

Abstract
Bacterial chondronecrosis with osteomyelitis (BCO) is recognized as an important cause of lameness in commercial broiler chickens (meat-type chickens). Relatively little is known about the microbial communities associated with BCO. This study was conducted to increase our understanding of the microbial factors associated with BCO using a culture-independent approach. Using Illumina sequencing of the hyper-variable region V6 in the 16S rRNA gene, we characterized the bacterial communities in 97 femoral or tibial heads from normal and lame broilers carefully selected to represent diverse variations in age, line, lesion type, floor type, clinical status and bone type. Our in-depth survey based on 14 million assembled sequence reads revealed that complex bacterial communities exist in all samples, including macroscopically normal bones from clinically healthy birds. Overall, Proteobacteria (mean 90.9%) comprised the most common phylum, followed by Firmicutes (6.1%) and Actinobacteria (2.6%), accounting for more than 99% of all reads. Statistical analyses demonstrated that there are differences in bacterial communities in different types of bones (femur vs. tibia), lesion types (macroscopically normal femora or tibiae vs. those with pathognomonic BCO lesions), and among individual birds. This analysis also showed that BCO samples overrepresented genera Staphylococcus, whose species have been frequently isolated in BCO samples in previous studies. Rarefaction analysis demonstrated the general tendency that increased severities of BCO lesions were associated with reduced species diversity in both femoral and tibial samples when compared to macroscopically normal samples. These observations suggest that certain bacterial subgroups are preferentially selected in association with the development of BCO lesions. Understanding the microbial species associated with BCO will identify opportunities for understanding and modulating the pathogenesis of this form of lameness in broilers.
Introduction
Generalized leg weakness and lameness are prominent economic and welfare concerns among poultry producers. Lameness in poultry can have several causes including viral and bacterial infections of bone and cartilage. Bacterial chondronecrosis with osteomyelitis (BCO, formerly known as femoral head necrosis, proximal femoral degeneration, or bacterial chondronecrosis, BCN) is recognized as an important cause of lameness in commercial broiler chickens (meat-type chickens). BCO commonly develops in the proximal femur and proximal tibiotarsus (tibia) where the wide, thick growth plates are susceptible to mechanical damage (osteochondrosis) that predisposes the cartilage to secondary bacterial infection. Physiological stress contributes to enhanced bacterial translocation through the gastrointestinal epithelium. Opportunistic bacteria pass through leaky epithelial tight junctions and spread hematogenously to osteochondrotic micro-fractures and clefts in the proximal femoral and tibial growth plates. Bacterial proliferation and the ensuing immunological response generate the necrotic abscesses and voids that are pathognomonic for BCO [1–5]. BCO has been diagnosed in broilers in Australia, Canada, Europe and the US, and is considered the most common cause of lameness in broilers [2,4,6–10]. Multiple opportunistic organisms in mixed cultures have been isolated from BCO lesions, including predominately Staphylococcus spp., Escherichia coli, and Enterococcus spp. [4]. However, no previous attempt has been made to investigate the overall composition and diversity of the bacterial communities associated with BCO.
In the past, methods for identifying pathogens relied overwhelmingly on culture techniques followed by biochemical testing using semi-automated platforms in the clinical laboratory [11]. Culture-independent approaches such as molecular profiling of the 16S rRNA gene sequences provide a powerful technique for comprehensively analyzing the structure and diversity of microbial communities [12]. The 16S rRNA gene sequences contain hyper-variable regions that can provide comprehensive information on bacterial communities, including diversity, composition, and community structure [13]. The development of next generation sequencing, 454 pyrosequencing, SOLID and Illumina platforms, provided a major advance by enabling hundreds of thousands of sequences to be collected from multiple samples. This powerful technique has been used to investigate the composition of complex microbial communities in diverse environments [14–18]. The two platforms that are most commonly used for microbial community analysis are 454 FLX and Illumina. The 454 FLX instrument generates ~1 million reads per instrument run, with the read lengths of 250–400bp, and the Illumina platform produces up to 1.5 billion reads per run, with the read lengths of 50–150 bp. The shorter Illumina reads may reduce phylogenetic resolution, both in terms of picking operational taxonomic units (OTUs) and determining phylogenetic distances between OTUs. The number of base pairs per reads for the Illumina platform can be doubled by the paired-end (PE) approach where each molecule is sequenced from both the 5’ and 3’ ends [19,20]. The regions of 16s rRNA gene, V1-V2 and V4, are longer than 200 bp in length, which cannot be easily covered by the PE overlap. However, the V3 [13] and V6 [21,22] regions can be easily covered by the overlapping PE reads.
One of the hindrance in studying BCO lameness in broilers is its low incidence, which makes it difficult to study BCO lameness in a systematic and reliable manner. Recently, Wideman et al. developed a wire-flooring model in which broilers raised on wire flooring reliably triggers high incidences of BCO consistently without purposefully exposing the birds to pathogenic bacteria [4]. The unstable footing created by wire flooring accelerates the formation of osteochondrotic micro-fractures and clefts in the proximal epiphyseal-physeal growth plates of the femora and tibiae. Wire flooring (or the lack of access to litter) also triggers physiological stress that can lead to immunosuppression and enhanced bacterial translocation from the gastrointestinal tract into the systemic circulation [3–5,23].
In this study, we used the Illumina paired-end sequencing method targeting V6 hyper-variable region of 16S rRNA gene in conjunction with wire flooring model of BCO lameness for comprehensive investigation of the bacterial communities associated with BCO.
Materials and Methods
Animal experiment and sample collection
Animal procedures were approved by the University of Arkansas Institutional Animal Care and Use Committee. For the present study we used environmental chambers 7 through 12 within the Poultry Environmental Research Laboratory at the University of Arkansas Poultry Research Farm. These chambers (3.7 × 2.5 × 2.5 m, length × width × height) utilize single-pass ventilation at a constant rate of 6 m3 per minute per chamber. Chambers 7 and 8 were set up with 3.05 m x 1.5 m pens having wood shavings litter flooring to serve as the baseline (non-challenge) control. Chambers 9 through 12 were set up with 3.05 m x 1.5 m pens and flat wire flooring wire panels. The wire flooring panels were constructed from 5 cm x 5 cm lumber and were 3 m long and 1.5 m wide, with 5 cm x 5 cm cross members added for support. Hardware cloth (1.3 cm x 2.54 cm mesh = 0.5 inch by 1 inch, 0.063 gauge, galvanized welded wire cloth) was fastened to the top of the frame and cross-members. The wire flooring was elevated on 30 cm high masonry blocks to permit manure to pass through and accumulate underneath the wire surface. Tube feeders were positioned at the front and nipple waterers were positioned at the rear of each pen, thereby forcing the chicks to traverse the length of the floor to eat and then drink [4]. The photoperiod was set for 23 h light:1 h dark throughout the experiment. Thermoneutral temperatures were maintained throughout, with target temperatures set at 32°C for d 1 to 3, 30°C for d 4 to 6, 28°C for d 7 to 10, 26°C for d 11 to 14, and 24°C thereafter. Feed and water were provided ad libitum. The diets were corn and soybean meal-based broiler starter (crumbles) and finisher (pellets) feeds formulated to meet minimum NRC [24] standards for all ingredients. Chicks from two commercial lines (B and D) were obtained from a commercial hatchery and were wing banded on the day of hatch. Line B is known to be more susceptible to BCO than Line D (Wideman, personal observations). All chicks initially were housed on litter flooring, with equal numbers of birds from the two lines comingled in chambers 7 and 8 (130 birds per line per chamber). On days 7 and 14, five birds per line were randomly selected and removed for analysis of bacterial communities from chambers 7 and 8 (n = 10 per line on days 7 and 14). On day 14 the remaining chicks in chambers 7 and 8 (120 birds per line per chamber) were redistributed evenly among all 6 chambers (2 lines per chamber; 40 birds per line per chamber). Thereafter at least five birds per line were randomly selected from all six chambers on days 21, 29, 36, 43 and 49 (n = 60 per line per sampling day: 20 from litter flooring and 40 from wire flooring). The selected birds were humanely euthanized by CO2 inhalation and, within 30 min post-mortem, the proximal femora and tibiae of both legs were exposed in a laminar flow hood for scoring based on macroscopic lesion appearance (see the following section) and sampling for bacterial community analysis. Forceps and shears that were dipped in 95% ethanol and flame sterilized were used to aseptically remove the upper metaphysis and physis (growth plate) of each proximal bone end. Forceps and shears were re-sterilized immediately before collecting each bone sample. The cut surfaces of the proximal femoral and tibial samples were dipped in 95% ethanol, flame sterilized, dropped into sterile culture tubes, and stored at -20°C for subsequent DNA isolation.
Birds in all chambers were “walked” and observed for lameness every two days beginning on day 15. Birds that developed clinical lameness were humanely euthanized and necropsied. Necropsy observations on dead and lame birds were recorded by day/date, gender and pen number. On day 56 all surviving birds were euthanized, weighed and necropsied. A total of 1,372 bone samples were collected from 343 broilers (4 samples per bird: right and left femora and tibiae) over the course of this experiment. From this massive sample reservoir, 97 individual samples were carefully selected for analysis, representing all 6 diagnostic categories [normal proximal femoral head (Normal Femur); proximal femoral head separation or epiphyseolysis (FHS); proximal femoral head necrosis (FHN); normal proximal tibia head (Normal Tibia); tibial head necrosis (THN); tibial head necrosis severe or caseous (THNsc)], all 7 ages, both flooring types (litter vs. wire), both genetic lines, and both body parts (right vs. left) (S1 Table).
Macroscopic classification of lesions
The femora and tibiae were categorized by macroscopic appearance according to the following diagnostic categories: Normal Femur; FHS; FHN; Normal Tibia; THN; THNsc. In all experiments the proximal femoral head lesions were categorized separately (FHS, or FHN), and the two levels of tibial head necrosis were categorized separately (THN, or THNsc) to emphasize the progressive severity of the BCO lesions [4], as represented in Fig 1.
Fig 1. Stages of proximal femoral head degeneration (Upper row) or tibial head necrosis (Low row) leading progressively to bacterial chondronecrosis with osteomyelitis (BCO).

1. Normal proximal femoral head; 2. Femoral head separation (FHS: epiphyseolysis); 3. Femoral head necrosis, FHN. 4. Normal proximal tibial head with struts of trabecular bone in the metaphyseal zone fully supporting the growth plate; 5. Tibial head necrosis (THN). Lytic channels (small arrows) penetrate from the necrotic voids into the growth plate. 6. Tibial head necrosis (THNsc). Bacterial infiltration and sequestrae (open arrows) provide macroscopic evidence of osteomyelitis.
DNA isolation, PCR amplification, and amplicon purification
For isolation of bacterial DNA, PBS buffer was added to each sample for 10X dilution, and the samples were homogenized using a tissue tearor. Two hundred microliter of each sample was transferred into a microcentrifuge tube for DNA isolation. DNA was extracted from this cell suspension using a QIAamp DNA Mini kit (Qiagen, Valencia, CA., USA). DNA concentrations were estimated spectrophotometrically using NanoDrop (Thermoscientific, USA). Because of its taxonomic resolution [25], conserved flanking regions [26], and length [21], the hyper-variable region 6 (V6) of the bacterial 16S rRNA gene was selected for this study. PCR amplification was carried out for each sample using Illumina paired-end primers in combination with unique barcode sequence at the 5’ end of each primer (S2 Table). Gloor et al. used paired-end sequencing in combination with unique barcode sequence tags at the 5′ end of each primer to perform microbiome analysis of the V6 region of 272 clinical samples using the Illumina sequencing technology [21]. We modified the primer design to use a different barcodes and added the random 5 nt sequence “NNNNN” in the first 5 nt positions of the forward reads of Illumina sequencing to facilitate more efficient and accurate analysis of clusters during processing of image data [27,28] (Fig 2). The PCRreactionss consisted of approximately 0.1 μg of purified genomic DNA, 1× cloned Pfu polymerase buffer, 5 U Pfu polymerase (Stratagene La Jolla, CA, USA), 1 mM dNTPs (GE Healthcare Bio-Sciences Corp., Piscataway, NJ), 1.2 μM each primer in a total volume of 50 μL. The DNA engine thermal cycler (Bio-Rad, Hercules, CA, USA) was used with the following amplification conditions: 94°C for 2 minutes; 30 cycles of 94°C sec for 30 sec, 58°C for 60 sec, 72°C for 90 sec; and 72°C for 10 minutes for final extension. PCR products of the correct size were gel-purified (Qiagen, Valencia, CA, USA). PCR products of each sample were mixed in equal quantities, based on measurement with Qubit 2.0 Fluorometer. Illumina sequencing was performed for 100 cycles in paired-end mode with Illumina HiSeq 2000.
Fig 2. Design of the PCR primers and the amplicon library.
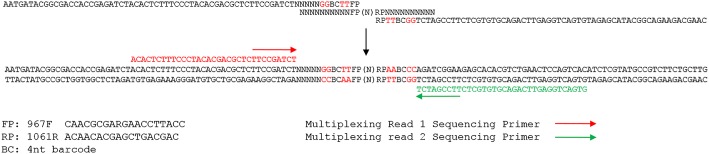
V6 rRNA region were amplified using the two primers with barcode tags and Illumina adaptors attached at 5’ ends.
Data Analysis
Illumina reads were binned according to barcode sequences using DNAStar software (DNASTAR, Inc). Overlapping regions within paired-end reads were then aligned to generate assembled reads. If a mismatch was discovered, the paired-end sequences involved in the assembly were discarded. The sequence reads with any ambiguous base calls or without a perfectly matching barcode were also discarded. Then the compiled dataset was analyzed with the software package Quantitative Insights Into Microbial Ecology (QIIME; http://qiime.sourceforge.net)[29] using default settings with one modification: the length cutoff was set at 130 instead of the default 150 bases, as the V6 region was expected to produce amplified fragments between 110 and 130 bp [21]. To match the input file format for QIIME, the assembled sequence reads were modified using a custom Python script.
For Operational taxonomic unit (OUT) analysis, we used the open-reference OTU picking method in QIIME with OTU definition at a level of 97% sequence similarity and Greengenes reference database (February 4th 2012) [30] as a reference sequence collection. Rarefaction curves, alpha (α) diversity and beta (β) diversity calculations were performed and plotted using QIIME. Similarity and differences in community membership were calculated based on the phylogenetic information within each sample with both the unweighted and weighted UniFrac analysis. The analysis of similarities (ANOSIM) [31] function in QIIME was used to test for differences in community composition among groups of samples. UPGMA trees of weighted and unweighted UniFrac values were calculated with QIIME and plotted with FigTree v 1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/). Principal coordinate analysis (PCoA) of weighted and unweighted UniFrac was also performed and plotted with QIIME [29]. LEfSe analysis (linear discriminant analysis effect size) was applied to discover the biomarkers related to BCO [32].
Statistical analysis
For comparisons of lameness incidences the individual bird was used as the experimental unit, and the SigmaStat Z-test procedure was used to compare proportions (Jandel Scientific, 1994). For comparison of α diversity expressed in Chao1 index, ANOVA was used to determine statistical difference and Duncan analysis was used for separation of the means showing statistical difference. Statistical difference was considered significant at the level of p≤0.05.
Results and Discussion
BCO is recognized as an important cause of lameness in rapidly growing broilers. BCO appears to be initiated by osteochondrotic micro-trauma to poorly mineralized columns of cartilage cells in the metaphyses and proximal growth plates of the leg bones, followed by colonization by hematogenously distributed opportunistic bacteria [4,5]. Bacteria that translocate into the chick's circulation can spread hematogenously and exit the bloodstream through the fenestrated capillaries supplying the growth plate. Once the osteochondrotic clefts and micro-fractures become infected, leukocytes migrate into the infected area, and, in their attempt to engulf the infectious organisms, release enzymes that lyse the bone. Pus spreads into the bone's blood vessels, impairing their flow, and areas of devitalized infected bone, known as sequestra, form the basis of a chronic infection. Terminal BCO presents as necrotic degeneration and bacterial infection primarily within the proximal head of the femora and tibiae, as well as in the growth plates of the flexible thoracic vertebrae [2,5]. Rearing broilers on wire flooring facilitates our ability to investigate the pathogenesis of BCO by consistently triggering substantially higher incidences than typically observed when broilers are reared on wood-shavings litter flooring [4]. In the present study significantly more broilers developed BCO lameness on wire flooring than on litter (27.2% vs. 4.4%, respectively; P = 0.002, z-test). More broilers from Line B developed lameness on wire flooring than broilers from Line D (33.8% vs. 20.6%, respectively; P = 0.011, z-test), whereas the lameness incidences for the two lines did not differ on litter flooring (7.5% vs. 1.3%, respectively; P = 0.126, z-test) (Fig 3).
Fig 3. The incidence of lameness in two lines (line B and line D) developed on wire flooring and on wood-shavings litter flooring from days 14 through 49.

High incidences of BCO are accompanied by the progressive development of pathognomonic lesions even in broilers that are not yet clinically lame [4,5]. Accordingly, we used the wire flooring model to enhance our ability to acquire suitable numbers of diverse bone samples for investigating the bacterial communities associated with BCO lesion progression. Using routine culture methods, multiple opportunistic organisms have been isolated from BCO lesions, including predominately Staphylococcus spp., Escherichia coli, and Enterococcus spp., often in mixed cultures with other microbes including Salmonella spp. [4]. However, our understanding of the bacteria present in the BCO lesions based on routine culture methods is limited, because non-culturable bacterial species that constitute a major portion of the bacterial communities remain undetected. With the development of the next generation sequencing technology, it is possible to comprehensively characterize bacterial communities by deep profiling of their 16S rRNA gene sequences. Recent studies have revealed the composition and diversity of the bacterial community in diverse environments, such as the sea [33], soil [34,35], chicken ceca [36], and salivary, fecal or nasal swabs from humans [37–39]. To our knowledge the present study is the first to evaluate microbial communities of non-mucosal tissue samples from animals. Additionally, BCO in broilers provides an excellent animal model for conducting translational research focused on developing effective prophylactic and therapeutic treatments that are relevant to osteomyelitis in young growing human children [5].
Data processing
By using combinatorial sequence-tagged PCR, we analyzed 97 bone samples by a single lane of Illumina paired-end sequencing run. We obtained 52,684,589 paired-end reads of exactly 100 nucleotides in length from each end before initial quality filtering. After the assembly using DNAStar, there were 15,598,270 assembled reads for the subsequent analysis. Using a custom Python script, the forward and reverse barcodes were extracted from the assembled reads, and combined together to generate new unique barcode for every read from each sample. The assembled reads were then sorted by the combinatorial barcodes and the file format was modified to be compatible for analysis using QIIME. After the quality filtering, 15,077,583 reads were retained for QIIME analysis. After an additional quality filtering, 1,040,076 short reads and 1,489 reads containing ambiguous nucleotides were discarded. The total number of input reads was 14,036,018 and the median sequence length of the assembled reads was 107 bp.
It is important to note that only 29.6% of the raw data were retained after assembly. This occurred because the length of V6 region is 110–130 and the paired end reads of 100 nucleotides are overlapped for almost their entire length. With this long overlapping region, a large portion of the paired reads with any mismatch between the two reads were discarded during assembly. Although the assembly under this high stringency reduced the number of the final assembled reads, it greatly increased the accuracy of the final assembled sequence reads used for downstream analysis.
Taxonomic analysis
Taxonomic analysis of the 16S rRNA gene V6 region amplicon reads yielded a total of 9 phyla across all samples with the dominant phylum being Proteobacteria (mean 90.9%) followed by Firmicutes (6.1%) and Actinobacteria (2.6%), together accounting for nearly 99.6% of all reads. Several other phyla, including Tenericutes, Bacteroidetes, Acidobacteria, Verrucomicrobia, Nitrospirae and Cyanobacteria, were detected at very low levels (less than 0.4% of the total), nevertheless these phyla were present in multiple samples (Fig 4). Sequence reads also were analyzed on the class, order, family, and genus levels using the QIIME pipeline. The dominant class in all of the bone samples was Gammaproteobacteria (90.5%), followed by Clostridia (3.5%), Bacilli (2.6%) and Actinobacteria (2.5%). The dominant order was Enterobacteriales (34.2%), followed by Vibrionales (27.3%), Pseudomonadales (14.6%), Oceanospirillales (7.9%), Pasteurellaceae (4.7%), Clostridiales (3.5%), Actinobacteriales (2.4%), Bacillales (1.4%), and Lactobacillales (1.1%) (S3 Table).
Fig 4. Taxonomic composition of the bone samples at phylum level.

Bacterial pathogens associated with BCO have been the focus of numerous studies. Most of these previous studies were based on traditional culturing methods. It is believed that BCO is most commonly caused by Staphylococcus aureus, but Escherichia coli, coagulase-negative staphylococci and Enterococcus spp. also have been identified [2,4]. However, not all cases of BCO have been associated with the presence of isolated bacteria [40–42]. Recently two studies used 16S rRNA technology to identify bacteria isolated by culturing infected bone samples [43,44]. Our results show that the bacterial communities in BCO samples are much more complex than previously reported or anticipated. The bacteria observed in the present study include most of the bacteria found in previous studies (S4 Table) at the phylum, class, order, family and genus levels.
Normal samples
Normal samples included proximal femoral and tibial heads that appeared macroscopically to be normal (Fig 1). A total of 28 normal samples (S1 Table) were collected during weeks 1–7, which includes 4 normal samples per week. The results of taxonomic analysis of the 16S rRNA gene V6 region amplicon reads from normal samples show that there were a total of 9 phyla across all normal samples with the dominant phylum being Proteobacteria (mean 84.7%), followed by Firmicutes (11.1%) and Actinobacteria (3.4%), accounting for nearly 99.2% of all normal samples reads (Fig 4). These results suggest that macroscopically normal bones from broilers that appeared to be clinically healthy are not “sterile” as traditionally assumed. Accordingly, the seemingly normal growth plates and metaphyses of broilers appear to archive multiple bacterial species that reached the bones probably through translocation and hematogenous distribution. Several plausible alternative explanations for this observation must be considered. First, since lesions are visible to the naked eye in only 40 to 67% of the BCO legs necropsied, histological examination has been recommended when no lesions are visible macroscopically. Because histological examination is often not carried out (as in the present study), BCO is almost certainly underdiagnosed [2]. Second, more than 99% of bacterial strains in the environments are resistant to cultivation in the laboratory and therefore would not previously have been identified without culture-independent method such 16S rRNA gene profling. Finally, the 16S rRNA gene profiling method is sensitive, but it cannot distinguish between live and dead bacteria.
There were general trends found in the normal bone samples that were very similar in both femur and tibia: the Proteobacteria, the most abundant phylum, increased gradually between 7 and 35 days of age, and then decreased from 35 to 49 days; Firmicutes decreased from 7 to 35 days of age; and Actinobacteria increased from 35 to 49 days of age in the normal bone samples (Fig 4). The prevalence of Proteobacteria in our normal bone samples contrasts with the report by Danzeisen et al. [36](2011) that Firmicutes comprise the dominant phylum of the chicken cecal microbiome. The cecum is located at the beginning of the large intestine, whose environments differ greatly from the other body sites, characterized primarily by interaction with the immune system, oxidative challenge, and hydration [45]. With regard to the intestinal tract as a potential site of bacterial translocation, this may suggest that the Proteobacteria may translocate more easily than Firmicutes through the gastrointestinal epithelium, or survive better than Firmicutes inside the blood stream or bones. Beside the gastrointestinal tract, the bacteria also can translocate across the integument or respiratory epithelium and spread hematogenously to the growth plates of the proximal femora and tibiae. Accordingly, translocation via the integument and respiratory system might also account for the differences in microbial communities between the bone and cecal samples.
Analysis of α and β diversities
Interestingly, there was a general trend that the diversity within a bacterial community (α diversity) was highest during the first week, and decreased with aging (Fig 5). The opposite trend was reported for chicken cecal samples [36]. These observations suggest selection processes are operating in favor of certain bacterial subgroups in bones in association with aging. The pathogenesis of progressive BCO lesion development, including increases in the size and caseous nature of the most severe bacterial sequestrae, undoubtedly would affect the diversity of the bacterial community, and would be expected to result in decreased diversity with aging. Indeed, a time-course analysis of lesion development demonstrates progressively greater incidences of the early macroscopic FHS and THN lesions over the seven week course of the present experiment. Age-related increases in incidences of the most severe lesions, FHN and THNsc, were less dramatic, likely because birds with BCO lesions in these categories rapidly succumbed to clinical lameness. In addition, the bones with more severe macroscopical lesions were generally associated with reduced diversity of the bacterial communities (Fig 5).
Fig 5. Comparison of the α-diversity in different groups of bone samples.

Diversity levels were estimated using Chao1 index with rarefied 180 V6 rRNA reads per sample. Means with the same letters in red color within the same row are not different significantly (p>0.05). Means with the same letters in blue within the same column are not different significantly (p>0.05).
Hierarchical clustering of the bacterial communities in our 6 BCO diagnostic categories (Normal Femur, FHS, FHN, Normal Tibia, THN and THNsc) was conducted by using the default β diversity metrics of un-weighted UniFrac. The resulting phylogenetic tree shows that most of the FHS, THN, FHN and THNsc samples tended to cluster together within a diagnostic category (Fig 6). The same phylogenetic tree also demonstrates that the Normal femur and Normal tibia samples grouped together in two different clusters that essentially were segregated from the BCO lesion samples. Evidently no distinctive difference existed between these “normal” sample groups from the two different bone types (femur and tibia) (p>0.05).
Fig 6. Hierarchical clustering analysis of the bacterial communities.

Phylogenetic tree shows that most of FHS (dark blue), THN (green), FHN (pink) and THNsc (red) samples can be clustered. Femur Normal (black), and Tibia Normal (light blue) were largely mixed together in two different clusters.
Comparison of microbial communities in different groups
ANOSIM analyses (Table 1) demonstrated the presence of distinctively different bacterial communities in different bones (femur vs. tibia, p≤0.001), different lesion categories (normal, FHS, FHN, THN, and THNsc, p≤0.001), different individuals (p≤0.001), and different ages (p≤0.005). However bacterial communities were not different between the samples from right vs. left legs (p = 0.156), between clinically healthy vs. lame broilers (p = 0.714), between broilers reared on litter vs. wire flooring (p = 0.248), or between different lines (Line B vs. Line D; p = 0.089). The lack of different communities between the two lines is important to note because the fact that broilers from Line B are more susceptible than broilers from Line D in developing BCO lameness on wire flooring cannot be attributed to the presence of (or harboring of) distinctively different bacterial communities. Instead other innate factors must contribute to the reproducible differences between these genetic lines in their relative susceptibilities to BCO lameness.
Table 1. Analysis of similarities (ANOSIM).
| Groups | R-value | P-value |
|---|---|---|
| Left vs. Right Leg | 0.033512634 | 0.156 |
| Femoral vs. Tibial Samples | 0.216799541 | 0.001* |
| Clinically Healthy vs. Lame | -0.041557977 | 0.714 |
| Individuals | 0.626107125 | 0.001* |
| Age (7 through 49 days) | 0.090237558 | 0.003* |
| Litter vs. Wire Flooring | 0.02794451 | 0.248 |
| Lesion Type (normal femur, normal tibia, FHS, FHN, THN, THNsc) | 0.4393 | 0.001* |
| Line B vs. Line D | 0.029131713 | 0.089 |
*significantly different (p≤0.05).
We selected 28 macroscopically normal samples and 69 samples with BCO lesions for analysis, including 23 FHS, 14 FHN, 23 THN and 9 THNsc samples (S1 Table). Principal coordinate analysis (PCoA) shown in Fig 7 visually illustrates the differences between the bacterial communities in these bone samples. The communities in macroscopically normal bones are more tightly clustered as compared to the dispersed communities in bones with BCO lesions. On the other hand, the LEfSe analysis of the communities from Normal and BCO samples shows that there are 28 differentially abundant taxonomic clades with an LDA score higher than 2.4 (Fig 8). The most differentially abundant bacterial taxa in BCO samples belong to phyla: Proteobacteria. The genera overrepresented in BCO samples include Staphylococcus, Enterobacter, Serrotia and Nitrincola. Interestingly, the genus Staphylococcus was detected repeatedly in BCO samples in previous studies using culture-dependent methods [2,8,41,46,47]. The result from our parallel study based on culturing method also showed that in most cases BCO was attributable to Staphylococcus species (data not shown). The Staphylococcus genus includes at least 40 species. Of these, nine have two subspecies and one has three subspecies [48]. One of the species, Staphylococcus aureus, is the most prominent musculoskeletal pathogen of men and animals. It has a particular propensity to infect tissues of the musculoskeletal system, as evidenced by the fact that it is the single leading cause of osteomyelitis in humans [49]. According to ultrastructural studies, adhesion of S. aureus on damaged growth plate cartilage plays an important role for infection and proliferation within a thick adherent glycocalyx. This situation allows the presentation of a fibrous exopolysaccharide bacterial cell surface to the defense mechanisms of the host and provides a thick barrier to the penetration of antibiotics [50]. Our recent study based on culture independent method indicated that the genus Staphylococcus is present in the blood samples of BCO birds (unpublished data). This observation supports the hypothesis that Staphyloccous cells enter the blood stream through translocation from gastrointestinal tract and transmitted to bones hematogenously. However, with prevalent nature of Staphylococcus on chicken feather and poultry litter [51], it may be possible that Staphylococcus enters into the bones through external infection through necrotic lesions on the bone surface.
Fig 7. Principal Coordinate analysis.

Unweighted PCoA on Qualitative measures and weighted PCoA on Quantitative measures illustrates the differences between Normal and BCO communities. Normal communities are tightly clustered as compared to the scattered BCO communities.
Fig 8. Comparison between Normal and BCO samples using LEfSe analysis.

Taxa abundance analysis determined 7 features were differentially abundant in BCO group (LDA score >2.4) and 21 features (LDA >2.4) in Normal cases. The genus Staphylococcus, which has been frequently isolated from BCO samples by previous researchers, is an abundant feature in BCO case.
The remaining three genera that were overrepresented in BCO samples, Enterobacter, Serrotia, and Nitrincola, have not been detected by culture-dependent methods. The genus Nitrincola is not common and is not well understood. It is a novel alkaliphilic bacterium isolated from an alkaline, saline lake [52]. Both of the genera, Enterobacter and Serrotia, are Gram-negative, facultative anaerobic, rod-shaped bacteria of the family Enterobacteriaceae. Several strains of Enterobacter are pathogenic and cause opportunistic infections in immunocompromised (usually hospitalized) hosts, and in patients who are on mechanical ventilation. The urinary and respiratory tracts are the most common sites of infection [53]. In the genus Serratia, the most common species, S. marcescens, is recognized as a pathogen and usually causes nosocomial infections. Biofilm formation, and the adherence of microbes to surfaces such as contact lenses, likely plays a role in S. marcescens pathogenesis [54]. However, rare strains of S. plymuthica, S. liquefaciens, S. rubidaea, and S. odoriferae have caused diseases through infection [55–58]. These two genera, Enterobacter and Serrotia, might be important for development of BCO. However, the other three previously reported genera that have been isolated by culture methods, Escherichia (Escherichia coli), Enterococcus (Enterococcus cecorum), and Salmonella (Salmonella spp.) were not overrepresented in BCO samples.
The community structure of femor associated bacteria differed significantly from tibial bacteria assemblages (Table 1). The observed difference may be due to the changes in physiological micro-environments caused by the different torque and shear stresses imposed on the femoral vs. tibial joints. For this analysis we selected 51 femur and 46 tibia samples (S1 Table). The results of taxonomic analysis of the V6 16S rRNA gene amplicon reads show that there are a total of 9 phyla across both bone locations, with the dominant phylum being Proteobacteria (mean 89.7% for the femur and 91.6% for the tibia) and followed by Firmicutes (6.8% for the femur and 5.7% for the tibia) and Actinobacteria (2.9% for the femur and 2.3% for the tibia), accounting for more 99% of all reads (Fig 4). We also compared the changes in bacterial communities in both femur and tibia at genus level. Fig 9 shows the distribution of major genera that belong to either Proteobacteria or Firmicutes. For the normal bones, there was a striking similarity in the changing patterns of the major genera over time between the femur and tibia. On the contrast, however, the development of the patterns was very distinctive between the femur and tibia with BCO lesions. It is interesting that the abundance of the genera Serratia and Vibrio increased to nearly 80% of total bacterial population at the later stages for THN and THNsc, respectively.
Fig 9. Taxonomic composition of the bone samples based at genus level.

Only 15 major genera of significant importance were selected and shown in the graphs.
ANOSIM analyses shows that individual chickens harbored distinctly different bacterial communities (Table 1). Recent research has demonstrated a high degree of inter-individual variability in the composition of bacterial communities at particular locations, such as human skin [59], and the chicken ceca [36]. Principal Coordinate Analysis showed that bacterial communities within the same bird are closely related, suggesting the presence of distinctive selective pressures in different individuals (Fig 10).
Fig 10. Comparison of samples using unweighted UniFrac.

Unweighted UniFrac was used to generate a matrix of pairwise distances between communities, then a scatterplot was generated from the matrix of distances using Principal Coordinate Analysis. Number = chicken ID, R = right, L = left, F = femur, T = tibia. A. Comparison of 4 samples from the same chicken, which has FHS on both femurs and THN on both tibias. B. Comparison of the samples from different individual chickens. Paired samples from same individual chickens have FHS on femurs and THN on tibias.
ANOSIM analyses showed that bacterial communities were similar among samples collected from birds reared on different flooring types (Table 1, litter vs. wire; p = 0.248). These data indicate that BCO triggered by wire flooring does not differ microbiologically from BCO that develops spontaneously in hatch-mates reared on wood shavings litter. This observation further validates the use of wire flooring model of BCO lameness to understand the etiology, pathogenesis, and treatment strategies for BCO lameness developed under normal rearing condition using a litter floor.
Conclusions
Significant differences were observed between the bacterial communities of bone samples that appeared to be macroscopically normal versus samples having obvious BCO lesions (p≤0.001). The previously detected genus Staphylococcus was overrepresented in samples with BCO lesions. Two other genera overrepresented in the BCO samples, Enterobacter and Serrotia, might contribute to the pathogenesis of BCO. Interestingly, significant differences were also observed between the bacterial communities of the femora and tibiae, and among individual birds (p≤0.001). Our study illustrates that even macroscopically normal samples host complex bacterial communities. Taxonomic analysis shows that Proteobacteria (mean 90.9%) is the most abundant phylum, followed by Firmicutes (6.1%) and Actinobacteria (2.6%), accounting for more than 99% of all assembled reads. The dominant phylum Firmicutes in chicken cecal samples was shifted to Proteobacteria in chicken bone samples. Understanding the reasons responsible for this shift might be helpful for detecting the routes used by bacteria to infect the bones. Rarefaction curves demonstrated that α diversity decreased in general with increasing severity of BCO lesions in both femur and tibia. This observation suggests that unidentified selection processes favor the survival of certain bacterial subgroups in association with progression of BCO lesions. One limitation of the short-read sequencing technology used in this study is that it does not provide sufficient sequence information for the classification at species level. Thus, the majority of the sequences could be classified only at the genus level. On other hand, the potential bias of PCR and error in Illumina sequencing might have affected the linear relationship between the number of sequences per OTU and the different taxonomic groups of bacteria in the population. In future studies, longer regions (such as 16S rRNA V1V2 region), previously shown to be most variable within genus Staphylococcus, should be chosen to identify overrepresented bacteria in BCO samples at species level. To our knowledge, this is the first comprehensive survey of bacterial communities associated with BCO lesions in broilers using culture-independent method. The results provided various insights on the possible association between the observed changes in bacterial communities and BCO pathogenesis. The results of this study warrants further investigation of the bacterial communities of BCO lesions along with different types of samples such as cecal and blood samples from the same individual birds using the similar 16S rRNA gene profiling as well as quantitative PCR analysis to estimate bacterial loads.
Supporting Information
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
Funding for this project was provided by Cobb-Vantress Inc., Siloam Springs, Arkansas, USA.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funding for this project was provided by Cobb-Vantress Inc., Siloam Springs, Arkansas, USA. The funder provided support in the form of a salary for author IP, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section.
References
- 1. McNamee PT, McCullagh JJ, Rodgers JD, Thorp BH, Ball HJ, et al. (1999) Development of an experimental model of bacterial chondronecrosis with osteomyelitis in broilers following exposure to staphylococcus aureus by aerosol, and inoculation with chicken anaemia and infectious bursal disease viruses. Avian Pathol 28: 26–35. [DOI] [PubMed] [Google Scholar]
- 2. McNamee PT, Smyth JA (2000) Bacterial chondronecrosis with osteomyelitis ('femoral head necrosis') of broiler chickens: A review. Avian Pathol 29: 253–270. 10.1080/03079450050118386 [DOI] [PubMed] [Google Scholar]
- 3. Wideman RF, Prisby RD (2012) Bone circulatory disturbances in the development of spontaneous bacterial chondronecrosis with osteomyelitis: A translational model for the pathogenesis of femoral head necrosis. Front Endocrinol (Lausanne) 3: 183 10.3389/fendo.2012.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wideman RF Jr, Hamal KR, Stark JM, Blankenship J, Lester H, et al. (2012) A wire-flooring model for inducing lameness in broilers: Evaluation of probiotics as a prophylactic treatment. Poult Sci 91: 870–883. 10.3382/ps.2011-01907 [DOI] [PubMed] [Google Scholar]
- 5. Wideman RF Jr, Pevzner I (2012) Dexamethasone triggers lameness associated with necrosis of the proximal tibial head and proximal femoral head in broilers. Poult Sci 91: 2464–2474. [DOI] [PubMed] [Google Scholar]
- 6. PATTISON M (1992) Impacts of bone problems on the poultry meat industry In: Whitehead C. C., editor. Bone Biology and Skeletal Disorders in Poultry.: Abingdon: Carfax Publishing Company; pp. 329–338. [Google Scholar]
- 7. McNamee PT, McCullagh JJ, Thorp BH, Ball HJ, Graham D, et al. (1998) Study of leg weakness in two commercial broiler flocks. Vet Rec 143: 131–135. [DOI] [PubMed] [Google Scholar]
- 8. Butterworth A (1999) Infectious components of broiler lam eness: A review. World's poultry Science Journal 56: 327–352. [Google Scholar]
- 9. Bradshaw RH, Kirkden RD, Broom DM (2002) A review of the aetiology and pathology of leg weakness in broilers in relation to welfare. Avian Poult Biol Rev 13: 45–103. [Google Scholar]
- 10. Dinev I (2009) Clinical and morphological investigations on the prevalence of lameness associated with femoral head necrosis in broilers. Br Poult Sci 50: 284–290. 10.1080/00071660902942783 [DOI] [PubMed] [Google Scholar]
- 11. Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J (2009) Metagenomic pyrosequencing and microbial identification. Clin Chem 55: 856–866. 10.1373/clinchem.2008.107565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Kessel MA, Dutilh BE, Neveling K, Kwint MP, Veltman JA, et al. (2011) Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (cyprinus carpio L.). AMB Express 1: 41-0855-1-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD (2011) Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl Environ Microbiol 77: 3846–3852. 10.1128/AEM.02772-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hong PY, Hwang C, Ling F, Andersen GL, LeChevallier MW, et al. (2010) Pyrosequencing analysis of bacterial biofilm communities in water meters of a drinking water distribution system. Appl Environ Microbiol 76: 5631–5635. 10.1128/AEM.00281-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vahjen W, Pieper R, Zentek J (2010) Bar-coded pyrosequencing of 16S rRNA gene amplicons reveals changes in ileal porcine bacterial communities due to high dietary zinc intake. Appl Environ Microbiol 76: 6689–6691. 10.1128/AEM.03075-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moya A, Pereto J, Gil R, Latorre A (2008) Learning how to live together: Genomic insights into prokaryote-animal symbioses. Nat Rev Genet 9: 218–229. 10.1038/nrg2319 [DOI] [PubMed] [Google Scholar]
- 18. Kip N, Dutilh BE, Pan Y, Bodrossy L, Neveling K, et al. (2011) Ultra-deep pyrosequencing of pmoA amplicons confirms the prevalence of methylomonas and methylocystis in sphagnum mosses from a dutch peat bog. Environ Microbiol Rep 3: 667–673. 10.1111/j.1758-2229.2011.00260.x [DOI] [PubMed] [Google Scholar]
- 19. Werner JJ, Zhou D, Caporaso JG, Knight R, Angenent LT (2012) Comparison of illumina paired-end and single-direction sequencing for microbial 16S rRNA gene amplicon surveys. ISME J 6: 1273–1276. 10.1038/ismej.2011.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT (2012) Direct comparisons of illumina vs. roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One 7: e30087 10.1371/journal.pone.0030087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gloor GB, Hummelen R, Macklaim JM, Dickson RJ, Fernandes AD, et al. (2010) Microbiome profiling by illumina sequencing of combinatorial sequence-tagged PCR products. PLoS One 5: e15406 10.1371/journal.pone.0015406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou HW, Li DF, Tam NF, Jiang XT, Zhang H, et al. (2011) BIPES, a cost-effective high-throughput method for assessing microbial diversity. ISME J 5: 741–749. 10.1038/ismej.2010.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El-Lethey H, Huber-Eicher B, Jungi TW (2003) Exploration of stress-induced immunosuppression in chickens reveals both stress-resistant and stress-susceptible antigen responses. Vet Immunol Immunopathol 95: 91–101. [DOI] [PubMed] [Google Scholar]
- 24. National Research Council (NRC) (1994) Nutrient requirements of poultry Washington D.C: National Academy Press. [Google Scholar]
- 25. Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, et al. (2008) Exploring microbial diversity and taxonomy using SSU rRNA hyper-variable tag sequencing. PLoS Genet 4: e1000255 10.1371/journal.pgen.1000255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59: 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith DR, Quinlan AR, Peckham HE, Makowsky K, Tao W, et al. (2008) Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res 18: 1638–1642. 10.1101/gr.077776.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whiteford N, Skelly T, Curtis C, Ritchie ME, Lohr A, et al. (2009) Swift: Primary data analysis for the illumina solexa sequencing platform. Bioinformatics 25: 2194–2199. 10.1093/bioinformatics/btp383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, et al. (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Australian Journal of Ecology 18: 117–143. [Google Scholar]
- 32. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, et al. (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12: R60-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, et al. (2004) Environmental genome shotgun sequencing of the sargasso sea. Science 304: 66–74. [DOI] [PubMed] [Google Scholar]
- 34. Ernebjerg M, Kishony R (2012) Distinct growth strategies of soil bacteria as revealed by large-scale colony tracking. Appl Environ Microbiol 78: 1345–1352. 10.1128/AEM.06585-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lang KS, Anderson JM, Schwarz S, Williamson L, Handelsman J, et al. (2010) Novel florfenicol and chloramphenicol resistance gene discovered in alaskan soil by using functional metagenomics. Appl Environ Microbiol 76: 5321–5326. 10.1128/AEM.00323-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ (2011) Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One 6: e27949 10.1371/journal.pone.0027949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, et al. (2009) A core gut microbiome in obese and lean twins. Nature 457: 480–484. 10.1038/nature07540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cephas KD, Kim J, Mathai RA, Barry KA, Dowd SE, et al. (2011) Comparative analysis of salivary bacterial microbiome diversity in edentulous infants and their mothers or primary care givers using pyrosequencing. PLoS One 6: e23503 10.1371/journal.pone.0023503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laufer AS, Metlay JP, Gent JF, Fennie KP, Kong Y, et al. (2011) Microbial communities of the upper respiratory tract and otitis media in children. MBio 2: e00245–10. 10.1128/mBio.00245-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Duff SR, Randall CJ (1987) Observations on femoral head abnormalities in broilers. Res Vet Sci 42: 17–23. [PubMed] [Google Scholar]
- 41. Thorp BH, Whitehead CC, Dick L, Bradbury JM, Jones RC, et al. (1993) Proximal femoral degeneration in growing broiler fowl. Avian Pathol 22: 325–342. [DOI] [PubMed] [Google Scholar]
- 42. Durairaj V, Okimoto R, Rasaputra K, Clark FD, Rath NC (2009) Histopathology and serum clinical chemistry evaluation of broilers with femoral head separation disorder. Avian Dis 53: 21–25. [DOI] [PubMed] [Google Scholar]
- 43. Cai H, Archambault M, Prescott JF (2003) 16S ribosomal RNA sequence-based identification of veterinary clinical bacteria. J Vet Diagn Invest 15: 465–469. [DOI] [PubMed] [Google Scholar]
- 44. Stalker MJ, Brash ML, Weisz A, Ouckama RM, Slavic D (2010) Arthritis and osteomyelitis associated with enterococcus cecorum infection in broiler and broiler breeder chickens in ontario, canada. J Vet Diagn Invest 22: 643–645. [DOI] [PubMed] [Google Scholar]
- 45. Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9: 799–809. 10.1038/nri2653 [DOI] [PubMed] [Google Scholar]
- 46. Andreasen JR Jr, Andreasen CB, Anwer M, Sonn AE (1993) Heterophil chemotaxis in chickens with natural staphylococcal infections. Avian Dis 37: 284–289. [PubMed] [Google Scholar]
- 47. Joiner KS, Hoerr FJ, van Santen E, Ewald SJ. (2005) The avian major histocompatibility complex influences bacterial skeletal disease in broiler breeder chickens. Vet Pathol 42: 275–281. [DOI] [PubMed] [Google Scholar]
- 48. Harris LG, Foster SJ, Richards RG (2002) An introduction to staphylococcus aureus, and techniques for identifying and quantifying S. aureus adhesins in relation to adhesion to biomaterials: Review. Eur Cell Mater 4: 39–60. [DOI] [PubMed] [Google Scholar]
- 49. Smeltzer MS, Gillaspy AF (2000) Molecular pathogenesis of staphylococcalosteomyelitis. Poult Sci 79: 1042–1049. [DOI] [PubMed] [Google Scholar]
- 50. Speers DJ, Nade SM (1985) Ultrastructural studies of adherence of staphylococcus aureus in experimental acute hematogenous osteomyelitis. Infect Immun 49: 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dumas MD, Polson SW, Ritter D, Ravel J, Gelb J Jr, et al. (2011) Impacts of poultry house environment on poultry litter bacterial community composition. PLoS One 6: e24785 10.1371/journal.pone.0024785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dimitriu PA, Shukla SK, Conradt J, Marquez MC, Ventosa A, et al. (2005) Nitrincola lacisaponensis gen. nov., sp. nov., a novel alkaliphilic bacterium isolated from an alkaline, saline lake. Int J Syst Evol Microbiol 55: 2273–2278. [DOI] [PubMed] [Google Scholar]
- 53. Cabral JP (2010) Water microbiology. bacterial pathogens and water. Int J Environ Res Public Health 7: 3657–3703. 10.3390/ijerph7103657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hume EB, Willcox MD (2004) Emergence of serratia marcescens as an ocular surface pathogen. Arch Soc Esp Oftalmol 79: 475–477. [PubMed] [Google Scholar]
- 55. Carrero P, Garrote JA, Pacheco S, Garcia AI, Gil R, et al. (1995) Report of six cases of human infection by serratia plymuthica. J Clin Microbiol 33: 275–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grohskopf LA, Roth VR, Feikin DR, Arduino MJ, Carson LA, et al. (2001) Serratia liquefaciens bloodstream infections from contamination of epoetin alfa at a hemodialysis center. N Engl J Med 344: 1491–1497. [DOI] [PubMed] [Google Scholar]
- 57. Ursua PR, Unzaga MJ, Melero P, Iturburu I, Ezpeleta C, et al. (1996) Serratia rubidaea as an invasive pathogen. J Clin Microbiol 34: 216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gorret J, Chevalier J, Gaschet A, Fraisse B, Violas P, et al. (2009) Childhood delayed septic arthritis of the knee caused by serratia fonticola. Knee 16: 512–514. 10.1016/j.knee.2009.02.008 [DOI] [PubMed] [Google Scholar]
- 59. Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, et al. (2010) Forensic identification using skin bacterial communities. Proc Natl Acad Sci U S A 107: 6477–6481. 10.1073/pnas.1000162107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.