10.1093/intimm/7.5.877. [DOI] [PubMed] [Google Scholar]
- 16.Reap EA, Sobel ES, Cohen PL, Eisenberg RA. Conventional B cells, not B-1 cells, are responsible for producing autoantibodies in lpr mice. J Exp Med. 1993;177:69–78. doi: 10.1084/jem.177.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reap EA, Sobel ES, Jennette JC, Cohen PL, Eisenberg RA. Conventional B cells, not B1 cells, are the source of autoantibodies in chronic graft-versus-host disease. J Immunol. 1993;151:7316–7323. [PubMed] [Google Scholar]
- 18.Matsiota P, Blancher A, Doyon B, Guilbert B, Clanet M, Kouvelas ED, Avrameas S. Comparative study of natural autoantibodies in the serum and cerebrospinal fluid of normal individuals and patients with multiple sclerosis and other neurological diseases. Ann Inst Pasteur Immunol. 1988;139:99–108. doi: 10.1016/0769-2625(88)90134-1. [DOI] [PubMed] [Google Scholar]
- 19.Xiao BG, Linington C, Link H. Antibodies to myelin-oligodendrocyte glycoprotein in cerebrospinal fluid from patients with multiple sclerosis and controls. J Neuroimmunol. 1991;31:91–96. doi: 10.1016/0165-5728(91)90014-x. [DOI] [PubMed] [Google Scholar]
- 20.Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: In situ evidence for antibody-and complement-mediated demyelination. Ann Neurol. 1998;43:465–471. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- 21.Miller DJ, Sanborn KS, Katzmann JA, Rodriguez M. Monoclonal autoantibodies promote central nervous system repair in an animal model of multiple sclerosis. J Neurosci. 1994;14:6230–6238. doi: 10.1523/JNEUROSCI.14-10-06230.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asakura K, Miller DJ, Pease LR, Rodriguez M. Targeting of IgMκ antibodies to oligodendrocytes promotes CNS remyelination. J Neurosci. 1998;18:7700–7708. doi: 10.1523/JNEUROSCI.18-19-07700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warrington AE, Asakura K, Bieber AJ, Ciric B, Van Keulen V, Kaveri SV, Kyle RA, Pease LR, Rodriguez M. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci USA. 2000;97:6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Correale J, Mix E, Olsson T, Kostulas V, Fredrikson S, Hojeberg B, Link H. CD5+ B cells and CD4−8−T cells in neuroimmunological diseases. J Neuroimmunol. 1991;32:123–132. doi: 10.1016/0165-5728(91)90004-q. [DOI] [PubMed] [Google Scholar]
- 25.Mix E, Olsson T, Correale J, Baig S, Kostulas V, Olsson O, Link H. B cells expressing CD5 are increased in cerebrospinal fluid of patients with multiple sclerosis. Clin Exp Immunol. 1990;79:21–27. doi: 10.1111/j.1365-2249.1990.tb05121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seidi OA, Semra YK, Sharief MK. Expression of CD5 on B lymphocytes correlates with disease activity in patients with multiple sclerosis. J Neuroimmunol. 2002;133:205–210. doi: 10.1016/s0165-5728(02)00360-0. [DOI] [PubMed] [Google Scholar]
- 27.Bongioanni P, Fioretti C, Vanacore R, Bianchi F, Lombardo F, Ambrogi F, Meucci G. Lymphocyte subsets in multiple sclerosis. A study with two-colour fluorescence analysis. J Neurol Sci. 1996;139:71–77. [PubMed] [Google Scholar]
- 28.Sellebjerg F, Jensen J, Jensen CV, Wiik A. Expansion of CD5− B cells in multiple sclerosis correlates with CD80 (B7-1) expression. Scand J Immunol. 2002;56:101–107. doi: 10.1046/j.1365-3083.2002.01108.x. [DOI] [PubMed] [Google Scholar]
- 29.Kasaian MT, Ikematsu H, Casali P. Identification and analysis of a novel human surface CD5− B lymphocyte subset producing natural antibodies. J Immunol. 1992;148:2690–2702. [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 31.Hayakawa K, Shinton SA, Asano M, Hardy RR. B-1 cell definition. Curr Top Microbiol Immunol. 2000;252:15–22. doi: 10.1007/978-3-642-57284-5_2. [DOI] [PubMed] [Google Scholar]
- 32.Tsunoda I, Kuang L-Q, Theil DJ, Fujinami RS. Antibody association with a novel model for primary progressive multiple sclerosis: Induction of relapsing-remitting and progressive forms of EAE in H2S mouse strains. Brain Pathol. 2000;10:402–418. doi: 10.1111/j.1750-3639.2000.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson LK, Tsunoda I, Masaki T, Fujinami RS. Polyreactive myelin oligodendrocyte glycoprotein antibodies: Implications for systemic autoimmunity in progressive experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;183:69–80. doi: 10.1016/j.jneuroim.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Libbey JE, Peterson LK, Tsunoda I, Fujinami RS. Monoclonal MOG-reactive autoantibody from progressive EAE has the characteristics of a natural antibody. J Neuroimmunol. 2006;173:135–145. doi: 10.1016/j.jneuroim.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 35.Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, Matthieu J-M, Baker D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153:4349–4356. [PubMed] [Google Scholar]
- 36.Papenfuss TL, Rogers CJ, Gienapp I, Yurrita M, McClain M, Damico N, Valo J, Song F, Whitacre CC. Sex differences in experimental autoimmune encephalomyelitis in multiple murine strains. J Neuroimmunol. 2004;150:59–69. doi: 10.1016/j.jneuroim.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Tsunoda I, Kuang L-Q, Tolley ND, Whitton JL, Fujinami RS. Enhancement of experimental allergic encephalomyelitis (EAE) by DNA immunization with myelin proteolipid protein (PLP) plasmid DNA. J Neuropathol Exp Neurol. 1998;57:758–767. doi: 10.1097/00005072-199808000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Greer JM, Sobel RA, Sette A, Southwood S, Lees MB, Kuchroo VK. Immunogenic and encephalitogenic epitope clusters of myelin proteolipid protein. J Immunol. 1996;156:371–379. [PubMed] [Google Scholar]
- 39.Tsunoda I, Kuang L-Q, Igenge IZM, Fujinami RS. Converting relapsing remitting to secondary progressive experimental allergic encephalomyelitis (EAE) by ultraviolet B irradiation. J Neuroimmunol. 2005;160:122–134. doi: 10.1016/j.jneuroim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Mi W, Prentice TW, Young CR, Johnson RR, Sieve AN, Meagher MW, Welsh CJ. Restraint stress decreases virus-induced pro-inflammatory cytokine mRNA expression during acute Theiler’s virus infection. J Neuroimmunol. 2006;178:49–61. doi: 10.1016/j.jneuroim.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Hayakawa K, Hardy RR, Herzenberg LA. Peritoneal Ly-1 B cells: Genetic control, autoantibody production, increased lambda light chain expression. Eur J Immunol. 1986;16:450–456. doi: 10.1002/eji.1830160423. [DOI] [PubMed] [Google Scholar]
- 42.Litzenburger T, Fassler R, Bauer J, Lassmann H, Linington C, Wekerle H, Iglesias A. B lymphocytes producing demyelinating autoantibodies: Development and function in gene-targeted transgenic mice. J Exp Med. 1998;188:169–180. doi: 10.1084/jem.188.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]