

Abstract
Synthetic nanoparticles coated with cellular membranes have been increasingly explored to harness natural cell functions toward the development of novel therapeutic strategies. Herein, we report on a unique bacterial membrane-coated nanoparticle system as a new and exciting antibacterial vaccine. Using Escherichia coli as a model pathogen, we collect bacterial outer membrane vesicles (OMVs) and successfully coat them onto small gold nanoparticles (AuNPs) with a diameter of 30 nm. The resulting bacterial membrane-coated AuNPs (BM-AuNPs) show markedly enhanced stability in biological buffer solutions. When injected subcutaneously, the BM-AuNPs induce rapid activation and maturation of dendritic cells in the lymph nodes of the vaccinated mice. In addition, vaccination with BM-AuNPs generates antibody responses that are durable and of higher avidity than those elicited by OMVs only. The BM-AuNPs also induce an elevated production of interferon gamma (INFγ) and interleukin-17 (IL-17), but not interleukin-4 (IL-4), indicating its capability of generating strong Th1 and Th17 biased cell responses against the source bacteria. These observed results demonstrate that using natural bacterial membranes to coat synthetic nanoparticles holds great promise for designing effective antibacterial vaccines.
Keywords: Nanomedicine, biomimetic nanoparticle, membrane coating, infectious disease, bacterial vaccine
Introduction
Despite the remarkable success in controlling former epidemics worldwide, vaccines effective against a number of serious infections, including those caused by pathogenic Escherichia coli (E. coli), Helicobacter pylori, and Staphylococcus aureus, remain largely unavailable.1, 2 Meanwhile, existing antibiotic regimens are increasingly threatened by the rapid emergence of bacterial drug resistance.3 Together, these challenges have motivated the search for novel antibacterial vaccine strategies.4–6 Among various reported approaches, integrating synthetic nanoparticles with cues from natural immunity has shown tremendous promise.7, 8 A plethora of nanoparticle-based vaccine systems have been developed to better manipulate immune responses and to potentially enhance antimicrobial immunity.9
On the front of nanotechnology development, combining synthetic nanoparticles with natural cellular materials has led to the creation of various biomimetic nanoparticles.10, 11 In particular, using natural cellular membranes to cloak synthetic nanoparticles through a top-down fabrication method has recently attracted much attention.12 The resulting cell membrane-coated nanoparticles preserve the highly tunable physicochemical properties of synthetic nanomaterials while harnessing complex cellular functions that are otherwise difficult to replicate. Based upon this new strategy, a variety of nanoparticle platforms mimicking natural cellular functions have been developed, including red blood cell (RBC) membrane-cloaked nanoparticles with long-circulating properties,13 leukocyte membrane-coated silica microparticles capable of traversing endothelium,14 and cancer cell membrane-coated nanoparticles with inherited homotypic cell binding as well as tumor-specific immune activation.15 Cell membrane-coated nanoparticles have also enabled novel therapeutics beyond traditional practices. For example, by exploiting particle-bound RBC membranes, these biomimetic nanoparticles can function as a toxin nanosponge to absorb and neutralize a broad spectrum of pore-forming toxins regardless of the toxins’ molecular structure.16, 17 These toxin-sequestered nanoparticles can further present the undisrupted toxins to the immune system as a safe and effective toxin vaccine.16, 17
Inspired by these technological advances, this work aims to utilize bacterial membranes to coat nanoparticles and investigate their potential as an antibacterial vaccine. Bacterial membranes are appealing vaccination materials as they contain a large number of immunogenic antigens with intrinsic adjuvant properties18 and exhibit various pathogen associated-molecular patterns that play a key role in stimulating innate immunity and promoting adaptive immune responses.19, 20 Coating bacterial membranes onto the surfaces of synthetic nanoparticles will preserve these complex biological characteristics of bacteria and mimic the natural antigen presentation by bacteria to the immune system. Meanwhile, the synthetic nanoparticle cores provide a wide range of tunable physicochemical properties such as particle size and shape for effective antigen presentation to the immune cells.21, 22 Therefore, the bacterial membrane coated nanoparticles marry the merits of two distinct materials and are expected to generate strong antibacterial immune responses.
Herein, we chose E. coli bacteria as a model pathogen and harnessed their outer membranes through the collection of their secreted outer membrane vesicles (OMVs). Originating from bacterial outer membranes, OMVs share a great similarity in biochemical profiles with their parent cells.19, 20 They are known to generate potent protective immune responses against the source pathogens, with particular success in treating Neisseria meningitides.23, 24 However, to treat a wider range of infections, novel strategies to manipulate OMVs and modulate the subsequent immune responses are highly desirable. As illustrated in Figure 1, the collected bacterial OMVs were coated onto the surfaces of small gold nanoparticles (AuNPs). Note that the AuNPs were chosen because their size and shape could be precisely tailored to favor immunization applications.25, 26 In the study, we demonstrated successful coating of bacterial membranes onto small AuNPs with a diameter of 30 nm. When subcutaneously injected into mice, the bacterial membrane-coated AuNPs (BM-AuNPs) traveled to adjacent draining lymph nodes and rapidly activated dendritic cells (DCs) residing in the lymph nodes. We further showed that the BM-AuNPs effectively elicited bacterium-specific B-cell and T-cell responses in the vaccinated animals. Collectively, these results indicate that coating natural bacterial membranes onto synthetic nanoparticles represent a promising approach to developing an antibacterial vaccine toward effective treatment of bacterial infection.
Figure 1.

A schematic illustration of modulating antibacterial immunity via bacterial membrane-coated nanoparticles. Briefly, bacterial membrane was first collected from source bacteria in the form of secreted outer membrane vesicles (OMVs) and then mixed with citrate-stabilized gold nanoparticles (AuNPs) with a diameter of approximately 30 nm. Through an extrusion process, OMVs were fused with AuNPs, translocating the entire bacterial membrane onto gold surfaces to form bacterial membrane-coated AuNPs (BM-AuNPs). When injected subcutaneously into mice, BM-AuNPs elicited bacterium-specific immunity against the source bacteria.
Results and Discussion
The process of functionalizing AuNPs with bacterial membranes consists of two steps: collecting bacterial OMVs and fusing the OMVs onto the surfaces of AuNPs. In the study, E. coli OMVs were collected and purified by following established protocols.19, 27 Dynamic light scattering (DLS) measurements showed that the collected vesicles had heterogeneous size distribution with diameters ranging from 30 to 300 nm (Figure 2a). For membrane fusion, 30 nm citrate-stabilized AuNPs were mixed with OMVs and the mixture was extruded through a 50 nm porous polycarbonate membrane to generate BM-AuNPs. The mechanical force facilitated the fusion of OMVs with AuNPs. Owing to the high density of gold, following the extrusion, the excess OMVs and soluble compounds were removed by repeated low speed centrifugation.
Figure 2.

Physicochemical characterization of BM-AuNPs. (a) Hydrodynamic sizes (diameter, nm) and (b) surface zeta potentials (mV) of AuNPs before coating and after coating, in comparison with those of OMVs. (c) A representative TEM image showing the spherical core–shell structure of the BM-AuNPs negatively stained with uranyl acetate (scale bar, 50 nm). Inset: a zoomed-in view of a single BM-AuNP (scale bar, 10 nm). (d) Quantification of protein concentration of AuNPs before and after the membrane coating by using a BCA assay. Protein loading yield is defined as the weight ratio of immobilized proteins to the gold nanoparticles.
DLS measurements showed that the diameter of AuNPs increased from 30.3 ± 0.2 nm to 41.9 ± 0.5 nm upon bacterial membrane coating (Figure 2a). This size increase is consistent with the addition of an approximately 6 nm thick lipid membrane, confirming the membrane coating onto the exterior surface of AuNPs.28 Meanwhile, zeta potential measurements also indicated a successful membrane coating, as the value changed from −38.6 ± 1.3 mV of bare AuNPs to −25.1 ± 0.9 mV following the coating, comparable to the value measured from the OMVs (Figure 2b). Next, the membrane coating was confirmed by examining the morphology of BM-AuNPs with transmission electron microscopy (TEM). Under the microscope, all nanoparticles showed a clear spherical core–shell structure, reflecting the enclosure of gold cores in a thin shell with a thickness of approximately 6 nm (Figure 2c). The membrane coating was further verified with a protein bicinchoninic acid (BCA) assay. While the bare AuNPs showed the absence of detectable protein content, tests on BM-AuNPs showed a significant increase in absorbance at 562 nm, implying the presence of protein content on the nanoparticles. Further quantification indicated a protein loading yield, defined as the weight ratio of immobilized proteins to the gold nanoparticles, of approximately 7.9±2.0 wt% (Figure 2d).
Membrane coating drastically improved the nanoparticle buffer stability. When bare AuNPs were transferred from 2 mM citrate storage buffer into 1X PBS, the characteristic cherry red color of the AuNPs faded immediately, suggesting a rapid destabilization and aggregation of AuNPs, likely due to the increase of buffer ionic strength (Figure 3a). On the contrary, when BM-AuNPs were transferred from 2 mM citrate into 1X PBS, the cherry red color remained unchanged, indicating the preservation of particle stability due to the membrane coating. Similar phenomena were also observed by using 100% fetal bovine serum instead of 1X PBS. Notably, vesicles generated from extruding OMVs without using AuNP cores was highly unstable, as their size increased rapidly from 50 nm to above 100 nm within 4 h. In contrast, size variation of BM-AuNPs was negligible, further confirming a higher stability conferred by membrane coating (Figure S1). Collectively, these results demonstrated the mutual benefits between the OMVs and the AuNPs. That is, the membrane coating effectively enhanced AuNP stability in biological buffers, while the AuNP cores template the OMVs into uniformly sized nanoparticles and also ensure adequate nanoparticle stability for downstream in vivo applications.
Figure 3.
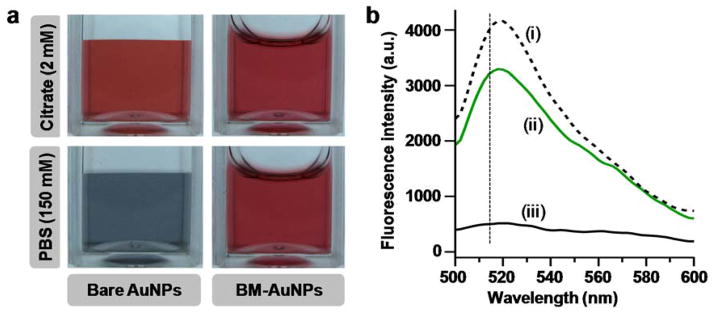
Stability and membrane-core affinity of BM-AuNP. (a) Both bare AuNPs and BM-AuNPs are stable in 2 mM citrate storage buffer. However, in 1X PBS, bare AuNPs rapidly aggregate while BM-AuNPs remain stable. (b) A fluorescence quenching assay shows the strong affinity between the coated bacterial membrane and the gold cores. Fluorescence spectra were taken from i) FITC-thiol alone, ii) FITC-thiol mixed with BM-AuNPs, and iii) FITC-thiol mixed with bare AuNPs. The concentrations of FITC-thiol and AuNP are 1 nM and 0.025 nM (equivalent to 50 μg mL−1), respectively.
In addition to buffer stability, we also examined the affinity of the coated membrane to the gold cores by using a fluorescein isothiocyanate (FITC)–thiol conjugate, which is a fluorescent probe that can be quenched when bound to the exposed surface of bare AuNPs.28 The fluorescence spectrum of the FITC–thiol conjugate in solution showed a peak at 520 nm, characteristic of FITC’s emission (Figure 3b). Upon mixing with bare AuNPs, the fluorescence was significantly quenched, indicating the probe’s close association with surfaces of the nanoparticles. In contrast, when BM-AuNPs were mixed with the conjugates under the same conditions, the quenching of FITC emission was largely absent, indicating that the highly reactive thiol groups were unable to replace the membrane coating. A slight decrease in fluorescence intensity observed upon the mixing of FITC-thiol and BM-AuNPs was attributable to the long-range fluorescence quenching effect of AuNPs. In addition, the sample showed no further decrease in fluorescence over a span of 72 h. Together, these results demonstrated a strong association of the membrane to the AuNPs, which effectively shielded the gold surfaces.
After having confirmed the coating of bacterial membranes onto AuNPs, we proceeded to evaluate the vaccine potential of BM-AuNPs. To this end, we first examined their efficiency in activating DCs in vivo. Nanoparticles were subcutaneously injected at the tail base of each mouse and then the upregulation of activation markers by DCs in the draining lymph nodes was examined.29 In the study, we divided the mice into three groups and injected them with BM-AuNPs, OMVs, and PBS, respectively. Herein, we chose natural OMVs as a control group as opposed to vesicles made by extruding OMVs because the latter had poor stability in PBS (Figure S1). Twelve hours after the injection, cells in the lumbar and sacral lymph nodes were collected and CD11c+ DC frequencies were counted. In the mice injected with BM-AuNPs and OMVs, DCs isolated from the lymph nodes displayed considerably higher frequencies of CD11c+ DCs than the mice injected with PBS, suggesting the elevated recruitment of DCs into these lymph nodes (Figure 4a). Between these two groups, a higher frequency was observed in the group injected with BM-AuNPs than the OMV-injected group (p = 0.0096), suggesting a higher efficacy of BM-AuNPs in boosting CD recruitment. We further compared the efficacy of BM-AuNPs and OMVs in eliciting DC maturation. In both groups, CD11c+ DCs isolated from lumbar and sacral lymph nodes displayed a shift from the immature to the mature phenotype through upregulation of the co-stimulatory molecules CD40, CD80, and CD86 (Figure 4b). Further quantification of the pencentage of CD11c+ DCs based on the histograms showed that the DC maturation level induced by BM-AuNPs was significantly higher than that induced by the OMVs alone (Figure 4c). It has been reported that the transport rate of macromolecles and nanoparticles injected through the mouse tail base to the lumbar and sacral lymph nodes as well as subsequent DC activation efficacy are inversely correlated to size.29, 30 Indeed, BM-AuNPs made with larger AuNPs (90 nm in diameter, Figure S2) showed a significantly reduced accumulation in the lymph nodes of mice as compared to smaller BM-AuNPs made with 30 nm AuNPs (Figure S3). Hence, the potent activation of DCs by BM-AuNPs indicates not only proper bacterial membrane coating on the particles but also highlights the advantage of using small sized nanoparticles for immune activation.
Figure 4.

BM-AuNPs activating DCs in the draining lymph nodes in vivo. (a) Percentage CD11c+ cells in a tatol of 20,000 lymphocytes from naive mice or mice vaccinated with either OMVs or BM-AuNPs (2.5 μg gold) 12 h after the subcutaneous injection. Data are shown with mean frequencies ± SEM (standard error of the mean) measured per lymph node (*p < 0.05). (b) A representative flow cytometry analysis of surface maturation markers (CD40, CD80, and CD86) on CD11c+ DCs from lumbar and sacral lymph nodes of the mice (n = 5). (c) Quantification of the pencentage of CD11c+ DCs based on the histograms in (b). Data are shown with mean percentage ± SEM (*p < 0.05, **p < 0.01). Three independent experiments were carried out.
We next assessed bacterium-specific B cell responses by examining the elicitation of E. coli-specific antibodies. In the study, we adopted a vaccination schedule that included a prime on day 0 and two booster vaccinations on day 7 and day 14, respectively.17 Naive mice and mice vaccinated with OMVs served as two control groups. To examine whether the response was dose-dependent, we administered the mice with either 0.2 or 0.02 μg antigen per injection. During the immunization process, sera were obtained from mice of all groups and E. coli-binding IgG titers were measured. Determination of antibody responses on day 21 showed that BM-AuNPs induced significantly higher E. coli-specific antibody titers in comparison to the OMVs (p = 0.0016 at 0.2 μg antigen/dose, p = 0.0148 at 0.02 μg antigen/dose, Figure 5a). Such enhancement in antibody response by BM-AuNPs was also observed in a time-course study. In the first 21 days of the immunization, a continuous rise in E. coli-binding IgG was detected in both BM-AuNP- and OMV-immunized mice, but not in naïve mice (Figure 5b). Afterward, the IgG titer levels remained stable in all groups throughout the study, where enhancements by approximately 3- and 5-fold (geometric means) were measured for the BM-AuNP groups as compared to the OMV groups at dosages of 0.2 and 0.02 μg antigen (n = 7), respectively. In addition to the titer levels, we also examined the quality of the antibody response raised against the source bacteria by assessing the avidity of sera from the mice. Compared to OMV vaccinated mice, antibody from BM-AuNP vaccinated mice showed a significantly higher avidity index to the E. coli bacteria (p = 0.027, n = 7, Figure 5c). Overall, vaccination with BM-AuNPs generates antibody responses that are durable and of higher avidity than those elicited by OMVs only, indicating the potential of BM-AuNPs for enhancing antibacterial immunity.
Figure 5.
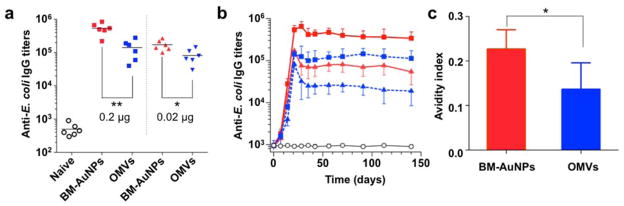
BM-AuNPs eliciting strong bacterium-specific antibody responses in vivo. (a) Anti-E. coli IgG titres at day 21 (n = 6). Black lines indicate geometric means. Naive mice were monitored as a negative control (open circles). (b) Time course of anti-E. coli IgG titres in naive mice (open circles) and mice immunized with 0.2 μg antigen/dose BM-AuNPs (red squares), 0.2 μg antigen/dose OMVs (blue squares), 0.02 μg antigen/dose BM-AuNPs (red triangles), and 0.02 μg antigen/dose OMVs (blue triangles), respectively (n = 6). (c) Quantified avidity index of the anti-sera from immunized mice (0.2 μg antigen/dose) binding to E. coli bacteria (n = 6). *p < 0.05, **p < 0.01.
Finally, we examined the effects of BM-AuNP immunization on activating bacterium-specific T cell responses. The role of T cell-mediated immunity has become increasingly recognized in generating effective protection against a variety of infections. In the study, mice were immunized with 0.2 μg antigen/dose of BM-AuNPs or OMVs, following the aforementioned schedules. We collected the splenic cells on day 21 and stimulated them with inactivated E. coli bacteria for 72 h. T cell activation was quantified by measuring levels of three cytokines in the cell culture: interferon gamma (IFNγ), interleukin 17 (IL-17), and interleukin 4 (IL-4). The results showed that the levels of IFN-γ and IL-17 production were higher in mice immunized with either BM-AuNPs or OMVs compared to the naïve mice, indicating successful E. coli-specific T cell activation (Figure 6a, b). The comparison also showed that mice immunized with BM-AuNPs produced significantly higher levels of IFN-γ (p = 0.0468, n = 10) and IL-17 (p = 0.0397, n = 10) than OMV-immunized mice, implying a higher efficacy of BM-AuNPs in activating T cells. Meanwhile, production of IL-4, the key cytokine of Th2 cells, was undetectable in all groups (Figure 6c). The elevated production of IFN-γ and IL-17 but not IL-4 demonstrated strong Th1 and Th17 biased cell responses against the bacterial infection.
Figure 6.
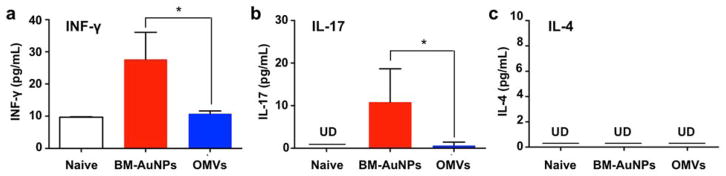
BM-AuNPs inducing pronounced bacterium-specific T cell activation in vivo. The mice (n = 10) were immunized with 0.2 μg antigen/dose BM-AuNPs or OMVs. Naive mice were tested in paralell as a negative control. On day 21, splenic cells were collected and stimulated with E. coli bacteria. After 72 h of co-culturing with the bacteria, the levels of (a) IFN-γ, (b) IL-17, and (c) IL-4 in the medium were quantified using an ELISA. *p < 0.05. Bars represent means ± SD (n=10). UD = undetectable by ELISA.
The design of BM-AuNPs reported in this study seeks synergy between bacterial membranes and synthetic nanoparticles to generate a unique and robust antibacterial vaccine. Using bacterial OMVs as membrane materials, BM-AuNPs contain a large number of immunogenic antigens with intrinsic adjuvant properties. In addition, the faithful translocation of the entire bacterial membranes via a top-down approach onto the nanoparticle surfaces preserves critical immune determinants such as the pathogen associated-molecular patterns. As a result, the BM-AuNPs closely mimic antigen presentation by bacteria to the immune cells. On the other hand, using AuNPs as coating template allows a range of nanoparticle physicochemical properties to be precisely tailored for desired immune responses. In this study, the AuNP-templated membrane coating transformed OMVs from widely polydispersed vesicles into uniformly distributed ultra small nanoparticles and subsequently resulted in rapid DC activation in vivo. Moreover, the AuNP-templated coating also led to strong association between the membrane and the core, which likely reinforced the multivalent display of epitopes on the bacterial membranes. Such strengthened interactions between the antigens and the nanoparticle architecture have been attributed to the enhanced antibody and endogenous T cell responses in other nanoparticle-based vaccine platforms.17, 31, 32 Taken together, the bacterial membranes and the AuNP cores mutually benefit each other, synergistically generating enhanced antibacterial immune responses.
Using bacterial membranes to coat synthetic nanoparticles opens many opportunities for designing effective antibacterial vaccines. For example, secretion of OMVs seems like a conserved process not only found on Gram-negative but also an increasing number of Gram-positive bacteria; therefore, the use of OMVs as membrane materials for nanoparticle coating is potentially applicable to a wide range of bacteria.33, 34 In addition, genetic engineering has been successful in developing OMVs that express multiple mutants or exogenous antigens.35, 36 These engineered OMVs can be also exploited as membrane materials for broadening immune protection. Meanwhile, the cellular membrane-coated nanoparticle, since its first development, has made significant progress. Particularly, a wide range of synthetic cores have been successfully coated, including those made of polymers,12, 16, 17, 37 silica,14 and gold.28, 38 These synthetic nanoparticles, already with intensive applications in drug delivery, offer exciting opportunities to manipulate nanoparticle characteristics for even broader biomedical applications. Together, these technological advances will benefit the development of bacterial membrane-coated nanoparticles with numerous implications toward effective and safe antibacterial vaccines.
Conclusions
We developed a new and robust antibacterial vaccine strategy by coating natural bacterial membranes collected from bacterium-secreted OMVs onto small AuNPs. The resulting BM-AuNPs showed significantly enhanced stability in biological buffer solutions, with the coated membrane strongly associated with the AuNP cores. When injected subcutaneously, BM-AuNPs induced rapid activation and maturation of DCs in lymph nodes. Furthermore, vaccination with BM-AuNPs generated strong and durable antibody responses with higher avidity than those elicited by OMVs only. Compared to OMVs, BM-AuNPs also induced an elevated production of IFN-γ and IL-17 but not IL-4, indicating their ability to generate Th1 and Th17 biased cell responses against the source bacteria. Taken together, the bacterial membranes and the AuNP cores mutually benefit each other, generating a synergy for enhanced immune responses. Overall, using bacterial membranes to coat synthetic nanoparticles holds a significant potential for designing effective antibacterial vaccines.
Materials and Methods
Bacterial culture and collection of bacterial outer membrane vesicles (OMVs)
Escherichia coli (E. coli) bacteria (strain DH5α) were cultured on Luria broth (LB) agar overnight at 37°C. Then a single colony was inoculated into LB medium. Following the inoculation, the medium was cultured in a rotary shaker at 37°C for 10 h and then refreshed with LB medium at a 1:100 dilution. The culture continued for another 3 h until the OD600 value of the medium reached approximately 1.0, indicating the logarithmic growth phase. E. coli OMVs were collected by following published protocols19, 27. Briefly, 250 mL bacterial culture was centrifuged at 4000 ×g for 10 min to remove the bacteria, followed by filtering through a 0.45-μm vacuum filter. The filtrate was then concentrated by using Amicon centrifugal filters with a molecular weight cutoff of 100 kDa (Millipore). The concentrated medium was then centrifuged with an SW 41 Ti rotor (Beckman Instruments) at 150,000 ×g for 2 h at 4°C. The OMV pellet was resuspended in water and stored at −80°C for further experiments.
BM-AuNP preparation and characterization
Citrate-stabilized gold nanoparticles (bare AuNPs) with a diameter of approximately 30 nm and a concentration of 50 μg mL−1 (nanoComposix, Inc) were used for all studies. To fuse with the OMVs, AuNPs were mixed with OMVs and then extruded 7 times through a 50-nm polycarbonate porous membrane with an Avanti mini extruder. Given the high density of gold, following the extrusion, we removed the excess OMVs and soluble compounds via repeated low speed centrifugation (approximately 9000 ×g). Particle size (diameter, nm), polydispersity, and surface charge (zeta potential, mV) of the resulting BM-AuNPs were measured by dynamic light scattering (DLS) on a Zetasizer Nano ZS (model ZEN3600 from Malvern Instruments). A FITC-thiol conjugate was synthesized and used to test the stability and the shielding effect of the bacterial membrane coating as previously described.28 In the study, the FITC-thiol was added into BM-AuNPs and bare AuNPs (without bacterial membrane coating), separately, and then the fluorescence intensity at 520 nm was measured by using a Tecan Infinite M200 microplate reader. Bacterial membrane coating was further examined by using transmission electron microscopy (TEM). Briefly, 1 mL of BM-AuNPs (50 μg mL−1) was carefully centrifuged to concentrate into a final volume of about 10 μL. Then 3 μL of concentrated particle suspension was deposited onto a glow-discharged carbon-coated copper grid. Five minutes after the sample was deposited, the grid was rinsed with 10 drops of distilled water, followed by staining with a drop of 1% uranyl acetate. The grid was subsequently dried and visualized using an FEI 200 kV Sphera microscope. Total protein content on the nanoparticles was determined by using a BCA kit (Thermo Scientific Pierce) to measure the absorbance at 562nm in comparison with bovine serum albumin protein standard.
Animal care and injections
6-week-old male CD-1 mice (Charles River Laboratories) were housed in the Animal Facility at the University of California San Diego under federal, state, local, and National Institutes of Health guidelines for animal care. In the study, BM-AuNPs, OMVs, and PBS were injected as a bolus of 50 μL into the base of the mouse tail through a 30-gauge needle29. No inflammation was observed at the sites of injection.
Flow cytometry analysis of dendritic cell (DC) activation
DC activation was tested by removing lumbar and sacral lymph nodes 24 h after injection of BM-AuNPs, OMVs, or PBS. Single cell suspensions were prepared by teasing the lymph nodes with 26-gauge needles and digesting in Collagenase D (Roche Applied Science) for 20 min at 37°C. The tissue suspension was passed through a 40-μm membrane (BD Biosciences) to produce a single cell suspension. Cells were then washed, blocked, and stained with antibodies against CD11c, CD40, CD80, and CD86. Flow cytometry analysis was performed on a BD Biosciences FACS Calibur HTS. To analyze frequency, a total of 20,000 cells were counted.
Immunization studies
Groups of mice (n = 6) were immunized subcutaneously at the tail base with the indicated doses of BM-AuNPs, OMVs, or PBS on day 0, 7 and 14, respectively. At pre-determined time points, sera were collected and quantified for anti-E. coli IgG antibodies by ELISA on E. coli bacterium-coated plates prepared by following a published protocol.17 Titers were defined as the lowest serum dilution at which the ELISA OD reading was ≥ 0.5. Avidity measurements were performed by incubating plates with 6 M urea for 15 min at 20°C to remove weakly bound IgG prior to addition of detection antibodies.17 The avidity index was defined as the ratio of IgG titer with 6 M urea to IgG titer without urea. Statistical analysis was performed with GraphPad Prism using an unpaired two-tailed t-test.
T cell response studies
Groups of mice (n=10) were immunized subcutaneously at the tail base with the indicated dosages of BM-AuNPs, OMVs, or PBS on day 0, 7 and 14, respectively. On day 21, the mice were sacrificed and their spleens were collected. To prepare single cell splenocyte suspensions, the collected spleens were minced and digested by Collagenase D. Then the tissue suspension was passed through a 40-μm cell strainer and red blood cells were removed by selected lysis. The collected cells were washed and resuspended with PBS. The cells in each suspension were then counted and seeded onto 12-well plates with a density of 2 × 105 cells/well. To each well, 1 × 107 CFU of E. coli bacteria, killed by formalin fixation and thoroughly washed with PBS, were added. The cells were incubated for 72 h at 37°C and 5% CO2. Following the incubation, supernatants from each well were collected and the concentrations of cytokines including IFNγ, IL-4, and IL-17 were quantified using ELISA Ready-SET-Go assays (eBiosciences). Statistical analysis was performed with GraphPad Prism using an unpaired two-tailed t-test.
Supplementary Material
Acknowledgments
This work is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK095168 and by the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under Grant Number HDTRA1-14-1-0064.
Footnotes
Author Contributions
W.G., R.F., S.T. and L.Z. conceived and designed the experiments; W.G., R.F., S.T., B.L., J.L., P.A., Q.Z., and C-M.H. performed all the experiments. W.G., R.F., S.T., and L.Z. analyzed and discussed the data and wrote the paper. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interests.
Supporting Information. Supplementary Figures S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mendoza N, Ravanfar P, Satyaprakah A, Pillai S, Creed R. Dermatol Ther. 2009;22:129–142. doi: 10.1111/j.1529-8019.2009.01225.x. [DOI] [PubMed] [Google Scholar]
- 2.Germain RN. Immunity. 2010;33:441–450. doi: 10.1016/j.immuni.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 3.Spellberg B, Bartlett JG, Gilbert DN. New Engl J Med. 2013;368:299–302. doi: 10.1056/NEJMp1215093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufmann SHE. Nat Rev Microbiol. 2007;5:491–504. doi: 10.1038/nrmicro1688. [DOI] [PubMed] [Google Scholar]
- 5.Rappuoli R, Aderem A. Nature. 2011;473:463–469. doi: 10.1038/nature10124. [DOI] [PubMed] [Google Scholar]
- 6.Koff WC, Burton DR, Johnson PR, Walker BD, King CR, Nabel GJ, Ahmed R, Bhan MK, Plotkin SA. Science. 2013;340:article 1232910. doi: 10.1126/science.1232910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swartz MA, Hirosue S, Hubbell JA. Sci Transl Med. 2012;4:article 148rv9. doi: 10.1126/scitranslmed.3003763. [DOI] [PubMed] [Google Scholar]
- 8.Irvine DJ, Swartz MA, Szeto GL. Nat Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leleux J, Roy K. Adv Healthcare Mater. 2013;2:72–94. doi: 10.1002/adhm.201200268. [DOI] [PubMed] [Google Scholar]
- 10.Balmert SC, Little SR. Adv Mater. 2012;24:3757–3778. doi: 10.1002/adma.201200224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gong Y-k, Winnik FM. Nanoscale. 2012;4:360–368. doi: 10.1039/c1nr11297j. [DOI] [PubMed] [Google Scholar]
- 12.Hu CMJ, Fang RH, Zhang L. Adv Healthcare Mater. 2012;1:537–547. doi: 10.1002/adhm.201200138. [DOI] [PubMed] [Google Scholar]
- 13.Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proc Natl Acad Sci U S A. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parodi A, et al. Nat Nanotechnol. 2013;8:61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang RH, Hu CMJ, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L. Nano Lett. 2014;14:2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu CMJ, Fang RH, Copp J, Luk BT, Zhang L. Nat Nanotechnol. 2013;8:336–340. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CMJ, Fang RH, Luk BT, Zhang L. Nat Nanotechnol. 2013;8:933–938. doi: 10.1038/nnano.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poetsch A, Wdlters D. Proteomics. 2008;8:4100–4122. doi: 10.1002/pmic.200800273. [DOI] [PubMed] [Google Scholar]
- 19.Lee EY, et al. Proteomics. 2007;7:3143–3153. doi: 10.1002/pmic.200700196. [DOI] [PubMed] [Google Scholar]
- 20.Kuehn MJ, Kesty NC. Genes Dev. 2005;19:2645–2655. doi: 10.1101/gad.1299905. [DOI] [PubMed] [Google Scholar]
- 21.Hu CMJ, Fang RH, Luk BT, Chen KNH, Carpenter C, Gao W, Zhang K, Zhang L. Nanoscale. 2013;5:2664–2668. doi: 10.1039/c3nr00015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luk BT, Hu CMJ, Fang RH, Dehaini D, Carpenter C, Gao W, Zhang L. Nanoscale. 2014;6:2730–2737. doi: 10.1039/c3nr06371b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Unal CM, Schaar V, Riesbeck K. Semin Immunopathol. 2011;33:395–408. doi: 10.1007/s00281-010-0231-y. [DOI] [PubMed] [Google Scholar]
- 24.Acevedo R, et al. Front Immunol. 2014;5:121–121. doi: 10.3389/fimmu.2014.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurst SJ, Lytton-Jean AKR, Mirkin CA. Anal Chem. 2006;78:8313–8318. doi: 10.1021/ac0613582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 27.Kim OY, et al. J Immunol. 2013;190:4092–4102. doi: 10.4049/jimmunol.1200742. [DOI] [PubMed] [Google Scholar]
- 28.Gao W, Hu CMJ, Fang RH, Luk BT, Su J, Zhang L. Adv Mater. 2013;25:3549–3553. doi: 10.1002/adma.201300638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neill CP, Lee LK, Swartz MA, Hubbell JA. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 30.Jewell CM, Lopez SCB, Irvine DJ. Proc Natl Acad Sci U S A. 2011;108:15745–15750. doi: 10.1073/pnas.1105200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moon JJ, et al. Nat Mater. 2011;10:243–251. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Proc Natl Acad Sci U S A. 2012;109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurung M, et al. PLoS One. 2011;6:article e27958. doi: 10.1371/journal.pone.0027958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thay B, Wai SN, Oscarsson J. PLoS One. 2013;8:article e54661. doi: 10.1371/journal.pone.0054661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen DJ, Osterrieder N, Metzger SM, Buckles E, Doody AM, DeLisa MP, Putnam D. Proc Natl Acad Sci U S A. 2010;107:3099–3104. doi: 10.1073/pnas.0805532107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gujrati V, Kim S, Kim SH, Min JJ, Choy HE, Kim SC, Jon S. ACS Nano. 2014;8:1525–1537. doi: 10.1021/nn405724x. [DOI] [PubMed] [Google Scholar]
- 37.Li LL, Xu JH, Qi GB, Zhao X, Yu F, Wang H. ACS Nano. 2014;8:4975–4983. doi: 10.1021/nn501040h. [DOI] [PubMed] [Google Scholar]
- 38.Piao JG, Wang L, Gao F, You YZ, Xiong Y, Yang L. ACS Nano. 2014;8:10414–10425. doi: 10.1021/nn503779d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.