

Abstract
Background
The accumulation of amyloid β plaques (Aβ) is a central feature of Alzheimer’s disease (AD). First reported in animal models, it remains uncertain if peripheral inflammatory/infectious conditions in humans can promote Aβ brain accumulation. Periodontal disease, a common chronic infection, has been previously reported to be associated with AD.
Methods
Thirty-eight cognitively normal, healthy, community residing elderly (mean age 61; 68% female) were examined in an Alzheimer’s Disease research center and a University-based Dental School. Linear regression models (adjusted for age, ApoE and smoking) were used to test the hypothesis that periodontal disease assessed by clinical attachment loss was associated with brain Aβ load using 11C-PIB PET imaging.
Results
After adjusting for confounders, clinical attachment loss (≥ 3mm), representing a history of periodontal inflammatory/infectious burden, was associated with increased 11C-PIB uptake in Aβ vulnerable brain regions (p=0.002).
Conclusion
We show for the first time in humans an association between periodontal disease and brain Aβ load. These data are consistent with prior animal studies showing that peripheral inflammation/infections are sufficient to produce brain Aβ accumulations.
Keywords: Alzheimer’s disease, infection, inflammation, periodontal disease, brain amyloid, PIB-PET, cognition
1. Introduction
Worldwide, more than 35 million persons suffer from dementia among which 50–60% are diagnosed with Alzheimer’s disease (AD) (Association, 2014). It is estimated these numbers will double by 2030 and double again by 2050. These statistics underline the enormous public health importance of identifying modifiable risk factors.
The accumulation of amyloid β plaques is a central feature of Alzheimer’s disease (AD) whose cause is poorly understood. Post mortem studies have shown that amyloid accumulation can start as early as 30 years of age and increases with age (Braak and Braak, 1997, Kok, et al., 2009). These findings have been confirmed by imaging studies (Jack, et al., 2009, Klunk, et al., 2004, Landau, et al., 2012). The results of clinical trials designed to remove brain amyloid from impaired individuals have been largely unsuccessful possibly due to the late intervention (Holmes, et al., 2008, Morgan, 2011, Ozudogru and Lippa, 2012). This has placed a great emphasis on identifying factors and mechanisms that promote brain amyloid deposition in advance of symptoms.
Both animal models and clinical evidence show that inflammation is involved in the pathogenesis of AD (Akiyama, et al., 2000, Griffin, et al., 1998, Holmes and Butchart, 2011, McGeer, et al., 2006, Tanzi, 2012), but it remains unknown which peripheral inflammatory and infectious conditions play a role and at which stage of AD development (Kamer, et al., 2008a, Kamer, et al., 2008b, Miklossy, 2011a, Miklossy, 2011b). We examined human periodontal disease as a model for testing the relationship between peripheral inflammation/infections and brain amyloid β. Periodontal disease is a chronic, peripheral, polymicrobial infection (Socransky and Haffajee, 1997) characterized by local and systemic inflammation. Periodontal disease is defined by the loss of the tissues surrounding the teeth, clinically defined by clinical attachment loss (CAL)(Demmer, et al., 2008).
The present cross-sectional study used positron emission tomography (PET) amyloid imaging and clinical periodontal examinations to test the hypothesis that in cognitively normal subjects the magnitude of periodontal disease burden is associated with the brain amyloid load.
2. Methods
2.1. Study subjects and design
Thirty-eight cognitively normal, healthy subjects were included in this study. All subjects were participants in NIH supported Alzheimer’s Disease studies at the NYU School of Medicine. Subjects were recruited from a random community sampling of voter registration lists. Among the 250 elderly individuals that were contacted and invited to participate, 70 subjects agreed to participate. Of these, 40 subjects had standardized medical and cognitive examinations consistent with the National Alzheimer Coordinating Center guidelines (Beekly, et al., 2007). The standardized diagnostic evaluation at the New York University School of Medicine consisted of medical, psychiatric, neuropsychological, ApoE genotyping, MRI examinations, and standardized periodontal examinations. Thirty-eight subjects also participated in 11C-PIB PET amyloid brain imaging performed at the Cornell Medical Center. All subjects provided written informed consent to participate in this Institutional Review Board (IRB)-approved study. The average interval between the PET scan and the periodontal examination was 1.29±0.89 years. All research measures were performed blinded to the clinical data.
Inclusion Criteria
All included subjects had at least 12 years of education and were fluent English speakers. Subjects were defined as cognitively normal if they had Clinical Dementia Rating (CDR) = 0 (Berg, 1984), Global Deterioration Scale (GDS) ≤ 2 (Reisberg, et al., 1982), and Mini–Mental State Examination (MMSE) ≥ 28 (Cockrell and Folstein, 1988).
All subjects were required to have a minimum 10 evaluable teeth (Stein, et al., 2007) and to have the physical capacity to manage their personal dental hygiene.
Exclusion Criteria
Individuals were excluded if they had history/medical conditions that could affect brain structure or function, such as clinical or MRI evidence of cortical stroke, uncontrolled hypertension, diabetes, head trauma with loss of consciousness, any manifest neurodegenerative disease, chronic depression, MRI evidence of hydrocephalus, or intracranial mass. Subjects taking anti-inflammatory medications for chronic conditions (i.e. NSAIDS, anti-TNFα), or antibiotics or having periodontal treatment 3 months prior to the periodontal evaluation were also excluded.
2.2. Clinical Evaluations
2.2.1 Measures of periodontal disease
The assessment for periodontal disease was conducted as follows: Teeth were counted and the presence of dental plaque on six surfaces of all teeth was recorded (Silness and Loe, 1964). Clinical Attachment Loss (CAL), the primary dependent variable, was measured using a Michigan probe (Demmer, et al., 2008) and recorded in millimeters at six sites per tooth. CAL defined the long-term periodontal inflammatory/infectious condition. CAL was obtained by adding the probing depth to the distance from the free gingival margin to the cemento-enamel junction (positive if the gingival margin is apical to the cement-enamel junction and negative if it is coronal. The probing depth (PD) was measured as the linear distance in millimeters from the gingival margin to the base of the periodontal pocket. Bleeding on probing (BOP) was assessed at each probing site.
The primary periodontal exposure was defined as the cumulative number of sites with CAL ≥ 3mm (referred to as CAL3) and provided a measure of periodontal disease burden (Tonetti, et al., 2005). The use of CAL ≥ 3mm was based on our a priori hypothesis proposing a linear relationship between the magnitude of periodontal destruction due to the history of periodontal inflammation and brain amyloid accumulation, a chronic process. The threshold of 3mm included also milder forms of the periodontal disease. These measures were used previously to define relationships between periodontal and cardiovascular disease and cognitive dysfunction (Beck, et al., 2001, Elter, et al., 2004). An additional consideration came from evidence showing that CAL associated better with a chronic systemic process (Demmer, et al., 2008) rather than an acute one. CAL are accepted measures of cumulative lifetime experience of periodontitis and using these measures, the 5th European Workshop in Periodontology proposed the following case definition for periodontitis: the presence of proximal attachment loss of ≥3 mm in at least 2 non-adjacent teeth. All our subjects had CAL ≥ 3mm on multiple teeth and thus they all fell within this case definition. To show consistency in the relationship between measures of periodontal disease and brain amyloid accumulation, other measures of periodontal disease were evaluated: CAL ≥ 4 mm (CAL4) (Page and Eke, 2007), PD ≥ 3mm (PD3) and BOP. By comparison to CAL, PD and BOP measure the present disease and inflammation. To further define periodontal disease, we combined historical with current measures of periodontal disease: the presence of CAL ≥ 3mm at ≥ 66% sites and concomitant PD ≥ 5mm was defined as Perio1 (Jonsson, et al., 2014).
2.2.2. Positron emission tomography (PET) outcome variables
Acquisition and preprocessing
Subjects received an11C-PIB-PET scan acquired in 3-D mode on a LS Discovery PET scanner (GE Medical Systems, Milwaukee, WI, USA) (Li, et al., 2008, Mosconi, et al., 2013a, Mosconi, et al., 2010). Briefly, as previously reported, subjects were injected with 15 mCi (550 MBq) of N-methyl[11C]2-(4′-methylaminophenyl)-6-hydroxy-benzothiazole, Pittsburgh compound B (PIB), followed by a 90 minute PET data acquisition (Mosconi, et al., 2010). Image analysis was carried out at NYU, blind to the clinical data. For each subject, summed PET images corresponding to the 60–90 minutes of PIB data were coregistered to the subject’s T1 Magnetic Resonance Imaging (MRI) scan using Statistical Parametric Mapping (SPM). Both the summed 60–90 minute PIB image and SPM2 segmented MRI gray matter (GM) and white matter (WM) images were reformatted into SPM’s standard template space. In standard space, regions of interest (ROI) were intersected with the GM to exclude all non-GM pixels. HIPMASK was used to for accurate ROI sampling (Li, et al., 2008, Mosconi, et al., 2005). A correction for partial volume effects (PVC) was done using Muller-Gartner’s 2-tissue method, which corrects for both CSF and white matter tracer uptake (Muller-Gartner, et al., 1992).
PET regions of interest
The average PIB intensity in each ROI was normalized by the average intensity from a cerebellar gray matter reference ROI, to create the standard uptake value ratio (SUVR). From our prior work (Li, et al., 2008, Mosconi, et al., 2013b), 5 bilateral ROIs known to be vulnerable to amyloid depositions were sampled to create a composite neocortical PIBAD mask (MaskAD), which was the primary outcome measure. The regions included in the AD mask were: prefrontal cortex. middle frontal gyrus, lateral temporal lobe, inferior parietal lobule, and posterior cingulate cortex/precuneus (Mosconi, et al., 2010).
2.3. Statistical methods
To determine whether measures of periodontal disease were associated with amyloid load in MaskAD (shown to be vulnerable to amyloid accumulation), hierarchical regression analyses were performed in which MaskAD SUVR was the dependent variable and measures of periodontal disease (i.e. CAL3) and the relevant covariates (Barnes and Yaffe, 2011, Heyanka, et al., 2010, Stein, et al., 2007, Stein, et al., 2010, Xu, et al.) were the independent variables. Demographic (age, gender, and education), systemic factors (co-morbidities), oral (brushing, flossing, dentist visits) and social (smoking) measures were obtained by a standardized examiner-conducted interview at the time of the oral examination. For high systemic (SBP) and diastolic blood pressure (DBP), BMI, cholesterol, high-density lipoproteins (HDL), standard cutoffs were used (Barba, et al., 2008, National Cholesterol Education Program Expert Panel on Detection and Treatment of High Blood Cholesterol in, 2002). ApoE genotype (carriers vs. non-carriers of ApoE ε4 allele) was obtained as previously reported (Mosconi, et al., 2012, Osorio, et al., 2014) and smoking was classified as never or former/current smokers. Cognitive performance was assessed by Logic2 of Wechsler Memory Scale-Revised test (De Santi, et al., 2008).
Our initial approach tested the above covariates and only significant covariates in at least one ROI were retained in the final models. These covariates included: age, ApoE and smoking. These analyses were repeated with additional measures of periodontal disease (CAL ≥4mm, PD ≥ 3mm, BOP, and Perio1 as independent variables.
The partial correlations, unstandardized β coefficients (β) and the 95% confidence interval (95%CI) of the β coefficients are obtained. All models were checked for linearity, independence, homoscedasticity and normality. The log transformation was used to normalize the distribution of the MaskAD SUVR. Regression plots are presented in which the axes represent the standardized residuals for the dependent (MaskAD SUVR) and independent variables (CAL3 and CAL4) adjusted for covariates. Statistical significance was set at p≤0.05. Statistical analyses were performed using IBM SPSS version 21, Chicago IL.
3. Results
3.1. Characteristics of the population
Table 1 summarizes the subject characteristics. Briefly, the mean years of age was 61.3 (sd=8.1; range 44–79; 5 subjects <55), mean years of education 17.6 (sd=2.2; range 13–22; 4 subjects <16 years of education), 68% were female, and 42% were carriers of an ApoE ε4 allele. The subjects were relatively healthy with 55% reporting no medical conditions and 73% no smoking history. Three subjects had high SBP and two had high DBP, five were obese (BMI>30) and none of the subjects had diabetes. Most subjects reported good oral hygiene practices and regular visits to the dentist (Table 1). The age, gender, education and ApoE adjusted means for Logic 2 scores were not different within the oral health categories (brushing: p=0.581; flossing: p=0.447; dental visits: p=0.727).
Table 1.
Characteristics of the subject population.
| Demographic data | ||
|---|---|---|
| Number Subjects (n) | 38 | |
| Female [n (%)] | 26 (68.4) | |
| Age [mean (SD)] | 61.3 (8.1) | |
| Years of education [mean (SD)] | 17.6 (2.2) | |
| ApoE carriers- total [n (%)] | 16 (42.1) | |
| Systemic health findings [Means (SD)]] | [n % high] | |
| BMI (n=38) | 24.8 (4.2) | 5 (13.2) |
| Systolic blood pressure (n=37) | 116.2 (16.6) | 3 (8.1) |
| Diastolic blood pressure (n=36) | 68.4 (9.1) | 2 (5.6) |
| Total Cholesterol (n=37) | 198.7 (33) | 20 (54.1) |
| HDL (n=37) | 65.4 (22.6) | 34 (91.1) |
| Smoking (n=38) | 10 (26.3) | |
| Oral Hygiene Behavior characteristics (n=38) | ||
| Brushing (%) | ||
| ≤once/day | 31.6 | |
| >once/day | 68.4 | |
| Flossing (%) | ||
| <once/day | 63.2 | |
| ≥once/day | 36.8 | |
| Visits to dentist (%) | ||
| ≤ 6 month | 71.1 | |
| > 6 month | 28.9 | |
| Periodontal exam findings [Means (SD)] | ||
| Tooth number | 25.86 (5.1) | |
| CAL≥3mm | 125.4 (30.2) | |
| CAL≥4mm | 79.4 (29.7) | |
| CAL≥5mm | 35.3 (23.6) | |
| CAL≥6mm | 10.2 (11.5) | |
| PD≥3mm | 96.3 (22.3) | |
| PD≥4mm | 26.3 (14.9) | |
| PD≥5mm | 6.9 (6.5) | |
| PD≥6mm | 1.7 (3.0) | |
| BOP | 36.6 (18.8) | |
| Perio1[n (%)] | 13.0 (36.1) | |
CAL=clinical attachment loss; PD=pocket depth; BOP=bleeding on probing; Perio1= CAL≥3mm at ≥66%sites/PD≥5mm); HDL=high-density lipoproteins.
3.2. Measures of periodontal disease associate with amyloid load in AD vulnerable areas
The mean CAL3 and CAL4 was 125.4 and 79.4 respectively (sd=30.2 and 29.7) meaning that 125.4 and 79.4 dental sites had a clinical attachment loss of 3mm and 4mm or more (maximum possible number of sites=192). The mean for PD3 was 96.3 (sd=22.3) and for BOP was 36.6 (sd=18.8). The range of CAL3, CAL4, PD3 and BOP was 60–159, 27–135, 44–139 and 6–76. Using the definition of Perio1 (CAL ≥ 3mm at ≥ 66%sites/PD ≥ 5mm), 13 subjects had extensive moderate to severe periodontitis. Among the covariates tested (SBP, DBP, obesity, cholesterol, HDL, co-morbidities, Logic2, plaque index, tooth loss, brushing, flossing, visits to the dentist and time difference between periodontal exam and PIB), none were found significant in any of the models. The regression analyses with each of these covariates are presented in Table S1 of the Supplementary Material.
The primary measure of exposure, CAL3 was significantly correlated with PIB retention in MaskAD after controlling for age, ApoE and smoking (adjusted partial correlation r=0.50, p=0.002; β=0.011, 95CI (0.004–0.017). Figure 1A shows the regression plot for CAL3 and MaskAD. Thus, addition of CAL3 to the model predicting PIB retention led to a statistically significant increase in R2 (ΔR2) of 0.22 (p=0.002), indicating that 22% of the variance of the PIB retention in the AD vulnerable regions could be attributed to CAL3. The significance of this observation is also supported by the consistency of the results in each of the regions comprising the MaskAD (Table 2S in the Supplementary Material). After controlling for age, ApoE and smoking, CAL4 and Perio1 also show consistent and significant results while PD3 correlations were not significant [CAL4: r=0.410, p=0.015, β=0.008, 95CI (0.002–0.015); Perio1: r=0.412, p=0.017, β =0.482, 95CI (0.084–0.881; PD3: r=0.299, p=0.085, β=0.009, 95CI (−0.001–0.019)] Figure 1B shows the regression plot for CAL4 and MaskAD. By comparison, BOP did not associate with PIB retention in MaskAD [r=−0.096, p=0.584, β=−0.003, 95CI (−0.003–0.008)]. Regression models showing the adjusted partial correlation coefficients and the unstandardized β coefficients with the 95%CI for age, ApoE and smoking adjusted partial correlation coefficients are presented in Table 2S and 3S in the Supplementary Material. No significant interaction was found between APOE genotype and any of the periodontal measures. Figure 2 illustrates the PIB PET scans of 4 normal individuals; 2 of them are ApoE E4 carriers with high vs. low CAL and two of them are non-carriers of ApoE E4 with high vs. low CAL.
Figure 1.
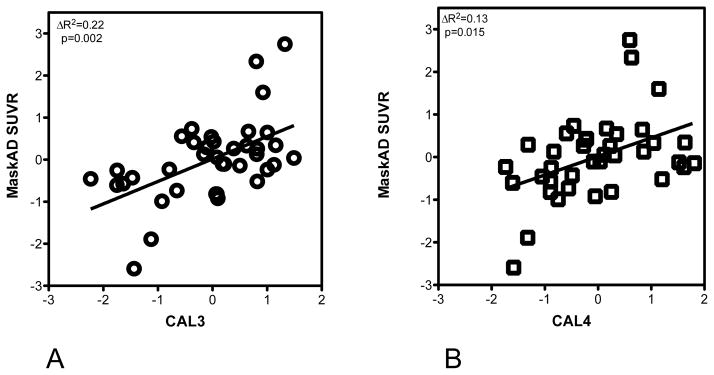
Partial regression plots show the relationships between CAL3 (A) and CAL4 (B) and PIB retention in MaskAD. The x and y axes are the residuals for CAL3 and CAL4 respectively and PIB retention adjusted for age, ApoE carrier status and smoking. The changes in R2 (ΔR2) and p values for these changes are shown. MaskAD is defined by combining the standardized uptake value ratios (SUVR) of inferior parietal lobule, lateral temporal lobe, middle frontal gyrus, posterior cingulate cortex/precuneus, and prefrontal cortex. Note, the CAL3-SUVR and CAL4-SUVR associations are significant.
Figure 2.
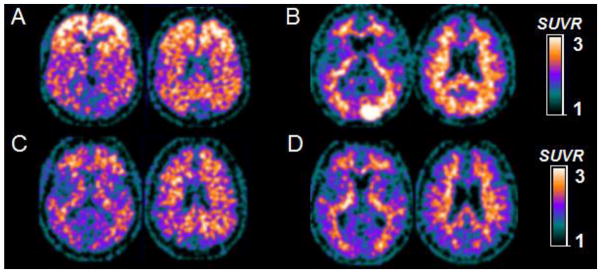
PIB-PET scans of four representative NL individuals: two of them age 72 and 75 are ApoE carriers with high CAL3 (A) and low CAL3 (C) and two of them age 79 and 76 are ApoE non-carriers with high CAL3 (B) and low CAL3 (D). Note: the subjects in the upper row have significant amyloid accumulation and high CAL3 measures while the subjects in the lower row have low brain amyloid accumulation and low CAL3. PIB-PET scans for each subject are displayed as axial sections encompassing basal ganglia (first slide) and inferior parietal level (second slide) respectively. PIB measures are standardized uptake value ratios (SUVR) to cerebellar gray matter. Note: the APOE4-positive subject presents with a positive PIB pattern while the APOE4-negative subject presents with an “emerging PIB pattern” in the right temporal-occipital cortex. A. PIB positive e4 carrier; B. PIB positive e4 non-carrier; C. PIB negative e4 carrier; D. PIB negative e4 non-carrier.
4. Discussion
To our knowledge, this is the first study to show that clinical measures of periodontal disease in cognitively normal healthy elders are positively associated with the magnitude of brain amyloid accumulation assessed by [11C]PIB-PET. This conclusion was reached after showing that neither medical confounds, smoking, oral health behaviors, tooth loss, memory performance, nor ApoE genotype accounted for this association. These results are consistent with the hypothesis that chronic periodontal inflammation/infection contributes to brain amyloid load and that these associations are not secondary to impaired cognition.
4.1 Periodontal Inflammation/Infection associate with amyloid load in AD-vulnerable amyloid areas
Periodontal disease is a chronic, peripheral, polymicrobial infection (Socransky and Haffajee, 1997) characterized by local and systemic inflammation ((Paraskevas, et al., 2008). Approximately 85% of the bacteria colonizing the subgingival biofilm are Gram negative (Socransky and Haffajee, 2002) which are rich in LPS. Among them, Aggregatibacter actinomycetemcomitans, Tannerella forsythus, Porphyromonas gingivalis (P.gingivalis) and Treponema denticola (T.denticola) are important periodontal pathogens (Haffajee and Socransky, 2006, Socransky and Haffajee, 1997). Periodontal disease is more prevalent in adulthood and elderly, but it can start in childhood. Approximately 64% of adults aged 65 or older have chronic moderate and severe periodontitis (Eke, et al., 2012). It is known that periodontal-derived pro-inflammatory molecules, bacteria and bacterial products can reach the brain via systemic circulation and/or neural pathways (Holmes and Cotterell, 2009, Kamer, et al., 2008b, Rivest, 2003) and increase brain cytokine levels. These types of inflammatory changes are separately known to contribute to brain amyloid accumulation, and cognitive dysfunction (Kamer, et al., 2008b, Perry, et al., 2003). Our prior clinical data as well as other studies have linked periodontal disease and AD with moderate odds ratios (Gatz, et al., 2006, Grabe, et al., 2009, Kamer, et al., 2012, Kaye, et al., 2010, Noble, et al., 2009, Stein, et al., 2007, Stewart and Hirani, 2007). For example, in a survey of over 6,000 subjects with a broad age range, CAL≥3mm associated with cognitive dysfunction (Stewart and Hirani, 2007). In a sample of 197 subjects, antibodies to known periodontal pathogens predicted the development of AD years before its clinical diagnosis (Sparks Stein, et al., 2012). However, prior clinical studies included subjects with a broad range of cognitive performance by including cognitively impaired subjects. Our study used robust standardized criteria to define medically and cognitively “normal” subjects thus minimizing the effect of cognitive and other confounds on periodontal and brain health. The strongest correlation between measures of periodontal disease and amyloid retention was achieved using 3 mm as the threshold for the clinical attachment loss. Although not as strong, CAL ≥ 4mm correlation to amyloid was also significant and PD ≥ 3mm approached statistical significance in some areas. BOP, a marker of current inflammation was not significant. When interpreting our results, it is important to bear in mind that our subject population is receiving good oral care. These findings are consistent with our hypothesis that long-term inflammatory burden is more important than current inflammation. These results also suggest that even mild cases of disease can have long terms effects as found previously (Demmer, et al., 2008).
Considerable in vitro, animal model, and clinical data show that peripheral inflammations and infections are sufficient to increase brain amyloid load possibly by: augmenting amyloid beta synthesis (Weintraub, et al., 2013), disrupting the brain blood barrier and/or amyloid beta trafficking (Erickson, et al., 2012). Further clinical evidence for the importance of inflammation in brain amyloid accumulation is suggested by data coming from the Alzheimer’s Disease Neuroimaging Initiative. This study reported that inflammatory molecules such as chemokine ligand 13, IL-17, fibrinogen, alpha-1-antitrypsin, complement C3, interleukin-3, interleukin-13 were associated with PIB retention (Kiddle, et al., 2012).
Infection-induced brain amyloid load has also been reviewed critically in the literature (Miklossy, 2008, Miklossy, 2011a, Miklossy, 2011b). It has been reported that spirochetal and chronic bacterial infections can cause cognitive decline and brain amyloid deposition (Miklossy, 2008, Miklossy, 2011a, Miklossy, et al., 2004, Miklossy, et al., 2006). A classic example of infection is the atrophic form of general paresis caused by Treponema pallidum, a spirochete that presents with progressive dementia and brain amyloid deposits. Miklossy proposed that oral spirochetes may be possible candidates to invade the brain and cause cognitive impairment in AD (Miklossy, 1993, Miklossy, et al., 1994). Riviere et al. detected 6 different periodontal pathogen Treponemes in the brains of more than 90% of the 16 AD cases analyzed (Riviere, et al., 2002). Moreover, P. gingivalis-derived LPS was also detected in the brains of AD patients (Poole, et al., 2013). It is accepted that periodontal bacteria can be found at distant sites (Cavrini, et al., 2005, Haraszthy, et al., 2000, Okuda, et al., 2001, Reichert, et al., 2013). These data indicate that peripheral inflammation/infections can lead to infection/inflammation in the brain and promote amyloid accumulation. Whether the amyloid constitutes an immune protective molecule or an injurious agent or both remains to be established (Castellani, et al., 2009, Soscia, et al., 2010, White, et al., 2014).
Our results can also have other explanations. Reduced masticatory abilities due to periodontal disease may result in dietary deficiencies and increased stress response, and these may lead to increased amyloid beta (Ekuni, et al., 2013). It is also possible that the relationship between CAL and amyloid load is related to a relationship between CAL and cognitive dysfunction. However, our subjects are cognitively normal and the inclusion of Logic2 was not significant in the models. It is also possible that people with periodontal disease also have poorer systemic health. However, our use of exclusion criteria mitigated this effect. And still another explanation may be related to host response (hyper-inflammatory) that could affect both periodontal disease and brain pathology (Kamer, et al., 2008a, Kamer, et al., 2008b).
Among the multiple covariates investigated, smoking associated with PIB retention in MaskAD but only at trend level (p=0.067). Smoking is a risk factor for periodontal disease and we expected that smoking would negatively confound the association between CAL and PIB retention. Contradictory to our prediction, smoking did not down-regulate the CAL effect on PIB retention. Controversy exists regarding the associations between smoking and AD. Some studies found that smoking may provide a protective effect (Brenner, et al., 1993), while the majority of studies showed a deleterious effect (Anstey, et al., 2007, Cataldo, et al., 2010, Reitz, et al., 2011). Moreover, in an animal model, it was shown that smoking was able to up-regulate brain amyloid possible through brain inflammation (Moreno-Gonzalez, et al., 2013). It is tempting to speculate that perhaps a common inflammatory mechanism for CAL and smoking leading to amyloid increase may explain these results. However, this sample size is small. Additional longitudinal studies are needed to untangle the relationship between smoking, periodontal disease and AD pathology.
4.2. Strengths and weaknesses
As in most cross-sectional studies, reverse causation should also be considered, as the direction of the associations observed in these studies cannot be determined. The observed association between CAL and brain amyloid accumulation and cognitive dysfunction may reflect the effects of amyloid load/cognition on periodontal health. Equally possible is that people with poor cognition have poorer oral health and several studies have shown that oral health measures such as tooth loss, caries level, and plaque control are impaired in subjects with cognitive impairment or dementia (Ellefsen, et al., 2008, Ellefsen, et al., 2009, Ship, 1992). However, our subjects are defined cognitively normal by robust tests. The presence of periodontal disease in individuals with lower cognitive function or dementia is contradictory (Ship, 1992, Yu and Kuo, 2008). However, the potential effect of cognition on periodontal condition cannot be ignored (Kaye, et al., 2010).
Our sample size was modest and therefore the results of this study should be considered in this context. We observed statistically significant associations between measures of periodontal disease and brain amyloid load even after controlling for covariates, because of our population characteristics. Our sample was quite homogeneous with respect to having excluded confounding measures. The cohort had high education, good systemic health, high cognitive function and lost few teeth. On the other hand, this homogeneity allowed us to detect statistically significant differences despite the limited number of subjects. All medical and dental exams were standardized and one trained periodontist performed all periodontal evaluations blind to both PIB retention data thus minimizing observer bias. A potential weakness is that some measurement misclassification of exposure variables was possible as oral health behaviors were assessed by subject report and recollection. An additional bias may be related to the participants themselves. Although, our sample was derived from the community, the participants were self-selected, thus introducing a potential bias. Notably, 95% of our subjects were white and 42% were ApoE carriers and most currently have good oral care. Finally, although homogeneity of the study population constituted a strength of our project, it also limited the generalizability of our findings.
In conclusion, we showed that after accounting for the relevant confounds, measures of periodontal disease were associated with amyloid accumulation in brain in areas that are prone to amyloid accumulation in patients with AD. Our results suggest that periodontal inflammation/infection may increase the risk for brain amyloid deposition. Future longitudinal and therapeutic studies involving changes in periodontal disease could potentially reveal what is the cause and what is the effect.
Acknowledgments
This study was supported by NIH/NIA grants AG035137, AG032554, AG12101, AG022374, and AG13616, NIH DE023139-02, Alzheimer’s Association NIRG-12-173937, NIH/NCATS 8 UL1 TR000038 and NYU College of Dentistry, Bluestone Center for Clinical Research.
Footnotes
Contributors
ARK, MJD, RGC, LG and DS designed the study. ARK and EP analyzed the data with assistance from MJD. ARK, MJD and LP interpreted the data. ARK wrote the manuscript with assistance from MJD, LG, RC and LM. PMH and SW performed medical examinations and collected the cognitive data. ARK performed the oral examinations assisted by PC, RL, SS and CS. HR, SV, RL, LM, LY and WT performed image analysis and assisted with data collection and interpretation. All authors reviewed the manuscript for intellectual content and approved the final draft.
Conflict of interest: No conflict of interest is reported for A. Kamer, P. Corby, R. Craig, D. Saxena, H. Rusinek, S. Vallabhajosula, S. Williams, R. Linker, S. Svetco and C. Shi. L. Mosconi, W. Tsui, and M. de Leon have a patent on an image analysis technology that was licensed to Abiant Imaging, Inc, by NYU, and have a financial interest in this license agreement, and NYU holds stock options on the company. Y. Li, L. Mosconi and M. de Leon have received compensation for consulting services from Abiant Imaging. Dr L. Glodzic was a Principal Investigator on an Investigator-Initiated project funded by Forest Laboratories, Inc, and received an honorarium for serving as a consultant to Roche Pharma.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiology of aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstey KJ, von Sanden C, Salim A, O’Kearney R. Smoking as a risk factor for dementia and cognitive decline: a meta-analysis of prospective studies. American journal of epidemiology. 2007;166(4):367–78. doi: 10.1093/aje/kwm116. [DOI] [PubMed] [Google Scholar]
- Association A.s. 2014 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2014;10(2):e47–e92. doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Barba R, Zapatero A, Losa JE, Valdes V, Todoli JA, Di Micco P, Monreal M, Riete I. Body mass index and mortality in patients with acute venous thromboembolism: findings from the RIETE registry. Journal of thrombosis and haemostasis : JTH. 2008;6(4):595–600. doi: 10.1111/j.1538-7836.2008.02907.x. [DOI] [PubMed] [Google Scholar]
- Beck JD, Elter JR, Heiss G, Couper D, Mauriello SM, Offenbacher S. Relationship of periodontal disease to carotid artery intima-media wall thickness: the atherosclerosis risk in communities (ARIC) study. Arteriosclerosis, thrombosis, and vascular biology. 2001;21(11):1816–22. doi: 10.1161/hq1101.097803. [DOI] [PubMed] [Google Scholar]
- Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, Hubbard JL, Koepsell TD, Morris JC, Kukull WA Centers N.I.A.A.s.D. The National Alzheimer’s Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer disease and associated disorders. 2007;21(3):249–58. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- Berg L. Clinical Dementia Rating. Br J Psychiatry. 1984;145:339. [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiology of aging. 1997;18(4):351–57. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Brenner DE, Kukull WA, van Belle G, Bowen JD, McCormick WC, Teri L, Larson EB. Relationship between cigarette smoking and Alzheimer’s disease in a population-based case-control study. Neurology. 1993;43(2):293–300. doi: 10.1212/wnl.43.2.293. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Lee HG, Siedlak SL, Nunomura A, Hayashi T, Nakamura M, Zhu X, Perry G, Smith MA. Reexamining Alzheimer’s disease: evidence for a protective role for amyloid-beta protein precursor and amyloid-beta. Journal of Alzheimer’s disease : JAD. 2009;18(2):447–52. doi: 10.3233/JAD-2009-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s Disease: an analysis controlling for tobacco industry affiliation. Journal of Alzheimer’s disease : JAD. 2010;19(2):465–80. doi: 10.3233/JAD-2010-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavrini F, Sambri V, Moter A, Servidio D, Marangoni A, Montebugnoli L, Foschi F, Prati C, Di Bartolomeo R, Cevenini R. Molecular detection of Treponema denticola and Porphyromonas gingivalis in carotid and aortic atheromatous plaques by FISH: report of two cases. Journal of medical microbiology. 2005;54(Pt 1):93–6. doi: 10.1099/jmm.0.45845-0. [DOI] [PubMed] [Google Scholar]
- Cockrell JR, Folstein MF. Mini-Mental State Examination (MMSE) Psychopharmacol Bull. 1988;24(4):689–92. [PubMed] [Google Scholar]
- De Santi S, Pirraglia E, Barr W, Babb J, Williams S, Rogers K, Glodzik L, Brys M, Mosconi L, Reisberg B, Ferris S, de Leon MJ. Robust and conventional neuropsychological norms: diagnosis and prediction of age-related cognitive decline. Neuropsychology. 2008;22(4):469–84. doi: 10.1037/0894-4105.22.4.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmer RT, Kocher T, Schwahn C, Volzke H, Jacobs DR, Jr, Desvarieux M. Refining exposure definitions for studies of periodontal disease and systemic disease associations. Community Dent Oral Epidemiol. 2008;36(6):493–502. doi: 10.1111/j.1600-0528.2008.00435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ, James Beck GDRP Cdc Periodontal Disease Surveillance workgroup. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012;91(10):914–20. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- Ekuni D, Endo Y, Tomofuji T, Azuma T, Irie K, Kasuyama K, Morita M. Effects of apoE deficiency and occlusal disharmony on amyloid-beta production and spatial memory in rats. PloS one. 2013;8(9):e74966–e74. doi: 10.1371/journal.pone.0074966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellefsen B, Holm-Pedersen P, Morse DE, Schroll M, Andersen BB, Waldemar G. Caries prevalence in older persons with and without dementia. Journal of the American Geriatrics Society. 2008;56(1):59–67. doi: 10.1111/j.1532-5415.2007.01495.x. [DOI] [PubMed] [Google Scholar]
- Ellefsen B, Holm-Pedersen P, Morse DE, Schroll M, Andersen BB, Waldemar G. Assessing caries increments in elderly patients with and without dementia: a one-year follow-up study. J Am Dent Assoc. 2009;140(11):1392–400. doi: 10.14219/jada.archive.2009.0076. [DOI] [PubMed] [Google Scholar]
- Elter JR, Champagne CM, Offenbacher S, Beck JD. Relationship of periodontal disease and tooth loss to prevalence of coronary heart disease. Journal of periodontology. 2004;75(6):782–90. doi: 10.1902/jop.2004.75.6.782. [DOI] [PubMed] [Google Scholar]
- Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid beta efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150–65. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, Mortimer JA, Fratiglioni L, Johansson B, Berg S, Reynolds CA, Pedersen NL. Potentially modifiable risk factors for dementia in identical twins. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2006;2(2):110–7. doi: 10.1016/j.jalz.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Grabe HJ, Schwahn C, Volzke H, Spitzer C, Freyberger HJ, John U, Mundt T, Biffar R, Kocher T. Tooth loss and cognitive impairment. Journal of clinical periodontology. 2009;36(7):550–57. doi: 10.1111/j.1600-051X.2009.01426.x. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain pathology. 1998;8(1):65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffajee AD, Socransky SS. Introduction to microbial aspects of periodontal biofilm communities, development and treatment. Periodontol 2000. 2006;42:7–12. doi: 10.1111/j.1600-0757.2006.00190.x. [DOI] [PubMed] [Google Scholar]
- Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. Journal of periodontology. 2000;71(10):1554–60. doi: 10.1902/jop.2000.71.10.1554. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Holmes C, Butchart J. Systemic inflammation and Alzheimer’s disease. Biochemical Society transactions. 2011;39(4):898–901. doi: 10.1042/BST0390898. [DOI] [PubMed] [Google Scholar]
- Holmes C, Cotterell D. Role of infection in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs. 2009;23(12):993–1002. doi: 10.2165/11310910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain : a journal of neurology. 2009;132(Pt 5):1355–65. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson D, Spinell T, Vrettos A, Stocklin-Wasmer C, Celenti R, Demmer RT, Kebschull M, Papapanou PN. Circulating Endothelial Progenitor Cells in Periodontitis. Journal of periodontology. 2014:1–14. doi: 10.1902/jop.2014.140153. [DOI] [PubMed] [Google Scholar]
- Kamer AR, Craig RG, Dasanayake AP, Brys M, Glodzik-Sobanska L, de Leon MJ. Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2008a;4(4):242–50. doi: 10.1016/j.jalz.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Kamer AR, Dasanayake AP, Craig RG, Glodzik-Sobanska L, Bry M, de Leon MJ. Alzheimer’s disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. Journal of Alzheimer’s disease : JAD. 2008b;13(4):437–49. doi: 10.3233/jad-2008-13408. [DOI] [PubMed] [Google Scholar]
- Kamer AR, Morse DE, Holm-Pedersen P, Mortensen EL, Avlund K. Periodontal Inflammation in Relation to Cognitive Function in an Older Adult Danish Population. Journal of Alzheimer’s disease : JAD. 2012;28(3):613–24. doi: 10.3233/JAD-2011-102004. [DOI] [PubMed] [Google Scholar]
- Kaye EK, Valencia A, Baba N, Spiro A, 3rd, Dietrich T, Garcia RI. Tooth loss and periodontal disease predict poor cognitive function in older men. Journal of the American Geriatrics Society. 2010;58(4):713–18. doi: 10.1111/j.1532-5415.2010.02788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiddle SJ, Thambisetty M, Simmons A, Riddoch-Contreras J, Hye A, Westman E, Pike I, Ward M, Johnston C, Lupton MK, Lunnon K, Soininen H, Kloszewska I, Tsolaki M, Vellas B, Mecocci P, Lovestone S, Newhouse S, Dobson R Alzheimers Disease Neuroimaging I. Plasma based markers of [11C] PiB-PET brain amyloid burden. PloS one. 2012;7(9):e44260–e70. doi: 10.1371/journal.pone.0044260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. 2009;65(6):650–57. doi: 10.1002/ana.21696. [DOI] [PubMed] [Google Scholar]
- Landau SM, Marks SM, Mormino EC, Rabinovici GD, Oh H, O’Neil JP, Wilson RS, Jagust WJ. Association of lifetime cognitive engagement and low beta-amyloid deposition. Archives of neurology. 2012;69(5):623–29. doi: 10.1001/archneurol.2011.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, Kemppainen N, Nagren K, Kim BC, Tsui W, de Leon MJ. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008;35(12):2169–81. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Rogers J, McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. Journal of Alzheimer’s disease : JAD. 2006;9(3 Suppl):271–6. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- Miklossy J. Alzheimer’s disease--a spirochetosis? Neuroreport. 1993;4(9):1069. [PubMed] [Google Scholar]
- Miklossy J. Chronic inflammation and amyloidogenesis in Alzheimer’s disease -- role of Spirochetes. Journal of Alzheimer’s disease : JAD. 2008;13(4):381–91. doi: 10.3233/jad-2008-13404. [DOI] [PubMed] [Google Scholar]
- Miklossy J. Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation. 2011a;8(1):90–106. doi: 10.1186/1742-2094-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011b;13:e30–e64. doi: 10.1017/S1462399411002006. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Kasas S, Janzer RC, Ardizzoni F, Van der Loos H. Further ultrastructural evidence that spirochaetes may play a role in the aetiology of Alzheimer’s disease. Neuroreport. 1994;5(10):1201–4. doi: 10.1097/00001756-199406020-00010. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Khalili K, Gern L, Ericson RL, Darekar P, Bolle L, Hurlimann J, Paster BJ. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. Journal of Alzheimer’s disease : JAD. 2004;6(6):639–49. doi: 10.3233/jad-2004-6608. discussion 73–81. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Kis A, Radenovic A, Miller L, Forro L, Martins R, Reiss K, Darbinian N, Darekar P, Mihaly L, Khalili K. Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiology of aging. 2006;27(2):228–36. doi: 10.1016/j.neurobiolaging.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nature communications. 2013;4:1495. doi: 10.1038/ncomms2494. [DOI] [PubMed] [Google Scholar]
- Morgan D. Immunotherapy for Alzheimer’s disease. J Intern Med. 2011;269(1):54–63. doi: 10.1111/j.1365-2796.2010.02315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Andrews RD, Matthews DC. Comparing brain amyloid deposition, glucose metabolism, and atrophy in mild cognitive impairment with and without a family history of dementia. Journal of Alzheimer’s disease : JAD. 2013a;35(3):509–24. doi: 10.3233/JAD-121867. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Rinne JO, Tsui WH, Berti V, Li Y, Wang H, Murray J, Scheinin N, Nagren K, Williams S, Glodzik L, De Santi S, Vallabhajosula S, de Leon MJ. Increased fibrillar amyloid-{beta} burden in normal individuals with a family history of late-onset Alzheimer’s. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(13):5949–54. doi: 10.1073/pnas.0914141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Rinne JO, Tsui WH, Murray J, Li Y, Glodzik L, McHugh P, Williams S, Cummings M, Pirraglia E, Goldsmith SJ, Vallabhajosula S, Scheinin N, Viljanen T, Nagren K, de Leon MJ. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiology of aging. 2013b;34(1):22–34. doi: 10.1016/j.neurobiolaging.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Tsui W, Murray J, McHugh P, Li Y, Williams S, Pirraglia E, Glodzik L, De Santi S, Vallabhajosula S, de Leon MJ. Maternal age affects brain metabolism in adult children of mothers affected by Alzheimer’s disease. Neurobiology of aging. 2012;33(3):624.e1–9. doi: 10.1016/j.neurobiolaging.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, Li Y, Boppana M, de Leon MJ. Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis. Neurology. 2005;64(11):1860–7. doi: 10.1212/01.WNL.0000163856.13524.08. [DOI] [PubMed] [Google Scholar]
- Muller-Gartner HW, Links JM, Prince JL, Bryan RN, McVeigh E, Leal JP, Davatzikos C, Frost JJ. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1992;12(4):571–83. doi: 10.1038/jcbfm.1992.81. [DOI] [PubMed] [Google Scholar]
- National Cholesterol Education Program Expert Panel on Detection E, Treatment of High Blood Cholesterol in A. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–421. [PubMed] [Google Scholar]
- Noble JM, Borrell LN, Papapanou PN, Elkind MS, Scarmeas N, Wright CB. Periodontitis is associated with cognitive impairment among older adults: analysis of NHANES-III. Journal of neurology, neurosurgery, and psychiatry. 2009;80(11):1206–11. doi: 10.1136/jnnp.2009.174029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K, Ishihara K, Nakagawa T, Hirayama A, Inayama Y, Okuda K. Detection of Treponema denticola in atherosclerotic lesions. J Clin Microbiol. 2001;39(3):1114–17. doi: 10.1128/JCM.39.3.1114-1117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio RS, Pirraglia E, Gumb T, Mantua J, Ayappa I, Williams S, Mosconi L, Glodzik L, de Leon MJ. Imaging and cerebrospinal fluid biomarkers in the search for Alzheimer’s disease mechanisms. Neuro-degenerative diseases. 2014;13(2–3):163–5. doi: 10.1159/000355063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozudogru SN, Lippa CF. Disease modifying drugs targeting beta-amyloid. Am J Alzheimers Dis Other Demen. 2012;27(5):296–300. doi: 10.1177/1533317512452034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RC, Eke PI. Case definitions for use in population-based surveillance of periodontitis. Journal of periodontology. 2007;78(7 Suppl):1387–99. doi: 10.1902/jop.2007.060264. [DOI] [PubMed] [Google Scholar]
- Paraskevas S, Huizinga JD, Loos BG. A systematic review and meta-analyses on C-reactive protein in relation to periodontitis. Journal of clinical periodontology. 2008;35(4):277–90. doi: 10.1111/j.1600-051X.2007.01173.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4(2):103–12. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. Journal of Alzheimer’s disease : JAD. 2013;36(4):665–77. doi: 10.3233/JAD-121918. [DOI] [PubMed] [Google Scholar]
- Reichert S, Haffner M, Keysser G, Schafer C, Stein JM, Schaller HG, Wienke A, Strauss H, Heide S, Schulz S. Detection of oral bacterial DNA in synovial fluid. Journal of clinical periodontology. 2013;40(6):591–98. doi: 10.1111/jcpe.12102. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Ferris SH, de Leon MJ, Crook T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry. 1982;139(9):1136–9. doi: 10.1176/ajp.139.9.1136. [DOI] [PubMed] [Google Scholar]
- Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nature reviews Neurology. 2011;7(3):137–52. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain, behavior, and immunity. 2003;17(1):13–9. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol. 2002;17(2):113–8. doi: 10.1046/j.0902-0055.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- Ship JA. Oral health of patients with Alzheimer’s disease. J Am Dent Assoc. 1992;123(1):53–8. doi: 10.14219/jada.archive.1992.0005. [DOI] [PubMed] [Google Scholar]
- Silness J, Loe H. Periodontal Disease in Pregnancy. Ii. Correlation between Oral Hygiene and Periodontal Condtion. Acta Odontol Scand. 1964;22:121–35. doi: 10.3109/00016356408993968. [DOI] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD. The nature of periodontal diseases. Ann Periodontol. 1997;2(1):3–10. doi: 10.1902/annals.1997.2.1.3. [DOI] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD. Dental biofilms: difficult therapeutic targets. Periodontol 2000. 2002;28:12–55. doi: 10.1034/j.1600-0757.2002.280102.x. [DOI] [PubMed] [Google Scholar]
- Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PloS one. 2010;5(3):e9505–e15. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks Stein P, Steffen MJ, Smith C, Jicha G, Ebersole JL, Abner E, Dawson D., 3rd Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2012;8(3):196–203. doi: 10.1016/j.jalz.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein PS, Desrosiers M, Donegan SJ, Yepes JF, Kryscio RJ. Tooth loss, dementia and neuropathology in the Nun Study. J Am Dent Assoc. 2007;138(10):1314–22. doi: 10.14219/jada.archive.2007.0046. [DOI] [PubMed] [Google Scholar]
- Stewart R, Hirani V. Dental health and cognitive impairment in an English national survey population. Journal of the American Geriatrics Society. 2007;55(9):1410–4. doi: 10.1111/j.1532-5415.2007.01298.x. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012;2(10):1–10. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonetti MS, Claffey N European Workshop in Periodontology group C. Advances in the progression of periodontitis and proposal of definitions of a periodontitis case and disease progression for use in risk factor research. Group C consensus report of the 5th European Workshop in Periodontology. Journal of clinical periodontology. 2005;32(Suppl 6):210–3. doi: 10.1111/j.1600-051X.2005.00822.x. [DOI] [PubMed] [Google Scholar]
- Weintraub MK, Bisson CM, Nouri JN, Vinson BT, Eimerbrink MJ, Kranjac D, Boehm GW, Chumley MJ. Imatinib methanesulfonate reduces hippocampal amyloid-beta and restores cognitive function following repeated endotoxin exposure. Brain, behavior, and immunity. 2013;33:24–8. doi: 10.1016/j.bbi.2013.05.002. [DOI] [PubMed] [Google Scholar]
- White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, Hartshorn KL. Alzheimer’s associated beta-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PloS one. 2014;9(7):e101364. doi: 10.1371/journal.pone.0101364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YH, Kuo HK. Association between cognitive function and periodontal disease in older adults. Journal of the American Geriatrics Society. 2008;56(9):1693–7. doi: 10.1111/j.1532-5415.2008.01847.x. [DOI] [PubMed] [Google Scholar]