

Abstract
Prognostic significance of histological anaplasia and BRAF V600E mutation were retrospectively evaluated in 74 patients with pleomorphic xanthoastrocytoma (PXA). Median age at diagnosis was 21.5 years (31 pediatric, 43 adult) and median follow‐up 7.6 years. Anaplasia (PXA‐AF), defined as mitotic index ≥ 5/10HPF and/or presence of necrosis, was present in 33 cases. BRAF V600E mutation was detected in 39 (of 60) cases by immunohistochemical and/or molecular analysis, all negative for IDH1 (R132H). Mitotic index ≥ 5/10HPF and necrosis were associated with decreased overall survival (OS; P = 0.0005 and P = 0.0002, respectively). In all cases except two, necrosis was associated with mitotic index ≥ 5/10HPF. Patients with BRAF V600E mutant tumors had significantly longer OS compared with those without BRAF V600E mutation (P = 0.02). PXA‐AF patients, regardless of age, had significantly shorter OS compared with those without (P = 0.0003). Recurrence‐free survival was significantly shorter for adult PXA‐AF patients (P = 0.047) only. Patients who either recurred or died ≤3 years from diagnosis were more likely to have had either PXA‐AF at first diagnosis (P = 0.008) or undergone a non‐gross total resection procedure (P = 0.004) as compared with patients who did not. This study provides further evidence that PXA‐AF behaves more aggressively than PXA and may qualify for WHO grade III “anaplastic” designation.
Keywords: anaplastic, BRAF V600E, glioma, IDH1 R132H, pleomorphic xanthoastrocytoma, WHO grade
Introduction
Pleomorphic xanthoastrocytoma (PXA) is a rare brain tumor, most commonly affecting children and young adults. Advances in our understanding of its natural history and prognosis have been hindered by its rarity and the lack of well‐studied large cohorts with adequate clinical information, including long‐term follow‐up. At 5 years, PXA has a relatively favorable prognosis when compared with diffusely infiltrative astrocytomas, with 30% recurrence rate and 75% to 80% overall survival rates following primary resection 20, 24, 50. In our previous study, a mitotic index (MI) ≥ 5/10 high‐power fields (HPF) was associated with significant decrease of both recurrence‐free and overall survival 24. The extent of surgical resection was an independent predictor of recurrence‐free survival. Based on these findings, we proposed that PXA cases with MI ≥ 5/10 HPF, with or without accompanying necrosis, be designated as “PXA with anaplastic features” (PXA‐AF) followed by a clear statement about the possibility of a potentially more aggressive clinical behavior 24. At that time, it was felt to be premature to classify PXA‐AF as an anaplastic (WHO grade III) glioma, as the term “anaplastic” could result in inappropriately aggressive treatment, especially when resectability more often affected the outcome than ordinary anaplastic astrocytoma. This view along with the “PXA‐AF” terminology was subsequently adopted in the 2007 World Health Organization Classification of Tumors of the Central Nervous System, in which PXA is graded as a WHO grade II tumor 23.
In the last 15 years, the rare nature of PXA has resulted in a slowly growing literature restricted to well‐documented case reports of classic examples 35, 63, classic examples with unusual clinical presentation/course 2, 8, 12, 21, 40, 48, 67 or in uncommon locations 3, 11, 19, 27, 45, 55, 60, 72, rare morphological variants 9, 18, 52, 54, 59, 65, 70, biphasic combined/collision tumors 1, 6, 15, 16, 25, 28, 31, 51, 61, 73, as well as a few small series 17, 20, 29, 38, 39, 42, 47, 53, 58, 62 and reviews 32, 42, 64, 68. Currently, WHO grading of so‐called “PXA‐AF” remains undefined, and it is still unclear whether these rare tumors should be termed “anaplastic” (WHO grade III). As a result, there is substantial heterogeneity on grading of PXA‐AF 68. In a recent study based on the National Cancer Institute's Surveillance, Epidemiology and End Results (SEER) database 50, of the 214 patients with PXA diagnosis (1981–2007), the percentage of unknown tumor grade was considerably high (54%), and the reported grading ranged from WHO grades I to IV. Therefore, a better understanding of the natural history and outcome of PXA and PXA‐AF remains a priority to guarantee consistent and homogeneous therapeutic strategies for these patients.
The characteristic genetic alterations of infiltrating gliomas are infrequent or absent in PXA 22, 33, 44, 49, 74. PXA frequently shows chromosomal imbalances 26, including recurrent 9p21.3 losses 56, 74, encompassing CDKN2A/2B tumor suppressor gene loci 69, 75, with loss of p16 expression 36. Recently, PXA has been found to harbor the highest frequencies of BRAF V600E mutation in primary central nervous system (CNS) neoplasms (up to 60%–78%) 5, 10, 13, 36, 57, 75. This mutation can be identified by immunohistochemistry using a BRAF V600E mutation‐specific monoclonal antibody 30. Conversely, IDH1‐2 mutations, which are frequently present in infiltrating gliomas, do not occur 58, with the exception of rare cases 71, which could potentially represent misclassified pleomorphic infiltrating gliomas rather than PXA.
Herein, we retrospectively reviewed 74 PXA cases, both of pediatric and adult age, from two highly active neuro‐oncology practices to further clarify the natural history and prognosis of this tumor. We reevaluated the prognostic significance of histologic features of anaplasia in addition to specific molecular alterations in order to determine whether PXA‐AF deserves a higher grade designation than the classic WHO grade II PXA.
Materials and Methods
Case selection
All studies were conducted according to Mayo Clinic‐ and Johns Hopkins Institutional Review Board‐approved protocols. Ninety‐eight cases with diagnosis that included the term “pleomorphic xanthoastrocytoma” upon first surgical resection, in which the patient had been clinically evaluated and/or treated at Mayo Clinic (n = 68) or Johns Hopkins University (n = 30), were identified from 1965 to 2013. All existing diagnostic slides were retrieved and reviewed by at least two of the authors (CG and CMI). According to previously described criteria 23, tumors showing a relatively solid growth pattern, composed of a combination of spindle‐shaped, xanthic and pleomorphic, multinucleated giant astrocytes, associated with both pale and bright eosinophilic granular bodies were diagnosed as PXA (Figure 1). Features of anaplasia, including mitotic index (MI) ≥ 5/10 HPF, necrosis (N) and endothelial proliferation (EP) were assessed (Figure 1). PXA cases were further classified as classic PXA (MI < 5/10 HPF, without N and EP) or PXA‐AF (MI ≥ 5/10 HPF and/or N and/or EP). Cases in which slides were unavailable for re‐review were included if the histologic description of the pathology report reflected the aforementioned diagnostic criteria.
Figure 1.
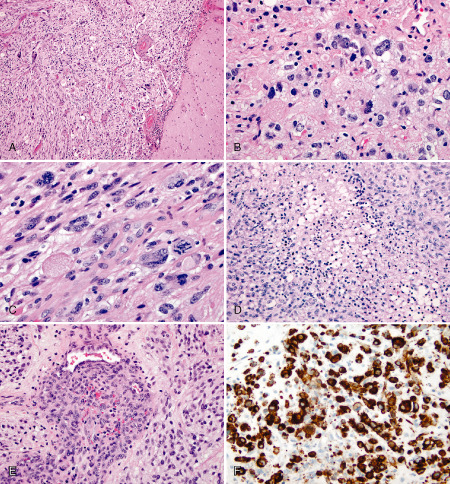
A,B. Characteristic histological features of PXA. A. Leptomeningeal involvement by pleomorphic, giant and spindle‐shaped tumor cells. H&E, 100×. B. Pale and intensively eosinophilic granular bodies with occasional xanthic tumor cells. H&E, 400×. C–E. Histological features of anaplasia. C. Mitotic index ≥ 5/10 HPF, including atypical mitotic figures. H&E. 400×. D. Necrosis and E. endothelial proliferation. H&E, 200×. F. BRAF V600E immunostain: strong labeling of tumor cells. 200×.
A total of 74 cases (76%) were selected and consisted of 54 (of 68) cases from Mayo Clinic, including 14 cases from our original series 24 and 20 (of 30) cases from Johns Hopkins University. Twenty‐four (of 98) cases were excluded: nine cases reclassified as “astrocytoma with pleomorphic/PXA‐like features” felt not to fulfill the diagnostic criteria for PXA; 12 cases previously diagnosed as “suggestive of” or “consistent with” PXA in which the original slides were unavailable for review; one case diagnosed as “combined PXA‐oligodendroglioma,” which had been previously reported 51; one case with multiple brain tumors, some of which had been diagnosed as PXA, and likely associated with an uncharacterized genetic syndrome other than neurofibromatosis type 1; and one case that was part of the original series in which the initial pediatric patient withdrew research consent at age 18 and could not be reached to discuss the possibility of a new research consent. Of the 74 selected patients, 28 cases had ≥2 resections, totaling 116 tumors. Slides were available for review in 95 (of 116) tumors from 72 (of 74) patients and included the first resection specimen in 65 cases.
Clinical and therapeutic data as well as follow‐up information were obtained from review of the medical records. Additional treatment generally followed recurrence and consisted of radiotherapy, chemotherapy, radiosurgery or a combination. In an attempt to update status for patients lost to follow‐up as of 2011 or earlier (n = 34), Accurint database (http://www.accurint.com/) was used to determine their current vital status, and contact letters were sent to a subset of patients from the Mayo Clinic (n = 23). Updated vital status information was obtained in all 34 cases. Twelve (of 23) patients that were contacted by letter responded, and updated follow‐up information was collected in 8 (of 12) cases.
BRAF V600E and IDH1 R132H mutation analyses
Four‐micron sections of formalin‐fixed, paraffin‐embedded (FFPE) tissue were available in 60 (of 74) cases (81%) and were stained with mouse monoclonal BRAF V600E antibody (1/100 titer; clone VE1, Spring Bioscience, Pleasanton, CA, USA) and with IDH1‐R132H monoclonal antibody (1/100 titer; clone H09, Dianova Anti‐Human IDH1‐R132H antibody, cat. # DIA H09), following previously described protocols 30, 66. Positive control included a known BRAF V600E mutant skin malignant melanoma and a known IDH1 R132H mutant infiltrating glioma. BRAF V600E immunostain was scored as “positive” when tumor cells showed non‐ambiguous cytoplasmic staining, as “indeterminate” for faint, weak granular staining or as “negative” 30. For IDH1‐R132H immunostain, cases were scored as either “positive” or “negative.” Thirty‐nine of these 60 cases (65%) had sufficient (≥20%) viable tumor for BRAF V600E molecular genetic analysis. In a single case, BRAF V600E mutation analysis had been performed at the primary referring institution. The remaining 38 cases were tested by BRAF allele‐specific PCR with fragment analysis 30. A subset of the cases, including 46 (of 60) cases evaluated for BRAF V600E immunostain and 37 (of 39) cases evaluated for BRAFV600E mutation by molecular genetic testing had been reported in our previous study 30.
Statistical analysis
Characteristics of the patients were summarized with means or medians and standard deviations or ranges for continuous data (as appropriate) and with frequencies and percentages for categorical data. Recurrence‐free survival and overall survival were compared between patient groups (ie, PXA vs. PXA‐AF) with log‐rank tests and were summarized with 5‐year survival estimates along with 95% confidence intervals using the Kaplan–Meier method. The survival analyses were performed in the full group as well as stratified by age in pediatric (≤18 years) and adult (>18) groups. Further analysis was undertaken to examine whether there was any difference between patients who had an “early event” (recurrence or death within ≤3 years from diagnosis) vs. those who did not have an early event. This analysis included all patients who had at least 5 years of follow‐up available or who may have died within the first 5 years (so that early recurrence resulting into death would not be missed). Patient characteristics were then compared between those “with” vs. those “without an early event” using chi‐square or Fisher's exact tests, as appropriate. All analyses were performed using SAS version 9 (Cary, NC, USA) or R (www.r‐project.org). P‐values < 0.05 were considered statistically significant. Because of the rare nature of PXA, no adjustments for multiple statistical testing were performed, and all P‐values presented are unadjusted so the reader may interpret at his or her own discretion.
Results
Clinicopathologic features
Clinical features are detailed in Table 1. Patients included 40 men and 34 women, with median age at diagnosis of 21.5 years (range 5–73). Of these, 31 patients were ≤18 years old (pediatric), and 43 patients were >18 years old (adult). Tumors were predominantly supratentorial (97%), with 43% of cases involving the temporal lobe (35% exclusively the temporal lobe, 8% >1 lobe including the temporal lobe). Radiologically, most tumors had contrast enhancement (94%), and cystic changes were present in 39% of cases. Seizures were the most frequent symptom at presentation (64%). Gross total resection (GTR) was achieved in 40 cases (57%), subtotal resection (STR) in 29 cases (41%), biopsy only in one case, and the type of surgery was unknown in four cases.
Table 1.
Clinical features
| Clinical features | ≤18 years | >18 years | All |
|---|---|---|---|
| Total number of cases | 31 (42%) | 43 (58%) | 74 |
| Male, n (%) | 13 (42%) | 27 (63%) | 40 (54%) |
| Age at diagnosis (years), median (range) | 14 (5–18) | 35 (19–73) | 21.5 (5–73) |
| Location, n (%) | |||
| Supratentorial | 31 (100%) | 41 (95.4%) | 72 (97%) |
| Temporal lobe | 9 (29.1%) | 17 (39.4%) | 26 (35%) |
| >1 lobe, including temporal | 3 (9.6%) | 3 (7%) | 6 (8%) |
| Other | 19 (61.3) | 21 (49%) | 40 (51%) |
| Infratentorial | 0 | 2 (4.6%) | 2 (3%) |
| Radiology, n (%) | |||
| Contrast enhancement | — | — | — |
| Present | 25 (93%) | 34 (94%) | 59 (94%) |
| Absent | 2 (7%) | 2 (6%) | 4 (6%) |
| Unknown | 4 | 7 | 11 |
| Cystic changes | — | — | — |
| Present | 12 (44%) | 13 (35%) | 25 (39%) |
| Absent | 15 (56%) | 24 (65%) | 39 (61%) |
| Unknown | 4 | 6 | 10 |
| Presenting symptoms, n (%) | |||
| Seizures only | 16 (57.1%) | 21 (51.2%) | 37 (54%) |
| >1 symptom, including seizures | 2 (7.2%) | 5 (12%) | 7 (10%) |
| Other | 10 (35.7%) | 15 (36.8%) | 25 (36%) |
| Unknown | 3 | 2 | 5 |
| Extent of surgery, n (%) | |||
| Gross total resection (GTR) | 18 (60%) | 22 (55%) | 40 (57%) |
| Subtotal resection (STR) | 11 (37%) | 18 (45%) | 29 (41%) |
| Biopsy only (BX) | 1 (3%) | 0 (0%) | 1 (1%) |
| Unknown | 1 | 3 | 4 |
At first diagnosis, 51 patients (69%) were diagnosed with PXA (WHO grade II) and 23 patients (31%) with PXA‐AF. Histologic features of anaplasia were present in 33 cases: at first diagnosis in 23 and upon recurrence in the remaining 10. Of these 33 cases, slides were available for review in 31 cases. Histological features of anaplasia were distributed as follows: MI ≥ 5/10 HPF in eight cases, N in two cases, MI ≥ 5/10 HPF + N in 11 cases and MI ≥ 5/10 HPF + N + EP in 10 cases (Table 2).
Table 2.
Histological features of anaplasia (PXA‐AF)
| All | At first diagnosis | At recurrence | |
|---|---|---|---|
| Total number of PXA‐AF cases | 33 | 23 | 10 |
| MI ≥ MI ≥ 5 | 8 | 5 | 3 |
| MI ≥ N | 2 | 2 | 0 |
| MI ≥ MI ≥ 5 + N | 13 | 8 | 5 |
| MI ≥ MI ≥ 5 + N + EP | 8 | 6 | 2 |
| SlideSlides unavailable | 2 | 2 | 0 |
EP = endothelial proliferation; MI = mitotic index; N = necrosis; PXA‐AF = pleomorphic xanthoastrocytoma with anaplastic features.
BRAF V600E and IDH1 R132H mutation status
All 60 cases with tissue available were evaluable by immunohistochemistry for BRAF V600E and IDH1 R132H mutations. BRAF V600E mutation molecular genetic testing was successful for all 39 cases with tissue available tested, including a single case tested at the primary referring institution.
BRAF V600E and IDH1 R132H mutation status results are shown in Table 3. Thirty‐nine (of 60) cases were positive for BRAF V600E immunostain (30 PXA; 9 PXA‐AF); 5 (of 60) cases (1 PXA and 4 PXA‐AF) were scored as “indeterminate”; and 16 (of 60) cases (10 PXA; 6 PXA‐AF) were negative. Of the cases tested for BRAF V600E mutation, 24 cases were mutant (18 PXA, 6 PXA‐AF), and 15 cases were nonmutant (8 PXA; 7 PXA‐AF). There was complete agreement between BRAFV600E mutation detection by immunohistochemistry and by molecular analysis, and 4 (of 5) cases scored as “indeterminate” for BRAF V600E immunostain lacked BRAF V600E mutation by molecular testing. The remaining fifth case did not have sufficient tissue for molecular analysis. Combining immunohistochemical and molecular genetics results demonstrated 39 BRAF V600E mutant (65%), 20 BRAF V600E nonmutant (33%) and one “indeterminate” for BRAF V600E mutation (2%). All 60 (100%) cases were negative for mutant IDH1 (IDH1‐R132H) immunostain.
Table 3.
BRAF V600E mutation status
| BRAF V600E mutation | First diagnosis | |
|---|---|---|
| PXA (n = 51) | PXA‐AF (n = 23) | |
| Final status | ||
| Mutant | 30 (73.2%) | 9 (47.4%) |
| Nonmutant | 10 (24.4%) | 10 (52.6%) |
| Indeterminate | 1 (2.4%) | 0 |
| NA | 10 | 4 |
| By immunohistochemistry | ||
| Positive | 30 (73.2%) | 9 (47.4%) |
| Negative | 10 (24.4%) | 6 (31.6%) |
| Indeterminate | 1 (2.4%) | 4 (21.1%) |
| NA | 10 | 4 |
| By molecular genetics | ||
| Mutant | 18 (69.2%) | 6 (46.2%) |
| Nonmutant | 8 (30.8%) | 7 (53.8%) |
| NA | 25 | 10 |
Survival analyses
Clinical follow‐up
Follow‐up data is detailed in Table 4. After first resection, 35 patients (17 PXA; 18 PXA‐AF) received postoperative therapy, which generally followed recurrence or progression and consisted of radiotherapy (RT) only (5 PXA and 3 PXA‐AF), radiosurgery (RS) only (1 PXA), chemotherapy +/− RT/RS (11 PXA and 14 PXA‐AF) and RT + RS (1 PXA‐AF). A single patient with recurrent PXA‐AF that harbored BRAF V600E mutation received BRAF V600E inhibitor vemurafenib for 3 days; therapy was discontinued because of side effects (rash and arthralgia). No other patient in this series was given BRAF V600E‐targeted therapy. Median follow‐up was 7.6 years (range 30 days to 31.9 years) for the entire group, 11.2 years (206 days–31.9 years) and 4.0 years (30 days–22.9 years) for PXA and PXA‐AF, respectively. Twenty‐one (of 51) PXA patients developed tumor recurrence; 11 cases recurred as classic PXA and 10 cases progressed to PXA‐AF at the time of first (n = 9) or subsequent (n = 1) recurrence. Among PXA‐AF patients, 11 (of 23) developed tumor recurrence: the recurrent tumor was histologically consistent with PXA‐AF in nine cases; in the remaining two cases, the recurrent tumor did not show definite histologic features of anaplasia, likely because of sampling and/or tumor heterogeneity. These 11 cases were grouped together for analysis purposes. The recurrence‐free 5‐year survival rate was 64.6% (95% CI: 53.1%–76.0%) for the entire (PXA + PXA‐AF) group. Recurrence‐free survival was not significantly longer for PXA patients in comparison with PXA‐AF patients (5‐year estimates 70.9% vs. 48.9%; P = 0.09; Figure 2). A total of 18 patients died, and the majority (n = 14) were known to have died of disease. Of 51 PXA patients, eight died (six died of disease, four of which had progressed from PXA to PXA‐AF). Of 23 PXA‐AF patients, 10 died (eight died of disease). For the remaining four patients, cause of death was car accident in one (unknown if the patient was a driver or a passenger) and unknown in three. The overall 5‐year survival rate was 80.6% (95% CI: 70.7%–90.5%) for the whole group of PXA + AF patients. The overall survival was significantly longer for PXA patients when compared with PXA‐AF patients (5‐year estimates 90.4% vs. 57.1%; P = 0.0003; Figure 2).
Table 4.
Additional therapy and follow‐up
| First diagnosis | |||
|---|---|---|---|
| PXA (n = 51) | PXA‐AF (n = 23) | ||
| Postoperative therapy, n (%) | — | — | — |
| Any type | 17 (33.3%) | 18 (78.3%) | — |
| Radiotherapy (RT) only | 5 (9.8%) | 3 (13.0%) | — |
| Radiosurgery (RS) only | 1 (2.0%) | 0 | — |
| Chemotherapy +/− RT/RS | 11 (21.6%) | 14 (60.9%) | — |
| RT + RS | 0 | 1 (4.3%) | — |
| Median follow‐up, years (range) | 11.2 (0.6–31.9) | 4.0 (0.1–22.9) | — |
| Recurrence, n | 21 | 11 | — |
| Outcome, n | — | — | — |
| Alive | 20 | 6 | (0.1–25.4 years)* |
| Alive with disease | 7 | 3 | (0.8–20.0 years)* |
| Died | 2 | 2 | (4.0–20.5 years)* |
| Died of disease | 6 | 8 | (0.8–27.7 years)* |
| No evidence of disease | 16 | 4 | (0.6–31.9 years)* |
| 5‐year survival % (95% CI) | — | — | — |
| Recurrence‐free | 70.9% (58.0–83.8) | 48.9% (26.5–71.3) | P = 0.092 |
| Overall | 90.4% (81.5–99.4) | 57.1% (34.2–80.0) | P = 0.0003 |
*Total available follow‐up.
PXA = pleomorphic xanthoastrocytoma; PXA‐AF = pleomorphic xanthoastrocytoma with anaplastic features.
Figure 2.
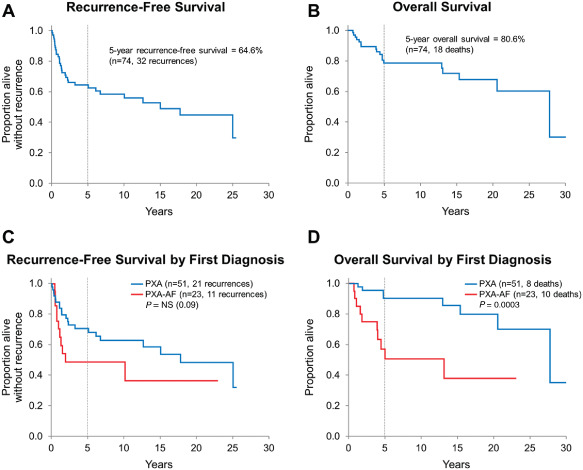
A,B. Recurrence‐free and overall survival for the entire PXA group. C,D. Recurrence‐free and overall survival stratified by diagnosis at first resection: recurrence‐free survival was slightly longer for patients with PXA when compared with patients with PXA‐AF (P = 0.09). PXA‐AF patients showed a significant decreased overall survival when compared with patients with PXA (P = 0.0003).
Survival in adult and pediatric patients
Survival was further evaluated in regard to age group (Figure 3), surgery type, histologic features of anaplasia and BRAF V600E mutation status. Recurrence occurred in 15 (of 31) pediatric patients and in 17 (of 43) adult patients, with similar recurrence‐free survival between the age groups (5‐year estimates 67.9% vs. 62.4%, respectively; P = 0.39). Similarly, overall survival was not significantly different between pediatric and adult groups (5‐year estimates 87.4% vs. 76.3%; P = 0.83). When stratified by diagnosis at first resection (PXA vs. PXA‐AF), overall survival was longer for PXA patients as compared with PXA‐AF patients within both pediatric and adult groups (P = 0.04 and P = 0.003, respectively). Recurrence‐free survival, however, was significantly longer only for adult patients with PXA when compared with adult patients with PXA‐AF (P = 0.047) but not for pediatric patients (P = 0.87; Figure 4).
Figure 3.
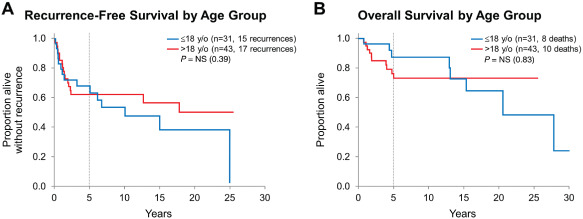
A,B. Recurrence‐free and overall survival by age group: similar recurrence‐free and overall survival for PXA pediatric vs. adult patients.
Figure 4.
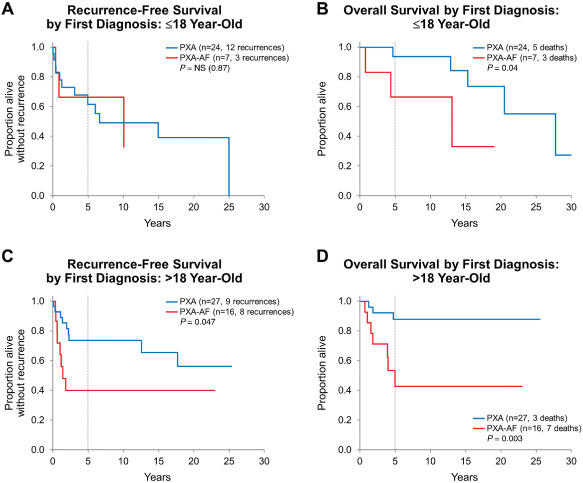
A,B. Recurrence‐free and overall survival stratified by diagnosis at first resection in the pediatric group: although recurrence‐free survival was similar between classic PXA and PXA‐AF patients, overall survival of patients with PXA‐AF was significantly shorter as compared with patients with PXA. C,D. Recurrence‐free and overall survival stratified by diagnosis at first resection in the adult group: PXA‐AF patients showed significantly decreased recurrence‐free and overall survival when compared with adult patients with PXA.
Survival by extent of resection
GTR was significantly associated with longer recurrence‐free survival when compared with STR/BX (5‐year estimates 84.9% vs. 45.4%; P = 0.0002) but not with overall survival. A similar finding was noted among pediatric patients, with 5‐year recurrence‐free survival rates of 92.3% and 41.7% (P = 0.0002) for GTR and STR/BX, respectively. Among adult patients, although recurrence‐free survival for those with GTR was slightly longer as compared with STR/BX (5‐year estimates 79.7% vs. 47.6%), this was not statistically significant (P = 0.10).
Survival by anaplasia
Among histological features of anaplasia, which are highly correlated with the diagnosis of PXA‐AF, MI ≥ 5/10 HPF and necrosis were significantly associated with decreased overall survival (Figure 5) but not with recurrence‐free survival in the overall group. In regard to recurrence‐free survival rate, it was slightly decreased for patients whose tumors had MI ≥ 5/10 HPF when compared with patients whose tumors showed MI < 5/10HPF. This association was statistically significant in the adult group (5‐year estimates 43.3% vs. 74.4%, P = 0.04) but not in the pediatric group (75.0% vs. 73.5%, P = 0.60) or in the overall group (51.6% vs. 73.7%; P = 0.06). Recurrence‐free survival was significantly higher among adult patients whose tumors did not have necrosis as compared with those with necrosis (5‐year estimates 70.8% vs. 41.1%, P = 0.03) but not among pediatric patients (73.5% vs. 75.0%, P = 0.69) and in the overall group (71.5% vs. 53.0%, P = 0.19). Overall survival for patients whose tumor had MI < 5/10HPF was better (5‐year estimate 89.4%) as compared with those whose tumor showed MI ≥ 5/10 HPF (55.6%, P = 0.0005), and the findings were similar among the pediatric (92.9% vs. 50.0%, P = 0.003) and adult groups (86.8% vs. 59.3%, P = 0.01). Overall survival for patients without tumor necrosis was better (5‐year estimate 90.2%) in contrast to patients whose tumor had necrosis (42.2%, P = 0.0002), and this finding was similar among pediatric (92.9% vs. 50.0%, P = 0.01) and adult patients (88.6% vs. 41.7%, P = 0.002). Presence of endothelial proliferation was not significantly associated with either overall or recurrence‐free survival (P = 0.21 and P = 0.12, respectively). Data was insufficient to detect a difference in survival or recurrence between PXA‐AF patients with MI ≥ 5 and necrosis (n = 14; seven died, six recurred), and those with MI ≥ 5 only (n = 5; two died, three recurred). Only two patients had necrosis but not increased mitotic activity; both patients are alive, one with limited follow‐up (1 month) and the other without evidence of disease at 10 years.
Figure 5.
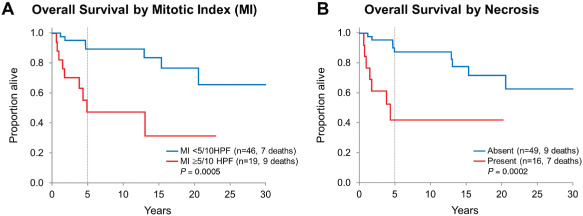
Overall survival for the entire PXA group stratified by (A) mitotic index (MI) and (B) tumor necrosis: MI ≥ 5/10HPF and presence of tumor necrosis are significantly associated with decreased overall survival.
Survival by BRAF V600E mutation
Patients with BRAF V600E mutant tumors had significantly longer overall survival when compared with those with BRAF V600E nonmutant tumors (P = 0.02; Figure 6). Adjusted multivariate analyses to explore this further were considered as the frequency of BRAF V600E mutation is higher among PXA patients (n = 30/40, 75.0%) than in PXA‐AF patients (n = 9/19, 47.4%). However, this could not be performed because of the small number of overall deaths and especially because of the even smaller number of cases with BRAF V600E mutation status data available. BRAF V600E mutation status was not significantly different between pediatric and adult groups: BRAF V600E mutation was present in 17 (of 26) pediatric cases and in 22 (of 33) adult cases (P = 0.92).
Figure 6.
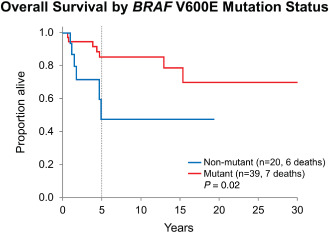
Overall survival for the entire PXA group stratified by BRAF V600E mutation status: patients with BRAF V600E mutant tumors had significantly longer overall survival when compared with those with BRAF V600E nonmutant tumors (P = 0.02). Adjusted multivariate analyses because of the higher frequency of BRAF V600E mutation among PXA patients in comparison with PXA‐AF patients could not be performed.
Survival by early vs. late events
Further analysis was undertaken to clarify whether there was any difference between patients with an “early event” (recurrence or death within ≤3 years from diagnosis) vs. those who did not have an early event as detailed in Table 5. This analysis included all patients (n = 56) who had at least 5 years of available follow‐up or who may have died within the first 5 years (so that early recurrence resulting into death would not be missed). Of these 56 patients, 21 cases (38%) had an early event (10 PXA and 11 PXA‐AF) and either died of disease (n = 7, five of which died following recurrence) or recurred (n = 14, all were still alive at 3 years); the remaining 35 cases (62%) had no early event within the first 3 years (29 PXA and 6 PXA‐AF). Of note, patients with an early event were more likely to have had PXA‐AF at first diagnosis as compared with those without an early event (11 of 21, 52.4% vs. 6 of 35, 17.1%; P = 0.008). In addition, patients with an early event were more likely to have undergone STR/BX and to have tumors with MI ≥ 5/10HPF when compared with the ones without an early event (P = 0.004 and P = 0.02, respectively). Postoperative therapy was more frequent amongst patients with early event, indicating that patients who received adjuvant treatment had clinically aggressive tumors (proxy for severity): radiotherapy was given to 57.1% (12 of 21) patients with an early event as opposed to 17.1% (6 of 35) patients without early event (P = 0.003); chemotherapy was received by 61.9% (13 of 21) patients with an early event in comparison with 22.9% (8 of 35) patients without early event (P = 0.005). Gender, age group, presence of necrosis or endothelial proliferation (not shown), and BRAF V600E mutation status were not statistically different between the two groups.
Table 5.
Analysis of “early event” (recurrence or death within ≤3 years from diagnosis) vs. no early event among those with at least 5 years of follow‐up available
| “Early event”* (n = 21) | No “early event” (n = 35) | P‐value | |
|---|---|---|---|
| First diagnosis | — | — | 0.008 |
| PXA | 10 (47.6%) | 29 (82.9%) | — |
| PX‐AF | 11 (52.4%) | 6 (17.1%) | — |
| Gender | — | — | 0.78 |
| Female | 8 (38.1%) | 16 (45.7%) | — |
| Male | 13 (61.9%) | 19 (54.3%) | — |
| Age | — | — | 0.26 |
| Pediatric (≤18) | 6 (28.6%) | 16 (45.7%) | — |
| Adult (>18) | 15 (71.4%) | 19 (54.3%) | — |
| Surgery type | — | — | 0.004 |
| N/A | 2 | 1 | — |
| Gross total resection (GTR) | 5 (26.3%) | 24 (70.6%) | — |
| Subtotal resection (STR)/biopsy only (BX) | 14 (73.7%) | 10 (29.4%) | — |
| Mitotic index (MI) | — | — | 0.02 |
| N/A | 3 | 4 | — |
| MI < 5/10HPF | 9 (50.0%) | 26 (83.9%) | — |
| MI ≥ 5/10 HPF | 9 (50.0%) | 5 (16.1%) | — |
| BRAF V600E mutation status | — | — | 0.07 |
| N/A | 3 | 11 | — |
| Nonmutant | 7 (38.9%) | 3 (12.5%) | — |
| Mutant | 11 (61.1%) | 21 (87.5%) | — |
| Radiotherapy | — | — | 0.003 |
| No | 9 (42.9%) | 29 (82.9%) | — |
| Yes | 12 (57.1%) | 6 (17.1%) | — |
| Chemotherapy | — | — | 0.005 |
| No | 8 (38.1%) | 27 (77.1%) | — |
| Yes | 13 (61.9%) | 8 (22.9%) | — |
*Defined as presence of recurrence or death of disease within first 3 years of diagnosis.
MI = mitotic index; N/A = not available; PXA = pleomorphic xanthoastrocytoma; PXA‐AF = pleomorphic xanthoastrocytoma with anaplastic features.
Discussion
The results of this extended follow‐up study of patients with PXA largely parallel the results of the original study 24, confirming GTR as the most important predictor of time to recurrence but not of overall survival. The present study also confirms MI ≥ 5/10 HPF as a significant predictor of overall survival and, most importantly, that overall survival is significantly decreased in PXA‐AF patients when compared with PXA patients, both in children and adults. The presence of necrosis is also a significant predictor of overall survival. Data was insufficient to detect a difference in survival or recurrence between PXA‐AF patients with MI ≥ 5 and necrosis and those with MI ≥ 5 only. In only two cases did the tumor show necrosis in the absence of increased mitotic activity, a number too small to evaluate its independent significance.
Taking into consideration the definition of WHO grade across CNS tumor entities 76, our study confirms that classic PXA truly behaves as a WHO grade II tumor, with a strong intrinsic tendency to recur, especially if incompletely excised. Significantly shorter overall survival of PXA‐AF patients in comparison with PXA patients supports the view that PXA‐AF tumors are definitively more aggressive than PXA. Therefore, it seems reasonable that PXA‐AF should be assigned a corresponding grade to reflect this difference in clinical behavior and inform appropriate therapeutic management.
Currently, although an official WHO grade has not been attributed to PXA‐AF tumors, they have been graded as either grade III 20, 38 or IV 50. Although the high‐grade component of PXA‐AF may be morphologically reminiscent of and has been reported as a “glioblastoma” 14, 41, 49, “small cell glioblastoma” 34 or “giant cell glioblastoma” 13, we believe that PXA‐AF should not be labeled or graded as a WHO grade IV tumor, as the overall clinical course of PXA‐AF does not parallel the behavior of glioblastoma.
Therefore, does PXA‐AF qualify for a WHO grade III tumor? Clearly, PXA‐AF is associated with histologic features of malignancy, frequently has an aggressive clinical behavior and, because of this, is frequently treated with adjuvant therapies. Outcome of these patients, however, is quite variable with some patients having early recurrence or death and others in whom these outcomes are quite delayed. The WHO grading system is based on a unifying classification criterion for tumors with distinct morphology and underlying molecular alterations. Within WHO grade III tumors, there are tumors with different natural history and survival (eg, anaplastic ependymoma, anaplastic astrocytoma, anaplastic oligodendroglioma and anaplastic meningioma). Therefore, it seems reasonable to consider that PXA‐AF would somewhat fit within this heterogeneous “anaplastic” WHO grade III group.
Conflicting reports suggest that PXA‐AF is more likely to occur primarily or to recur mostly in adults 13, 46, 58, 64. Among the 23 patients with PXA‐AF at first diagnosis in our series, adult patients were overrepresented in relation to pediatric patients (16 vs. 7), but neither recurrence‐free nor overall survival significantly differed between these age groups.
We also evaluated the presence and significance of two potential diagnostically relevant molecular genetic alterations: BRAF V600E and IDH1 R132H mutations. Our data confirm the high frequency of BRAF V600E mutation in PXA. Diffusely infiltrating gliomas represent an important morphologic diagnostic differential diagnosis of PXA and have been shown to frequently harbor the IDH1 R132H mutation 71. In our current series, 65% of the cases demonstrated BRAF V600E mutation by immunohistochemistry and/or molecular genetics testing, whereas all cases were negative for IDH1 R132H mutation by immunohistochemistry. Therefore, the finding of a BRAF V600E‐positive IDH1‐R132H‐negative immunohistochemical profile for a morphologically pleomorphic astrocytoma in a clinical setting of a young patient with a contrast‐enhancing tumor would suggest the diagnosis of a PXA. In contrast with other studies suggesting that the frequency of BRAF V600E mutation in PXA‐AF is considerably lower in adults (17%–50%) 13, 57, 58 than in the pediatric population (100%) 57, frequency of BRAF V600E mutation did not differ significantly between pediatric and adult patients in our series. We found evidence that patients with BRAF V600E mutant tumors had significantly longer overall survival when compared with those with BRAF V600E nonmutant tumors (P = 0.02). However, the frequency of BRAF V600E mutation is higher among PXA patients than in PXA‐AF patients. Adjusted multivariate analyses to clarify the real significance of this association could not be performed.
Finally, our findings support the view that additional nonsurgical therapies may be a consideration when treating patients with PXA. The role of adjuvant treatment for this tumor type, however, is not well established in the literature, and both chemotherapy and radiotherapy have been used mainly for PXA‐AF tumors with reports of good response 2, 8, 14. Recently, a small retrospective series reported adult patients with BRAF V600E mutant recurrent PXA that were radiation‐ and alkylator‐refractory who were given salvage therapy using BRAF V600E inhibitor vemurafenib. Of the four patients evaluated, one had partial response, two had stable disease and one showed tumor progression; the authors concluded that vemurafenib appeared to show a single‐agent activity with manageable toxicity but noted that confirmation in a larger series of similar patients was warranted 7. A single case of successful treatment of a progressive BRAF V600E‐mutated anaplastic pleomorphic xanthoastrocytoma with vemurafenib monotherapy has just been reported 37. Marucci et al identified MGMT promoter methylation in 18% (2 of 11) of PXA (9 PXA and 2 PXA‐AF), and all MGMT promoter methylated tumors were histologically WHO grade II 43. Although based in a very small number of cases, the authors discussed the limited role of temozolomide treatment for PXA‐AF, which only infrequently harbors MGMT promoter methylation. Of note, primary cell cultures obtained from a recurrent PXA‐AF were treated with five chemotherapeutic drugs, of which, temozolomide was the most effective in reducing tumor cell viability in vitro 4.
In summary, this study expands our knowledge on the natural history and molecular features of PXA and provides additional evidence that PXA‐AF, as defined by MI ≥ 5/10 HPF with or without necrosis, behaves more aggressively than PXA and, accordingly, may qualify for a WHO grade III “anaplastic” designation. Further studies to determine optimal therapeutic strategies for PXA‐AF patients are certainly needed, and establishing an official WHO grade may be the very first step toward this goal.
Acknowledgments
Dedicated to the memory of Dr. Bernd W. Scheithauer, under whose guidance, the first PXA project was developed. This subject was very close to Bernd's heart and, to him, we would like to dedicate this paper.
References
- 1. Aisner DL, Newell KL, Pollack AG, Kleinschmidt‐Demasters BK, Steinberg GK, Smyth LT et al (2014) Composite pleomorphic xanthoastrocytoma‐epithelioid glioneuronal tumor with BRAF V600E mutation—report of three cases. Clin Neuropathol 33:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexiou GA, Moschovi M, Stefanaki K, Prodromou C, Sfakianos G, Prodromou N (2010) Malignant progression of a pleomorphic xanthoastrocytoma in a child. Neuropediatrics 41:69–71. [DOI] [PubMed] [Google Scholar]
- 3. Arita K, Kurisu K, Tominaga A, Sugiyama K, Sumida M, Hirose T (2002) Intrasellar pleomorphic xanthoastrocytoma: case report. Neurosurgery 51:1079–1082. discussion 82. [DOI] [PubMed] [Google Scholar]
- 4. Bagriacik EU, Baykaner MK, Yaman M, Sivrikaya G, Durdag E, Emmez H et al (2012) Establishment of a primary pleomorphic xanthoastrocytoma cell line: in vitro responsiveness to some chemotherapeutics. Neurosurgery 70:188–197. [DOI] [PubMed] [Google Scholar]
- 5. Bettegowda C, Agrawal N, Jiao Y, Wang Y, Wood LD, Rodriguez FJ et al (2013) Exomic sequencing of four rare central nervous system tumor types. Oncotarget 4:572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chacko G, Chacko AG, Dunham CP, Judkins AR, Biegel JA, Perry A (2007) Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol 84:217–222. [DOI] [PubMed] [Google Scholar]
- 7. Chamberlain MC (2013) Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neurooncol 114:237–240. [DOI] [PubMed] [Google Scholar]
- 8. Chang HT, Latorre JG, Hahn S, Dubowy R, Schelper RL (2006) Pediatric cerebellar pleomorphic xanthoastrocytoma with anaplastic features: a case of long‐term survival after multimodality therapy. Childs Nerv Syst 22:609–613. [DOI] [PubMed] [Google Scholar]
- 9. Chapman EM, Ranger A, Lee DH, Hammond RR (2009) A 15 year old boy with a posterior fossa tumor. Brain Pathol 19:349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chappe C, Padovani L, Scavarda D, Forest F, Nanni‐Metellus I, Loundou A et al (2013) Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol 23:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das S, Yip S, Hukin J, Cochrane D, Dunham C (2014) Pleomorphic xanthoastrocytoma of the spinal cord: case report and literature review. Clin Neuropathol 33:190–196. [DOI] [PubMed] [Google Scholar]
- 12. Delgado‐Alvarado M, Gomez‐Roman J, Sanchez‐Salmon E, Rodriguez‐Rodriguez E, Polo JM, Garcia‐Castano A et al (2014) Nonanaplastic pleomorphic xanthoastrocytoma with meningeal dissemination presenting with bilateral visual loss. J Neuroimaging 24:533–535. [DOI] [PubMed] [Google Scholar]
- 13. Dias‐Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR et al (2011) BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS ONE 6:e17948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dickerman RD (2006) Malignant transformation of a pleomorphic xanthoastrocytoma. Acta Neurochir (Wien) 148:98. [DOI] [PubMed] [Google Scholar]
- 15. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ et al (2010) Activating mutations in BRAF characterize a spectrum of pediatric low‐grade gliomas. Neuro‐Oncol 12:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evans AJ, Fayaz I, Cusimano MD, Laperriere N, Bilbao JM (2000) Combined pleomorphic xanthoastrocytoma‐ganglioglioma of the cerebellum. Arch Pathol Lab Med 124:1707–1709. [DOI] [PubMed] [Google Scholar]
- 17. Fouladi M, Jenkins J, Burger P, Langston J, Merchant T, Heideman R et al (2001) Pleomorphic xanthoastrocytoma: favorable outcome after complete surgical resection. Neuro‐Oncol 3:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frank S, Cordier D, Tolnay M, Rosenblum MK (2009) A 28‐year‐old man with headache, visual and aphasic speech disturbances. Brain Pathol 19:163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu YJ, Miyahara H, Uzuka T, Natsumeda M, Okamoto K, Hirose T et al (2010) Intraventricular pleomorphic xanthoastrocytoma with anaplastic features. Neuropathology 30:443–448. [DOI] [PubMed] [Google Scholar]
- 20. Gallo P, Cecchi PC, Locatelli F, Rizzo P, Ghimenton C, Gerosa M et al (2013) Pleomorphic xanthoastrocytoma: long‐term results of surgical treatment and analysis of prognostic factors. Br J Neurosurg 27:759–764. [DOI] [PubMed] [Google Scholar]
- 21. Gardiman MP, Fassan M, Orvieto E, Iaria L, Calderone M, Mardari R et al (2012) A 14‐year‐old girl with multiple tumors. Brain Pathol 22:865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giannini C, Hebrink D, Scheithauer BW, Dei Tos AP, James CD (2001) Analysis of p53 mutation and expression in pleomorphic xanthoastrocytoma. Neurogenetics 3:159–162. [DOI] [PubMed] [Google Scholar]
- 23. Giannini C, Paulus W, Louis DN, Liberski PP (2007) Pleomorphic xanthoastrocytoma. In: The World Health Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler DO, Cavenee WK (eds), Chapter 1, pp. 22–24. IARC: Lyon. [Google Scholar]
- 24. Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B et al (1999) Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer 85:2033–2045. [PubMed] [Google Scholar]
- 25. Giannini C, Scheithauer BW, Lopes MB, Hirose T, Kros JM, VandenBerg SR (2002) Immunophenotype of pleomorphic xanthoastrocytoma. Am J Surg Pathol 26:479–485. [DOI] [PubMed] [Google Scholar]
- 26. Grau E, Balaguer J, Canete A, Martinez F, Orellana C, Oltra S et al (2009) Subtelomeric analysis of pediatric astrocytoma: subchromosomal instability is a distinctive feature of pleomorphic xanthoastrocytoma. J Neurooncol 93:175–182. [DOI] [PubMed] [Google Scholar]
- 27. Hamlat A, Le Strat A, Guegan Y, Ben‐Hassel M, Saikali S (2007) Cerebellar pleomorphic xanthoastrocytoma: case report and literature review. Surg Neurol 68:89–94. discussion 5. [DOI] [PubMed] [Google Scholar]
- 28. Hattab EM, Martin SE, Shapiro SA, Cheng L (2011) Pleomorphic xanthoastrocytoma and oligodendroglioma: collision of 2 morphologically and genetically distinct anaplastic components. J Neurosurg 114:1648–1653. [DOI] [PubMed] [Google Scholar]
- 29. Hirose T, Ishizawa K, Sugiyama K, Kageji T, Ueki K, Kannuki S (2008) Pleomorphic xanthoastrocytoma: a comparative pathological study between conventional and anaplastic types. Histopathology 52:183–193. [DOI] [PubMed] [Google Scholar]
- 30. Ida CM, Vrana JA, Rodriguez FJ, Jentoft ME, Caron AA, Jenkins SM et al (2013) Immunohistochemistry is highly sensitive and specific for detection of BRAF V600E mutation in pleomorphic xanthoastrocytoma. Acta Neuropathol Commun 1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ishizawa K, Terao S, Kobayashi K, Yoshida K, Hirose T (2007) A neuroepithelial tumor showing combined histological features of dysembryoplastic neuroepithelial tumor and pleomorphic xanthoastrocytoma—a case report and review of the literature. Clin Neuropathol 26:169–175. [DOI] [PubMed] [Google Scholar]
- 32. Kahramancetin N, Tihan T (2013) Aggressive behavior and anaplasia in pleomorphic xanthoastrocytoma: a plea for a revision of the current WHO classification. CNS Oncol 2:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaulich K, Blaschke B, Numann A, von Deimling A, Wiestler OD, Weber RG et al (2002) Genetic alterations commonly found in diffusely infiltrating cerebral gliomas are rare or absent in pleomorphic xanthoastrocytomas. J Neuropathol Exp Neurol 61:1092–1099. [DOI] [PubMed] [Google Scholar]
- 34. Kepes JJ, Rubinstein LJ, Ansbacher L, Schreiber DJ (1989) Histopathological features of recurrent pleomorphic xanthoastrocytomas: further corroboration of the glial nature of this neoplasm. A study of 3 cases. Acta Neuropathol 78:585–593. [DOI] [PubMed] [Google Scholar]
- 35. Kobayashi S, Hirakawa E, Haba R (1999) Squash cytology of pleomorphic xanthoastrocytoma mimicking glioblastoma. A case report. Acta Cytol 43:652–658. [DOI] [PubMed] [Google Scholar]
- 36. Koelsche C, Sahm F, Wohrer A, Jeibmann A, Schittenhelm J, Kohlhof P et al (2014) BRAF‐mutated pleomorphic xanthoastrocytoma is associated with temporal location, reticulin fiber deposition and CD34 expression. Brain Pathol 24:221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee EQ, Ruland S, LeBoeuf NR, Wen PY, Santagata S (2014) Successful treatment of a progressive BRAF V600E‐mutated anaplastic pleomorphic xanthoastrocytoma with vemurafenib monotherapy. J Clin Oncol. [DOI] [PubMed] [Google Scholar]
- 38. Lim S, Kim JH, Kim SA, Park ES, Ra YS, Kim CJ (2013) Prognostic factors and therapeutic outcomes in 22 patients with pleomorphic xanthoastrocytoma. J Korean Neurosurg Soc 53:281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lim SC, Jang SJ, Kim YS (1999) Cerebellar pleomorphic xanthoastrocytoma in an infant. Pathol Int 49:811–815. [DOI] [PubMed] [Google Scholar]
- 40. Lubansu A, Rorive S, David P, Sariban E, Seligmann R, Brotchi J et al (2004) Cerebral anaplastic pleomorphic xanthoastrocytoma with meningeal dissemination at first presentation. Childs Nerv Syst 20:119–122. [DOI] [PubMed] [Google Scholar]
- 41. Macaulay RJ, Jay V, Hoffman HJ, Becker LE (1993) Increased mitotic activity as a negative prognostic indicator in pleomorphic xanthoastrocytoma. Case report. J Neurosurg 79:761–768. [DOI] [PubMed] [Google Scholar]
- 42. Marton E, Feletti A, Orvieto E, Longatti P (2007) Malignant progression in pleomorphic xanthoastrocytoma: personal experience and review of the literature. J Neurol Sci 252:144–153. [DOI] [PubMed] [Google Scholar]
- 43. Marucci G, Morandi L (2011) Assessment of MGMT promoter methylation status in pleomorphic xanthoastrocytoma. J Neurooncol 105:397–400. [DOI] [PubMed] [Google Scholar]
- 44. Matsumoto K, Suzuki SO, Fukui M, Iwaki T (2004) Accumulation of MDM2 in pleomorphic xanthoastrocytomas. Pathol Int 54:387–391. [DOI] [PubMed] [Google Scholar]
- 45. Nakamura M, Chiba K, Matsumoto M, Ikeda E, Toyama Y (2006) Pleomorphic xanthoastrocytoma of the spinal cord. Case report. J Neurosurg Spine 5:72–75. [DOI] [PubMed] [Google Scholar]
- 46. Ng WH, Lim T, Yeo TT (2008) Pleomorphic xanthoastrocytoma in elderly patients may portend a poor prognosis. J Clin Neurosci 15:476–478. [DOI] [PubMed] [Google Scholar]
- 47. Oh T, Kaur G, Madden M, Bloch O, Parsa AT (2014) Pleomorphic xanthoastrocytomas: institutional experience of 18 patients. J Clin Neurosci 21:1767–1772. [DOI] [PubMed] [Google Scholar]
- 48. Okazaki T, Kageji T, Matsuzaki K, Horiguchi H, Hirose T, Watanabe H et al (2009) Primary anaplastic pleomorphic xanthoastrocytoma with widespread neuroaxis dissemination at diagnosis—a pediatric case report and review of the literature. J Neurooncol 94:431–437. [DOI] [PubMed] [Google Scholar]
- 49. Paulus W, Lisle DK, Tonn JC, Wolf HK, Roggendorf W, Reeves SA et al (1996) Molecular genetic alterations in pleomorphic xanthoastrocytoma. Acta Neuropathol 91:293–297. [DOI] [PubMed] [Google Scholar]
- 50. Perkins SM, Mitra N, Fei W, Shinohara ET (2012) Patterns of care and outcomes of patients with pleomorphic xanthoastrocytoma: a SEER analysis. J Neurooncol 110:99–104. [DOI] [PubMed] [Google Scholar]
- 51. Perry A, Scheithauer BW, Szczesniak DM, Atkinson JL, Wald JT, Hammak JE (2001) Combined oligodendroglioma/pleomorphic xanthoastrocytoma: a probable collision tumor: case report. Neurosurgery 48:1358–1361. [DOI] [PubMed] [Google Scholar]
- 52. Primavera J, Nikas DC, Zamani AA, Shafman T, Alexander E III, De Girolami U et al (2001) Clear cell pleomorphic xanthoastrocytoma: case report. Acta Neuropathol 102:404–408. [DOI] [PubMed] [Google Scholar]
- 53. Rao AA, Laack NN, Giannini C, Wetmore C (2010) Pleomorphic xanthoastrocytoma in children and adolescents. Pediatr Blood Cancer 55:290–294. [DOI] [PubMed] [Google Scholar]
- 54. Rogerio F, Queiroz Lde S, de Lima MS, Kaleff PR, Vargas AA (2008) Temporal pleomorphic xanthoastrocytoma with glycogen accumulation—case report. Clin Neuropathol 27:234–240. [DOI] [PubMed] [Google Scholar]
- 55. Rosemberg S, Rotta JM, Yassuda A, Velasco O, Leite CC (2000) Pleomorphic xanthoastrocytoma of the cerebellum. Clin Neuropathol 19:238–242. [PubMed] [Google Scholar]
- 56. Sawyer JR, Roloson GJ, Chadduck WM, Boop FA (1991) Cytogenetic findings in a pleomorphic xanthoastrocytoma. Cancer Genet Cytogenet 55:225–230. [DOI] [PubMed] [Google Scholar]
- 57. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 58. Schmidt Y, Kleinschmidt‐DeMasters BK, Aisner DL, Lillehei KO, Damek D (2013) Anaplastic PXA in adults: case series with clinicopathologic and molecular features. J Neurooncol 111:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sharma MC, Arora R, Khanna N, Singh VP, Sarkar C (2001) Pigmented pleomorphic xanthoastrocytoma: report of a rare case with review of the literature. Arch Pathol Lab Med 125:808–811. [DOI] [PubMed] [Google Scholar]
- 60. Srinivas BH, Uppin MS, Panigrahi MK, Vijaya Saradhi M, Jyotsna Rani Y, Challa S (2010) Pleomorphic xanthoastrocytoma of the pineal region. J Clin Neurosci 17:1439–1441. [DOI] [PubMed] [Google Scholar]
- 61. Sugita Y, Irie K, Ohshima K, Hitotsumatsu T, Sato O, Arimura K (2009) Pleomorphic xanthoastrocytoma as a component of a temporal lobe cystic ganglioglioma: a case report. Brain Tumor Pathol 26:31–36. [DOI] [PubMed] [Google Scholar]
- 62. Sugita Y, Shigemori M, Okamoto K, Morimatsu M, Arakawa M, Nakayama K (2000) Clinicopathological study of pleomorphic xanthoastrocytoma: correlation between histological features and prognosis. Pathol Int 50:703–708. [DOI] [PubMed] [Google Scholar]
- 63. Tan TC, Ho LC, Yu CP, Cheung FC (2004) Pleomorphic xanthoastrocytoma: report of two cases and review of the prognostic factors. J Clin Neurosci 11:203–207. [DOI] [PubMed] [Google Scholar]
- 64. Tekkok IH, Sav A (2004) Anaplastic pleomorphic xanthoastrocytomas. Review of the literature with reference to malignancy potential. Pediatr Neurosurg 40:171–181. [DOI] [PubMed] [Google Scholar]
- 65. Vajtai I, Stibal A, von Gunten M, Kappeler A, Vassella E, Frank S (2011) Glycogen‐rich pleomorphic xanthoastrocytoma with clear‐cell features: confirmatory report of a rare variant with implications for differential diagnosis. Pathol Res Pract 207:256–261. [DOI] [PubMed] [Google Scholar]
- 66. van den Bent MJ, Hartmann C, Preusser M, Strobel T, Dubbink HJ, Kros JM et al (2013) Interlaboratory comparison of IDH mutation detection. J Neurooncol 112:173–178. [DOI] [PubMed] [Google Scholar]
- 67. Vizcaino MA, Caccamo DV, Fox E, Rodriguez FJ (2014) Pleomorphic xanthoastrocytoma: report of two cases with unconventional clinical presentations. Clin Neuropathol 33:380–387. [DOI] [PubMed] [Google Scholar]
- 68. Vu TM, Liubinas SV, Gonzales M, Drummond KJ (2012) Malignant potential of pleomorphic xanthoastrocytoma. J Clin Neurosci 19:12–20. [DOI] [PubMed] [Google Scholar]
- 69. Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B et al (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26:1088–1097. [DOI] [PubMed] [Google Scholar]
- 70. Xiong J, Chu SG, Mao Y, Wang Y (2011) Pigmented pleomorphic xanthoastrocytoma: a rare variant and literature review. Neuropathology 31:88–92. [DOI] [PubMed] [Google Scholar]
- 71. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yeaney GA, O'Connor SM, Jankowitz BT, Hamilton RL (2009) A 16‐year‐old male with a cerebellar mass. Brain Pathol 19:167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yeh DJ, Hessler RB, Stevens EA, Lee MR (2003) Composite pleomorphic xanthoastrocytoma‐ganglioglioma presenting as a suprasellar mass: case report. Neurosurgery 52:1465–1468. discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 74. Yin XL, Hui AB, Liong EC, Ding M, Chang AR, Ng HK (2002) Genetic imbalances in pleomorphic xanthoastrocytoma detected by comparative genomic hybridization and literature review. Cancer Genet Cytogenet 132:14–19. [DOI] [PubMed] [Google Scholar]
- 75. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B et al (2013) Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet 45:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) WHO Classification of Tumors of the Central Nervous System, 4th edn. International Agency for Research on Cancer: Lyon. [Google Scholar]