

Abstract
Chronic glial activation and neuroinflammation induced by the amyloid‐β peptide (Aβ) contribute to Alzheimer's disease (AD) pathology. APOE4 is the greatest AD‐genetic risk factor; increasing risk up to 12‐fold compared to APOE3, with APOE4‐specific neuroinflammation an important component of this risk. This editorial review discusses the role of APOE in inflammation and AD, via a literature review, presentation of novel data on Aβ‐induced neuroinflammation, and discussion of future research directions. The complexity of chronic neuroinflammation, including multiple detrimental and beneficial effects occurring in a temporal and cell‐specific manner, has resulted in conflicting functional data for virtually every inflammatory mediator. Defining a neuroinflammatory phenotype (NIP) is one way to address this issue, focusing on profiling the changes in inflammatory mediator expression during disease progression. Although many studies have shown that APOE4 induces a detrimental NIP in peripheral inflammation and Aβ‐independent neuroinflammation, data for APOE‐modulated Aβ‐induced neuroinflammation are surprisingly limited. We present data supporting the hypothesis that impaired apoE4 function modulates Aβ‐induced effects on inflammatory receptor signaling, including amplification of detrimental (toll‐like receptor 4‐p38α) and suppression of beneficial (IL‐4R‐nuclear receptor) pathways. To ultimately develop APOE genotype‐specific therapeutics, it is critical that future studies define the dynamic NIP profile and pathways that underlie APOE‐modulated chronic neuroinflammation.
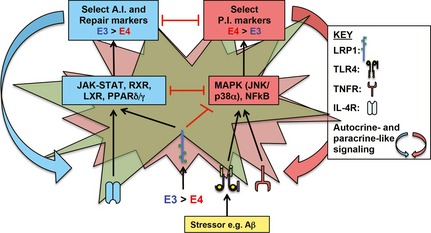
In this editorial review, we present data supporting the hypothesis that impaired apoE4 function modulates Aβ‐induced effects on inflammatory receptor signaling, including amplification of detrimental (TLR4‐p38α) and suppression of beneficial (IL‐4R‐nuclear receptor) pathways, resulting in an adverse NIP that causes neuronal dysfunction. NIP, Neuroinflammatory phenotype; P.I., pro‐inflammatory; A.I., anti‐inflammatory.
Keywords: Alzheimer's disease, amyloid‐β, apolipoprotein E, interleukin‐4 receptor, neuroinflammation, toll‐like receptor 4
Abbreviations used
- 5xFAD
mice expressing 5 FAD mutations
- AD
Alzheimer's disease
- ADAPT
Alzheimer's disease Anti‐inflammatory Prevention Trial
- apoE
apolipoprotein E
- APOE‐TR
APOE‐targeted replacement mice
- Aβ
amyloid‐β
- BBB
blood–brain barrier
- BEC
brain endothelial cells
- BMT
bone marrow transplant
- CCL3
chemokine (C‐C motif) ligand 3
- CNS
central nervous system
- COX
cyclooxygenase
- CR1
complement receptor 1
- CX
cortex
- EFAD
transgenic mice generated by crossing 5xFAD mice with APOE‐TR mice
- FAD
familial AD
- FAD‐Tg mice
mice containing transgenes for FAD mutations
- h‐apoE
human apoE
- ICV
intracerebroventricular
- IHC
immunohistochemistry
- IL‐4R
IL‐4 receptor
- IL
interleukin
- KO
knockout
- LDLR
low‐density lipoprotein receptor
- LPS
lipopolysaccharide
- LPS‐RS
LPS from Rhodobacter sphaeroides
- LRP1
low‐density lipoprotein receptor‐related protein 1
- m‐apoE
mouse apoE
- MCI
mild cognitive impairment
- MD‐2
myeloid differentiation protein 2
- MMP
matrix metalloproteinase
- NIP
neuroinflammatory phenotype
- NO
nitric oxide
- NSAIDs
non‐steroidal anti‐inflammatory drugs
- oAβ
oligomeric Aβ
- TLR4
toll‐like receptor 4
- TNFα
tumor necrosis factor α
- TREM2
triggering receptor expressed on myeloid cells 2
- VLDLR
very LDL receptor
Introduction
This review discusses the role of APOE in inflammation and Alzheimer's disease (AD), via a literature review (Fig. 1), presentation of novel data on APOE‐modulated Aβ‐induced neuroinflammation (Figs 2 and 3), and discussion of future research directions (Fig. 4).
Figure 1.
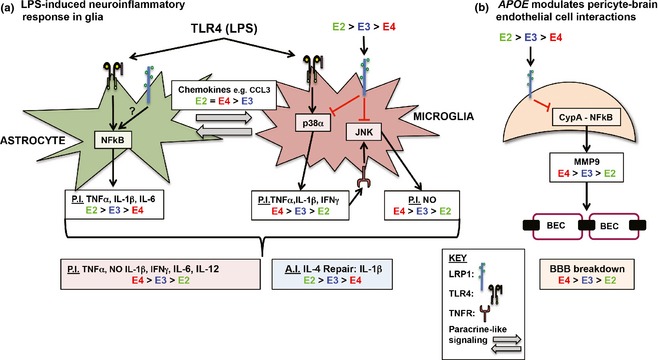
Proposed model of APOE‐modulated lipopolysaccharide (LPS)‐induced glia and pericyte activation. (a) Published data support that in astrocytes, toll‐like receptor (TLR)4/LPS‐induced secretion of pro‐inflammatory (PI) (tumor necrosis factor α, TNFα, IL‐1β, IL‐6) secretion follows the pattern: APOE2 ≥ APOE3 ≥APOE4, via differential NF‐κB activation (Maezawa et al. 2006a), and CCL3 levels follow the pattern: APOE2 = APOE4 > APOE2 (Cudaback et al. 2015). In microglia, LPS‐induced p38α‐dependent microglial secretion of PI cytokines (TNFα, nitric oxide, NO, IL‐1β, or IL‐6) is higher with APOE4 compared to APOE3 (Maezawa et al. 2006c). Paracrine‐ and autocrine‐like signaling will also impact the inflammatory response (gray arrows), e.g., CCL3‐dependent recruitment of microglia. Red box under glial diagram = overall PI effects for astrocytes and microglia, blue box = overall anti‐inflammatory (AI) effects. (b) Apolipoprotein E (ApoE)3 and apoE2, but not apoE4 signal via low density lipoprotein receptor‐related protein 1 (LRP1) to suppress NF‐κB‐dependent secretion of matrix metalloproteinase 9. Thus, MMP9 levels are higher with apoE4, resulting in brain endothelial cell (BEC) dysfunction and blood–brain barrier (BBB) breakdown (Bell et al. 2012).
Figure 2.
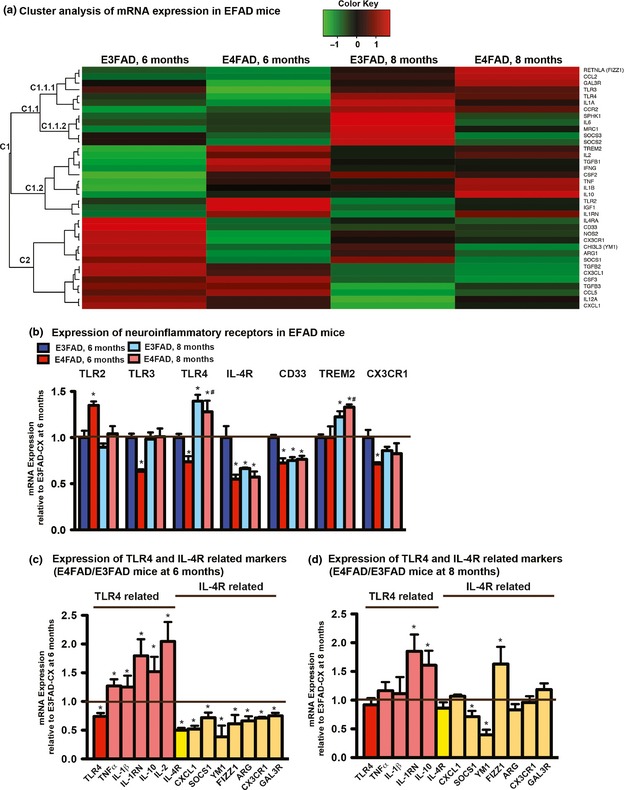
APOE4 modulates Aβ‐induced neuroinflammation in vivo. (a) The expression of inflammatory markers (see Table 1) was assessed in the CX of EFAD mice at 6 and 8 months at the mRNA level by multiplex analysis, and analyzed by cluster analysis. (b) Inflammatory receptor expression in 6‐ and 8‐month‐old EFAD mice was plotted relative to 6‐month‐old E3FAD mice. *p < 0.05 versus 6‐month‐old E3FAD, # p < 0.05 versus 6‐month‐old E4FAD mice. The expression of toll‐like receptor (TLR)4‐ and IL‐4R‐related proteins in (c). 6‐month‐old E4FAD mice plotted relative to 6‐month‐old E3FAD mice and in (d). 8‐month‐old E4FAD mice plotted relative to 8‐month‐old E3FAD mice. *p < 0.05 versus E3FAD at each age. n = 6 for (a–d). Horizontal line = no change i.e., E4FAD/E3FAD = 1.
Figure 3.
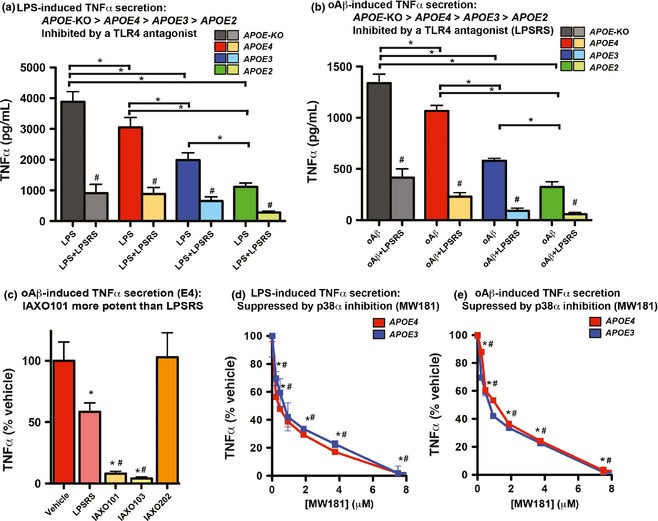
Lipopolysaccharide (LPS)‐ and oAβ‐induced tumor necrosis factor α (TNFα) secretion is higher with apolipoprotein E (apoE)4 than apoE3 and is blocked by toll‐like receptor (TLR)4 antagonism and p38α inhibition. Mixed glial cultures isolated from APOE‐ TR and APOE‐knock‐out (KO) mice were incubated with (a) LPS (100 ng/mL), or (b) oAβ (10 μM) +/− LPS‐RS (10 μg/mL) for 24 h, and TNFα levels were measured in the media by ELISA. *p < 0.05. (c) Mixed glial cultures isolated from APOE4‐ TR mice were incubated with oAβ (10 μM) and LPS‐RS, IAXO101, IAXO103, or controls (vehicle, IAXO202) for 24 h, and TNFα was measured in the media by ELISA. *p < 0.05 versus vehicle, # p < 0.05 versus LPS‐RS. Mixed glial cultures isolated from APOE3‐TR and APOE4‐TR mice were incubated with MW181, a p38α inhibitor, at the indicated concentrations for 30 min, followed by (d) LPS (100 ng/mL), or (e) oAβ (10 μM) for 24 h, and TNFα levels were measured in the media. Data are expressed as % vehicle control for each APOE genotype. *p < 0.05 versus vehicle for APOE3, # p < 0.05 versus vehicle for APOE4. n = 5–9.
Figure 4.
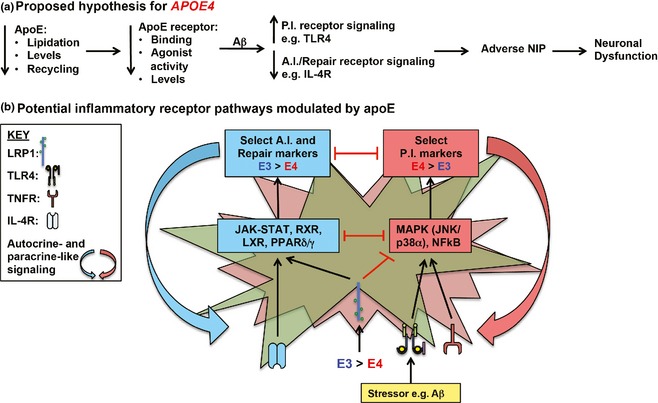
Proposed hypothesis of APOE‐modulated Aβ‐induced neuroinflammation. (a and b) Compared to APOE3, with APOE4: Lower lipoprotein lipidation and apolipoprotein E (apoE)4 levels result in lower apoE receptor signaling and levels, which with Aβ‐induction amplifies detrimental and suppresses beneficial inflammatory receptor signaling. This imbalance leads to an adverse neuroinflammatory phenotype (NIP) and downstream neuronal dysfunction. ApoE signaling via the apoE receptors may suppress Aβ‐induced activation of toll‐like receptor (TLR)4‐p38α signaling and potentiate IL‐4R signaling through pathways that converge on nuclear receptor signaling. PI, pro‐inflammatory; AI, anti‐inflammatory; Green, astrocyte; red, microglia.
Neuroinflammation from aberrantly activated glia is re‐emerging as an important mechanism that contributes to AD progression (for review Akiyama et al. 2000; Griffin & Mrak 2002; Eikelenboom & van Gool 2004; Van Eldik et al. 2007; Landreth 2009; Tobinick 2009; Vitek et al. 2009; Bachstetter & Van Eldik 2010; Gorelick 2010; Imbimbo et al. 2010; Ferretti & Cuello 2011; Keene et al. 2011; Sardi et al. 2011; Butchart & Holmes 2012; Rubio‐Perez & Morillas‐Ruiz 2012; Wyss‐Coray & Rogers 2012; McGeer & McGeer 2013; Medeiros & LaFerla 2013; Meraz‐Rios et al. 2013; Wilcock & Griffin 2013; Cherry et al. 2014; Nuzzo et al. 2014). The initial observations of activated microglia and astrocytes in pathologically relevant AD brain regions (Griffin et al. 1989), including within and surrounding amyloid plaques (Perlmutter et al. 1990; Edison et al. 2008), are now complemented by recent genome‐wide association studies and experimental data suggesting that several AD‐genetic risk factors function through modulating neuroinflammation. Examples of such AD risk genes include triggering receptor expressed on myeloid cells 2 (TREM2) (Guerreiro et al. 2013; Jonsson et al. 2013), complement receptor 1 (CR1) (Brouwers et al. 2012), CD33 (Hollingworth et al. 2011; Naj et al. 2011; Green et al. 2014), and toll‐like receptor 4 (TLR4) (Minoretti et al. 2006; Wang et al. 2011; Yu et al. 2012). However, it is important to note that data conflict on the role of CD33 and TLR4 on AD risk, highlighting the complexity of neuroinflammation in AD.
Increased levels of the amyloid‐β peptide (Aβ), including Aβ42, oligomeric (oAβ), and Aβ present in amyloid plaques, are considered a major component of neuronal dysfunction in AD. In AD, glial activation may be independent of the Aβ peptide, via damaged cells (e.g., via damaged‐associated molecular patterns) or other as yet unidentified factors (e.g., via pathogen‐associated molecular patterns), and also Aβ dependent. Although knowledge of the mechanistic contribution of glial cells to disease progression remains incomplete, the overproduction of select detrimental inflammatory mediators and/or decreased levels of beneficial mediators is postulated as a driving force for cognitive impairment (Butchart & Holmes 2012). Identified functions modulated by neuroinflammation include: stem cell replication and differentiation; neuronal and glial metabolism; neuronal plasticity and toxicity; tau phosphorylation; axon myelination; blood–brain barrier (BBB) permeability; extracellular matrix production/maintenance; and the removal of debris, foreign cells, and host‐cells targeted for removal (de Vries et al. 1996; de Vries et al. 1997; Krishnaswamy et al. 1999; Reyes et al. 1999; Rosenberg 2002; Vadeboncoeur et al. 2003; Persson et al. 2005; Simard et al. 2006; Tilleux & Hermans 2007; Russo et al. 2011; Britschgi et al. 2012; Lazarov & Marr 2013). Therefore, it is critical to dissect the role of AD risk factors on Aβ‐induced neuroinflammation.
APOE4 is the greatest genetic risk factor for sporadic AD, increasing risk approximately 12‐fold with two copies of the ε4 allele compared to APOE3, whereas APOE2 reduces risk twofold (Verghese et al. 2011). Furthermore, APOE4 carriers respond differently in clinical trials, often negatively (Farlow et al. 1998; Risner et al. 2006; Sperling et al. 2012; Qiu et al. 2013). Although the impact of apolipoprotein E (apoE) on brain function is multifactorial, evidence indicates that the apoE isoforms differentially modulate neuroinflammation (Keene et al. 2011). Indeed, epidemiological data indicate that non‐steroidal anti‐inflammatory drugs (NSAIDs) may lower AD risk, particularly for APOE4 carriers (Gorelick 2010; Imbimbo et al. 2010). While multiple lines of evidence demonstrate a heightened inflammatory response with APOE4 compared to APOE3, surprisingly little data address APOE modulation of Aβ‐induced neuroinflammation. Thus, until the APOE‐specific neuroinflammatory phenotype (NIP) is defined and the underlying mechanistic pathways dissected, the development of therapeutics targeting AD‐relevant, apoE‐modulated neuroinflammatory pathways is limited.
Neuroinflammation in AD
Investigations into the role of neuroinflammation in AD have raised important issues surrounding neuroinflammation‐directed clinical trials, the type of Aβ that induces neuroinflammation, how to classify the functional NIP and how to target neuroinflammatory receptors.
NSAIDs as a therapeutic for neuroinflammation
For virtually every inflammatory mediator in AD, reports conflict on the functional consequences of targeting these mediators, e.g., tumor necrosis factor α (TNFα), interleukin‐1β (IL‐1β), IL‐4, and IL‐10. (Bettoni et al. 2008; Leon et al. 2008; Shimizu et al. 2008; Ennaciri & Girard 2009; Song et al. 2011; Wang et al. 2011; Chakrabarty et al. 2012; Fenn et al. 2012; Ma et al. 2012; Peri & Piazza 2012; Opal et al. 2013). This issue is exemplified by data on the use of NSAIDs for the prevention of AD. Epidemiological data indicate that NSAIDs, which inhibit the cyclooxygenase (COX) enzymes, lower the risk of developing AD, an effect greater for APOE4 (Gorelick 2010; Imbimbo et al. 2010). Therefore, the AD Anti‐inflammatory Prevention Trial (ADAPT) was initiated (Martin et al. 2002; Meinert et al. 2009) with the goal of determining whether naproxen (non‐selective) or celecoxib (COX2 selective) delayed the onset of dementia in individuals considered high risk for sporadic AD. Unfortunately, ADAPT was suspended after ~ 4 years (2001–2004) because of cardiovascular safety concerns identified in the Adenoma Prevention with Celecoxib trial (Solomon et al. 2005). While there are confounding factors with the ADAPT trial, including the short treatment duration (7 years planned vs. median duration of ~ 1.3 years), the relatively low AD incidence rate (2.5% predicted for year 1 vs. 1.12% for the entire trial as conducted), and age of enrollment (≥ 70), extensive follow‐up data has been published on the ADAPT participants. Initial data demonstrated that neither NSAID prevented conversion to AD, or slowed cognitive decline (Lyketsos et al. 2007; Martin et al. 2008), including for APOE4 carriers (Drye & Zandi 2012), although some evidence indicated a decreased AD risk with naproxen (Breitner et al. 2011; Leoutsakos et al. 2012). However, the main conclusion from the ADAPT‐Follow‐up study (conducted for 7 years post trial) was that neither naproxen nor celecoxib prevent AD in adults with a family history of dementia (ADAPT‐Research‐Group 2013, ADAPT‐FS‐Research‐Group 2014). In contrast, CHF5074, a novel flurbiprofen analog with no anti‐COX activity, has recently been reported to improve executive function in APOE4 carriers with mild cognitive impairment (Ross et al. 2013). Therefore, identifying appropriate neuroinflammatory targets and pathways is an important focus of current research.
Aβ‐dependent activation of neuroinflammation
An important consideration for interpreting functional data on Aβ‐induced neuroinflammation in AD is the type of the peptide aggregate that induces neuroinflammation. While genetic and experimental evidence indicates that Aβ42 is necessary for AD pathology, it is difficult to identify the actual neurotoxic species, if there is, indeed, only one. These different forms include soluble Aβ (Lue et al. 1999; McLean et al. 1999; Wang et al. 1999), oAβ (Kuo et al. 1996; Tomic et al. 2009; Jin et al. 2011; Selkoe 2011) and Aβ present in amyloid plaques (Klein 2002; Thal et al. 2006; Haass & Selkoe 2007; Thal et al. 2008; Youmans et al. 2012). In addition, a dynamic compartmentalization between the different types of Aβ may exist, e.g., between plaques and soluble Aβ (Hong et al. 2011; Hudry et al. 2013), and the different forms of Aβ may contribute to neurodegeneration at different stages of the disease (Lesne et al. 2013). Accumulating evidence over the last decade suggests that soluble Aβ42 (Lue et al. 1999; McLean et al. 1999; Wang et al. 1999), specifically soluble oAβ (Kuo et al. 1996; Tomic et al. 2009; Jin et al. 2011; Selkoe 2011; Hayden & Teplow 2013; Teplow 2013; Sebollela et al. 2014), rather than insoluble Aβ (Hardy 2006; Haass & Selkoe 2007) may be the proximal neurotoxin in AD. Although the exact stoichiometry of the oAβ aggregates that are most active remains unclear (Hayden & Teplow 2013; Teplow 2013; Sebollela et al. 2014), oAβ correlates with cognitive decline and disease severity in humans, and oAβ levels are associated with memory decline in transgenic mice (Tg) expressing familial AD (FAD) mutations (FAD‐Tg mice) (Kuo et al. 1996; Tomic et al. 2009; Jin et al. 2011; Selkoe 2011; Hayden & Teplow 2013; Teplow 2013; Sebollela et al. 2014). In the context of neuroinflammation, compared to fibrillar assemblies, oAβ preparations induce greater or differential pro‐inflammatory cytokine production by microglia and astrocytes in vitro (Hashioka et al. 2005; Lindberg et al. 2005; White et al. 2005; Parvathy et al. 2009; Sondag et al. 2009; Hayden & Teplow 2013). Indeed, our previous data in primary glia demonstrate that the oAβ‐induced increase in pro‐inflammatory cytokine (nitric oxide, NO, TNFα, β) secretion occurs earlier and is greater than with fibrillar assemblies of Aβ (White et al. 2005). Thus, for different forms of Aβ (particularly oAβ subtypes), identifying their levels at different stages of AD, the inflammatory response they produce, and their underlying mechanisms (e.g., receptor mediated) may provide critical information for therapeutic development.
Neuroinflammatory phenotype
Although peripheral inflammation has been studied extensively over the last 20 years, studies of chronic neuroinflammatory responses in AD are less advanced (Cherry et al. 2014). As a simple model of peripheral inflammation, exogenous or endogenous molecules activate both innate and adaptive immunity to induce a pro‐inflammatory response that can also activate and recruit additional immune cells to destroy/remove the initiator of the signal. Typically, there is also an anti‐inflammatory/repair response to promote debris removal and restoration of the damaged area. However, the cell types, inflammatory mediators, and extent that this model translates to the brain in AD remain unclear. At the cellular level, neuroinflammation is a coordinated response among neurons, astrocytes, microglia, brain endothelial cells (BECs), oligodendrocytes, pericytes, ependymal cells, and peripheral immune cells recruited to the brain (Akiyama et al. 2000; Griffin & Mrak 2002; Eikelenboom & van Gool 2004; Van Eldik et al. 2007; Landreth 2009; Tobinick 2009; Vitek et al. 2009; Bachstetter & Van Eldik 2010; Gorelick 2010; Imbimbo et al. 2010; Ferretti & Cuello 2011; Keene et al. 2011; Sardi et al. 2011; Butchart & Holmes 2012; Rubio‐Perez & Morillas‐Ruiz 2012; Wyss‐Coray & Rogers 2012; McGeer & McGeer 2013; Medeiros & LaFerla 2013; Meraz‐Rios et al. 2013; Wilcock & Griffin 2013; Cherry et al. 2014; Nuzzo et al. 2014; Prinz & Priller 2014). In AD, Aβ‐ and non‐Aβ‐induced activation of each cell type can produce an equally complex profile of cytokines and chemokines, which in turn produce a positive or negative feedback loop through autocrine‐ and paracrine‐like signaling. Furthermore, with chronic neuroinflammation, the function of each inflammatory mediator and receptor can vary by cell type and at different stages of AD pathology. Within this milieu, identifying the effect of AD risk factors on the multiphasic expression of the NIP at different stages of AD pathology may ultimately address when and how to target neuroinflammation in AD, consistent with approaches attributed to personalized medicine. For example, the efficacy of NSAIDs will be dependent on the contribution and functional consequence of COX signaling on the NIP at different stages of AD, which may be further modulated by other risk factors. Indeed, in‐depth analysis of ADAPT has raised the intriguing possibility that NSAID efficacy, either beneficial or detrimental for AD risk, may be dependent on whether individuals experience fast or slow rates of cognitive decline (Leoutsakos et al. 2012).
One approach to identify the NIP and pathway interactions is using non‐biased ‘omic’ approaches, which has and will continue to provide valuable data. An alternative approach is to measure select candidate markers in functional NIP classifications. An increasingly adopted classification is based on applying the peripheral T‐cell/macrophage inflammatory phenotype nomenclature to microglia, namely, M1 (pro‐inflammatory), M2A (anti‐inflammatory, phagocytic), M2B (unclear function, general M2 response), and M2C (tissue remodeling, matrix deposition) (Mantovani et al. 2002; Gordon 2003; Mantovani et al. 2004; Mosser & Edwards 2008; Aguzzi et al. 2013; Cherry et al. 2014). One hypothesis proposes that a prolonged activation of the M1 and suppression of the M2 states underlies the detrimental inflammatory phenotype in AD and other chronic neurodegenerative disorders (Cherry et al. 2014). However, there are several potential confounding factors with this classification including (i) the assumption that peripheral inflammation directly translates to the NIP, (ii) the assumption that microglia produce all cytokines and are the only functionally relevant neuroinflammatory cell type in the brain, and (iii) the restriction of the inflammatory phenotype to two to four potential classifications, i.e., in some cases features of both an M1 and M2 phenotype will be observed, and not all markers within an M class will be equally affected through the course of AD. Indeed, the latter point was initially described by Colton et al. (2006) using mouse and human tissue, with data suggesting that (when using the microglial classification system) innate immune cells in the AD brain exhibit a hybrid of activation states. An alternative perspective is to divide inflammatory mediators by function into the subclassifications pro‐inflammatory, anti‐inflammatory, repair, and immunoregulatory, as presented by Chhor et al. (2013), incorporate the M1/2A/2B/2C classifications (Mantovani et al. 2002; Gordon 2003; Mantovani et al. 2004; Mosser & Edwards 2008) and include additional inflammatory receptors (Table 1). Table 1 lists the candidate neuroinflammatory markers for the genomic analysis described in Novel in vivo and in vitro analysis of APOE‐modulated Aβ‐induced neuroinflammation, as this type of classification has the advantage that it is not specific to microglial cells, thus enabling subsequent assignment of function for pathway and cell‐specific mechanistic studies. Potential limitations of this classification include an oversimplification of mediator function and the pre‐selection of mediators. Indeed, if assessed using unbiased approaches, the functional network genetic profile of the NIP in AD will be much broader than the genes in Table 1, as observed in vitro (Hickman et al. 2013; Butovsky et al. 2014). Nonetheless, unbiased and candidate approaches, which can both become targeted depending on their interpretation and application, are currently being pursued by the field to further define the AD‐NIP and track the expression of the NIP at different stages of AD pathology. Critical considerations include the functional classification system used to define the NIP, recognition that not all markers within a class will be modulated equally, and that during AD progression, the multiphasic expression of the NIP may help explain conflicting therapeutic data. Thus, once the NIP profile is identified, detailed cellular pathway analysis can be conducted, including the effect of known risk factors.
Table 1.
Select markers of the neuroinflammatory phenotype
| Inflammatory markers | Inflammatory receptors | ||||
|---|---|---|---|---|---|
| Pro‐inflammatory | Anti‐inflammatory | Repair | Immuno regulatory | ||
| M1 classical cytotoxic | iNOS (NOS2), TNFα, IL‐1β, IL‐6, IL‐12, IFNγ, CXCL1, IL‐1α, IL‐2, CCL5 | COX‐2 |
CD33 TREM‐2 LRP1 TLR2 TLR3 TLR4 CX3CR1 CCR2 (CD192) IL‐4R |
||
| M2A alternate repair | IL‐10, IL‐1R, TGFβ(1‐3), CX3CL1, IL‐4 | IL‐4, YM1, FIZZ1, ARG1, G‐CSF (CSF3), GM‐CSF (CSF‐2), IGF‐1 | CD206 (MRC1), GALR3, CCL2 (MCP‐1) | ||
|
M2B Type II |
iNOS,TNFα, IL‐1β, | IL‐10 | Sphk1/2 | ||
|
M2C tissue remodeling |
TGFβ, IL‐4, IL‐10, SOCS1‐3 | ARG1, IL‐4, Sphk1 | |||
Adapted from Chhor et al. (2013).
Neuroinflammatory receptors in AD
Experimental data have established that the neuroinflammatory receptors TREM2, CD33, and CR1 are genetic risk factors for AD. Neuroinflammatory receptor activation can modulate both Aβ phagocytosis and the levels of inflammatory mediators in the NIP. TREM2, expressed by microglia, is hypothesized to function as a phagocytic receptor for neuronal debris and Aβ, and to suppresses a pro‐inflammatory response, particularly after TLR activation, e.g., TLR4 (reviewed in Ford & McVicar 2009; Chouraki & Seshadri 2014; Hickman & El Khoury 2014; Lue et al. 2014). Thus, loss of function single‐nucleotide polymorphisms (SNPs) in TREM2 that increase AD risk are thought to decrease Aβ phagocytosis and promote a pro‐inflammatory response. For CD33, data conflict as to whether identified SNPs increase AD risk (reviewed in Bradshaw et al. 2013; Jiang et al. 2014). For example, the rs3865444 SNP in CD33 has been demonstrated to both increase and decrease AD risk in different populations (Lambert et al. 2013; Chouraki & Seshadri 2014; Li et al. 2014). Hypotheses to account for the differences include linkage disequilibrium across populations, and/or modulation of the rs3865444 SNP effect on AD risk by population‐specific environmental and lifestyle factors. However, the minor allele of the CD33 rs3865444 SNP is thought to decrease AD risk through reducing microglial expression of CD33, thus increasing Aβ phagocytosis. Less data are available on how SNPs in CR1 may impact AD risk compared to TREM2 or CD33, although the complement system in general has, and remains, a focus of AD research (reviewed in Crehan et al. 2012; Shen et al. 2013). Functionally, the SNPs in CR1 may modulate microglial phagocytosis of C3b‐opsonized Aβ, and also remove increased unbound C3b levels to limit complement‐mediated inflammation in AD.
Additional inflammatory receptors activated by Aβ, particularly aggregated forms, i.e., oAβ, have also been identified in genetic or experimental studies. In astrocytes, these receptors include members of the low‐density lipoprotein receptor (LDLR) family, scavenger receptors, formyl peptide receptor‐like 1 (FPRL1), and leucine‐rich glioma inactivated protein 3 (LGL3). In microglia, Aβ receptors include scavenger receptors, CD14, CD47, CD36, TLR2, and TLR4 (Mohamed & Posse de Chaves 2011; Meraz‐Rios et al. 2013; Yu & Ye 2014). Furthermore, the ability of microglia, and other immune cells, to phagocytose Aβ has led to the development of both active and passive Aβ immunization strategies for AD (reviewed in Lambracht‐Washington & Rosenberg 2013; Hardy et al. 2014; Karran & Hardy 2014). Passive immunization with Aβ antibodies, which in part clear Aβ via Fc receptors expressed on microglia, is the most actively pursued approach. In general, passive immunization has failed to stop or reverse cognitive decline when used as a treatment for AD, although there is hope that the approach will prove efficacious in prevention paradigms. Although there is evidence of extracellular Aβ clearance with passive immunization, side effects include edema and cerebral microhemorrhage, identified by amyloid‐related imaging abnormalities (Sperling et al. 2011; Sperling et al. 2012). It has also been proposed that some of the negative side effects from Fc‐dependent Aβ phagocytosis may be the result of a pro‐inflammatory response (Fuller et al. 2014; Liu et al. 2014). Thus, an important issue when assessing the functional role of neuroinflammatory receptors in AD is whether Aβ‐binding causes Aβ degradation (i.e., phagocytosis) and/or an inflammatory response. Indeed, although Aβ phagocytosis will be beneficial for AD, an associated chronic pro‐inflammatory response and decreased anti‐inflammatory/repair response may lower the expression of phagocytic receptors (Hickman et al. 2008), induce neuronal damage, and prevent neuronal recovery. In addition, the functional effect of inflammatory receptors in AD results from a combination of responses by multiple cell types that express the same inflammatory receptor (e.g., Fc receptors are expressed by BECs and glia, TLR4 is expressed by neurons, microglia, and BECs). Finally, inflammatory receptor signaling may be species specific (e.g., TLR4, CD33), potentially limiting the relevance of FAD‐Tg mice as a preclinical model.
In summary, a major goal for determining the effect of neuroinflammation on AD progression is the identification of novel therapeutic targets. However, the complex nature of neuroinflammation has resulted in conflicting data on the role of specific inflammatory mediators and receptors. Important issues facing the field include dissecting the neuroinflammatory activity of the different forms of Aβ, identifying the multiphasic expression of the NIP during AD pathology, and assessing neuroinflammatory receptor pathways as therapeutic targets.
APOE‐modulated inflammation: current perspective
Human apoE (h‐apoE) has three isoforms that differ by a single amino acid substitution at residues 112 or 158: apoE2 contains Cys112, 158 apoE3 contains Cys112Arg158 and apoE4 contains Arg112,158 (for review Weisgraber et al. 1982). ApoE is the major apolipoprotein present on lipoproteins in the central nervous system (CNS) (for review LaDu et al. 2000). Indeed, apoE‐containing CNS lipoproteins are separate from apoE‐containing lipoproteins in the periphery. This compartmentalization was definitively demonstrated in human liver transplantation studies, as the liver is the primary source of peripheral apoE. If the donor liver was from an APOE4/4 individual and the recipient was an APOE3/3, apoE4/4 became the peripheral isoform while apoE3/3 remained the isoform in the CNS (Linton et al. 1991). In the CNS, apoE is produced primarily by glia (astrocytes and microglia), although under certain conditions of stress, neuronal apoE expression may occur (Xu et al. 2006). The function(s) of apoE are, in part, mediated via members of the LDLR family expressed throughout the CNS, with astrocytes, microglia, and neurons expressing LDLR, LDLR‐related protein 1 (LRP1), very LDL receptor, and ApoER2. LDLR and LRP1 are endocytic receptors, while very LDL receptor and ApoER2, as well as LRP1, are signaling receptors (Holtzman et al. 2012). Although APOE genotype/apoE isoform‐specific effects have been described for virtually all the proposed pathogenic AD pathways (Bu 2009; Liu et al. 2013), accumulating evidence supports a role for h‐APOE in modulating neuroinflammation (Keene et al. 2011). These data are derived from in vivo and in vitro experiments in the absence of Aβ, both in the periphery and the CNS, and limited data for Aβ‐induced neuroinflammation.
APOE‐modulated peripheral inflammation
APOE modulates the peripheral immune response to stressors that mimic infection
Extensive data demonstrate that, in response to a peripheral inflammatory stimulus, pro‐inflammatory cytokine production is higher with APOE4 compared to APOE3. In a groundbreaking study by Gale and co‐workers, healthy volunteers were subjected to intravenous (IV) administration of lipopolysaccharide (LPS), a TLR4 agonist and bacterial endotoxin used to elicit a general inflammatory response (Gale et al. 2014). LPS‐induced hyperthermia and increased plasma levels of pro‐inflammatory markers (TNFα, IL‐6) were higher in APOE3/4 carriers compared to APOE3/3 carriers (Gale et al. 2014), data recapitulated in Tg mice. Indeed, in APOE‐targeted replacement mice (APOE‐TR) (Sullivan et al. 1997,1998), which express the h‐APOE genotypes (see ‘APOE‐modulated inflammation: current perspective'), peritoneal LPS injection results in thermal dysregulation (in this case hypothermia), higher plasma levels of TNFα, and higher markers of liver toxicity with APOE4 compared to APOE3 (Gale et al. 2014). In vitro data support this APOE4 inflammatory phenotype in the periphery. In whole blood samples isolated from healthy volunteers, TLR4/LPS‐induced pro‐inflammatory cytokine (TNFα, IL‐1β, IL‐6, IFNγ) secretion is higher with APOE3/4 compared to APOE3/3 (Gale et al. 2014). In addition, LPS‐induced TNFα levels are higher, and anti‐inflammatory markers (IL‐10) are lower, with APOE4 compared to APOE3 in a monocyte–macrophage cell line (Jofre‐Monseny et al. 2007). Furthermore, efferocytosis (phagocytosis of dead/dying cells) is decreased in macrophages isolated from APOE4‐TR mice compared to APOE3‐TR mice (Cash et al. 2012).
Despite these findings, data are conflicted on whether the increased levels of select pro‐inflammatory markers and decreased levels of anti‐inflammatory markers with APOE4 are beneficial or detrimental for peripheral infection. Indeed, although APOE‐knock out (KO) mice demonstrate an impaired immune response to various bacteria, including Listeria monocytogenes and Klebsiella pneumonia (Roselaar & Daugherty 1998; de Bont et al. 1999; Zhu et al. 2012), young APOE4 humans have an increased pathogen resistance for hepatitis C‐induced liver damage compared to APOE3 (Wozniak et al. 2002), and APOE4 protects against damage caused by Cryptosporidium infection (Azevedo et al. 2014). The beneficial effect with APOE4 may be related to the enhanced pro‐inflammatory response (TNFα and NO) and/or effects on cellular arginine uptake to produce NO, ultimately disrupting pathogen function (Brown et al. 2002; Vitek et al. 2009).
APOE modulation of peripheral T‐cell response
APOE modulation of peripheral immunity may also play a role in APOE4‐induced AD risk. Independent of disease status, APOE4 carriers have lower levels of CXCL9 (a T‐cell attractant), and higher levels of IL‐13 (anti‐parasitic) in the plasma compared to APOE3 carriers (Soares et al. 2012). The data for CXCL9 may be significant, as apoE can suppress T‐cell levels (Kelly et al. 1994; Mistry et al. 1995) and there is an age‐ and APOE‐dependent decrease in Aβ‐specific CD4+ T‐cell activity in humans (Begum et al. 2014). Indeed, Aβ‐specific CD4+ T‐cell activity follows the order: APOE4‐negative males > APOE4‐positive males > APOE4‐negative females > APOE4‐positive females (Begum et al. 2014). However, the significance of this T‐cell response for AD pathogenesis remains unclear. APOE genotype also affects the therapeutic efficacy of bone marrow transplant (BMT) in vivo. After BMT from APOE‐TR mice to an FAD‐Tg mouse model (specifically the APP/PS1 model, bearing APP K670N/M671L and PS1 L166P, both under the control of Thy1), APOE4‐recipient FAD‐Tg mice demonstrate significantly impaired spatial working memory, higher levels of Aβ and pro‐inflammatory cytokines (TNFα), and lower levels of anti‐inflammatory cytokines (IL‐10) compared to APOE3‐recipient FAD‐Tg mice receiving BMT from APOE3‐TR mice (Yang et al. 2013). Thus, APOE impacts peripheral immunity and inflammation, although further research is required to determine whether and how these effects apply to CNS function.
APOE modulation of Aβ‐independent neuroinflammation
TLR4/LPS‐induced pro‐inflammatory cytokine secretion in vivo: APOE4 > APOE3
The effects of APOE on Aβ‐independent neuroinflammation have been investigated in APOE‐TR mice, likely the most physiologically relevant model for the study of h‐apoE isoforms (Sullivan et al. 1997,1998). In these mice, the coding domain for one of the h‐apoE isoforms replaces the coding domain for mouse apoE (m‐apoE). Thus, the h‐apoE is expressed in a native conformation at physiologically regulated levels, and in the same temporal and spatial pattern as endogenous m‐apoE. In APOE‐TR mice, intraperitoneal injection of LPS results in higher cortical TNFα and IL‐12 (pro‐inflammatory) expression with APOE4 compared to APOE3 (Vitek et al. 2009). However, as LPS demonstrates low brain bioavailability (Banks & Robinson 2010), this effect is likely due to peripheral APOE genotype‐specific effects or BEC inflammatory responses. Direct intracerebroventricular (ICV) injection of LPS into the CNS of APOE‐TR mice (4 months) demonstrate that after 24 and 72 h, synaptic protein levels decrease, microglia and astrocyte activation increase, and pro‐inflammatory cytokine (TNFα, IL‐6, IL‐1β) levels increase, and follow the order: APOE4 > APOE3 = APOE2. Furthermore, dendrites fail to recover with APOE4 but not APOE2 or APOE3 (72 h post‐ICV LPS injection) in 6‐week‐old APOE‐TR mice (Maezawa et al. 2006c). These data indicate that TLR4 activation via LPS impairs the repair/recovery function of APOE4 compared to APOE2/3. However, it is not clear whether these effects are due to direct TLR4 activation of neurons, or through neuroinflammatory mediators secreted by activated glia. Thus, for APOE modulation of acute neuroinflammation, the overall response is likely a combination of the inflammatory profile prior to inflammatory insult, the magnitude of response to the inflammatory insult, and the inflammatory response following inflammatory insult, all of which may be relevant for determining the AD‐NIP.
TLR4/LPS‐induced signaling and cytokine secretion is cell‐type specific
In vitro data using microglia isolated from APOE‐TR mice validate APOE genotype‐specific modulation of TLR4/LPS‐induced inflammation (reviewed in Keene et al. 2011). In microglia, LPS‐induced pro‐inflammatory cytokine (TNFα, IL‐6, and IL‐1β) levels are higher with APOE4 than APOE3, with a delayed but similar response for NO (pro‐inflammatory) (Maezawa et al. 2006b). Furthermore, after combined LPS/IFNγ treatment of microglia, pro‐inflammatory cytokine (TNFα, IL‐6, and IL‐12) levels are higher and anti‐inflammatory/repair cytokine (IL‐4) levels are lower with APOE4 compared to APOE3 (Vitek et al. 2009). Importantly, the effect of TLR4 activation on neuronal function is recapitulated in vitro, with microglial cytokine secretion hypothesized as the major effector. Indeed, in microglia/neuron co‐cultures, LPS‐induced microglia‐mediated decreases in synaptic proteins follow the order: APOE4 > APOE3 > APOE2, results repeated in hippocampal slice cultures (Keene et al. 2011). The decreased synaptic protein levels are correlated with the secretion of TNFα, IL‐1β, or IL‐6, rather than NO by microglia (Maezawa et al. 2006b). Interestingly, pathway analysis supports that LPS‐induced activation of the p38α pathway in microglia mediates the secretion of the pro‐inflammatory cytokines, particularly TNFα, that cause decreases in synaptic proteins (Maezawa et al. 2006b; Xing et al. 2011). LPS‐induced NO secretion by microglia in vitro is suppressed by an apoE peptide that consists of a tandem repeat of the apoE‐receptor‐binding domain, residues 136–142 (Pocivavsek et al. 2009a,b). This suppression of NO secretion is attributed to LRP1‐dependent activation of the c‐Jun N‐terminal kinase (JNK) pathway (Pocivavsek et al. 2009a,b), although the relevance of these data to full‐length apoE peptide remains unclear. A potential overall mechanism for these data is that TLR4‐p38α pro‐inflammatory cytokine production activates the JNK pathway (e.g., TNFα signaling through the TNF receptor) to induce NO secretion. ApoE could suppress p38α and/or JNK via the LDLR family in an apoE isoform‐specific manner: apoE2 > apoE3 > apoE4 (Fig. 1a). Additional study of the intracellular signaling cascades modulated by apoE may provide therapeutic opportunities for modulating acute neuroinflammation.
In astrocytes, TLR4/LPS‐induced secretion of TNFα, IL‐1β, or IL‐6 follows the pattern: APOE2 ≥ APOE3 ≥ APOE4, via differential NF‐κB activation (Maezawa et al. 2006a) (Fig. 1a). This is in direct contrast to microglia, where LPS‐induced pro‐inflammatory cytokine secretion is: APOE4 > APOE3 > APOE2. These data, and the paracrine‐like signaling that occurs between inflammatory cells (Landreth & Reed‐Geaghan 2009; Okun et al. 2009; Buchanan et al. 2010; Cameron & Landreth 2010; Hennessy et al. 2010; Okun et al. 2011; Hanamsagar et al. 2012), indicate that incorporating both astrocytes and microglia in vitro may represent a more physiologically relevant model. Astrocyte activation may also modulate microglial migration. Indeed, in astrocytes isolated from APOE‐TR mice, TLR4/LPS‐ and TLR3/Poly I:C‐induced production of chemokine (C‐C motif) ligand 3 (CCL3) follows the order: APOE2 = APOE4 > APOE3 (Cudaback et al. 2015). Furthermore, CCL3 levels are higher with APOE4 compared to APOE3 in the cortex (CX) of APOE‐TR mice (after peritoneal LPS injection) and in the parietal CX of AD patients (Cudaback et al. 2015).
APOE also modulates BBB function via differential pericyte activation in a process that could be considered neuroinflammatory (Bell et al. 2012). Compared to APOE2‐ and APOE3‐TR mice, APOE4‐TR and APOE‐KO mice demonstrated BBB impairments that include reduced cerebral blood flow, reduced microvascular length, and increased BBB permeability (Bell et al. 2012). At the cellular level, the APOE‐modulated changes in BBB function are associated with DNA fragmentation in pericytes and BECs, and diminished microvascular coverage by pericytes. Furthermore, pharmacological and genetic analyses of APOE‐TR mice support the idea that apoE3 and apoE2, but not apoE4, signal via LRP1 to suppress the cyclophilin A‐NF‐κB‐matrix metalloproteinase 9 (MMP9) pathway in pericytes (Bell et al. 2012). Thus, higher MMP9 levels with APOE4 compromise BEC function, including decreased levels of tight junction proteins, leading to BBB dysfunction (Bell et al. 2012) (Fig. 1b).
As discussed previously, whether pro‐inflammatory cytokines are considered detrimental or beneficial likely depends (see Neuroinflammation in AD and APOE modulates the peripheral immune response to stressors that mimic infection) on the disease state. For example, in tissue from epilepsy patients, neuronal resilience, astrocyte and microglial activation, IL‐1α, and IL‐1β levels are all higher with APOE3 compared to APOE4 (Wilcock & Griffin 2013). This increased pro‐inflammatory response with APOE3, potentially the result of neuronal hyperactivation and increased Aβ production, is considered protective, in contrast with increased pro‐inflammatory cytokine production with APOE4 after LPS‐induced activation (Maezawa et al. 2006b; Zhu et al. 2012). However, for both epilepsy and LPS, overall neuronal function is worse with APOE4 than APOE3, highlighting the context specificity of neuroinflammatory responses.
APOE modulation of Aβ‐dependent neuroinflammation
In contrast to APOE modulation of acute TLR4/LPS‐induced neuroinflammation, data are limited on APOE modulation of Aβ‐induced neuroinflammation in vivo and in vitro. Indeed, the data described above for peripheral inflammation in AD patients and FAD‐Tg mice, and the increased CCL3 levels with APOE4 in AD patients, are almost the extent of published research in this field. Further in vivo data have demonstrated that hippocampal astrocyte and microglial activation are increased in APOE4‐TR, but not APOE3‐TR mice after inhibition of the Aβ‐ degrading enzyme neprilysin (Belinson & Michaelson 2009). In addition, in novel EFAD transgenic mice (further described in Novel in vivo and in vitro analysis of APOE‐modulated Aβ‐induced neuroinflammation), which express 5xFAD mutations and h‐APOE (Youmans et al. 2012), cortical microglial activation is greater surrounding plaques in E4FAD (APOE4) compared to E3FAD (APOE3) mice (Rodriguez et al. 2014).
While in vitro data demonstrate that m‐apoE and h‐apoE affect Aβ‐induced pro‐inflammatory cytokine secretion, there are apparent contradictions among studies. Indeed, Aβ42‐induced increases in IL‐6, inducible NO synthase (iNOS), and IL‐1β levels are greater in mixed glial cultures (95% astrocytes, 5% microglia) isolated from APOE‐KO mice compared to wild‐type mice (LaDu et al. 2001). ApoE3 and apoE4, isolated from the conditioned media of human embryonic kidney cells transfected with h‐apoE cDNA (HEK‐apoE), suppress Aβ‐induced glial inflammatory responses, with no apoE isoform effects (Hu et al. 1998; Guo et al. 2004). However, HEK‐apoE3 and HEK‐apoE4 induce inflammation (IL‐1β) in the absence of Aβ42 (Guo et al. 2004), confounding data interpretation. Dorey and co‐workers have also reported that the Aβ‐induced inflammatory response in astrocytes is in the order: APOE4 > APOE3 > APOE2, although no published evidence is included (Dorey et al. 2014). In addition, Aβ degradation via MMP9 is greater in macrophages expressing APOE2 than APOE3 or APOE4 and this effect appears to be dependent on LDLR signaling (Zhao et al. 2009). Section Neuroinflammation in AD outlines the various factors that influence Aβ‐induced inflammation and phagocytosis as well as the receptors and signaling pathways involved. These variables also apply to any APOE modulation of Aβ‐induced neuroinflammatory processes.
In summary, there is a growing body of work demonstrating that APOE4 exerts a negative effect on peripheral and Aβ‐independent neuroinflammation, compared to APOE3. However, data on APOE modulation of Aβ‐induced chronic neuroinflammation in vivo and in vitro are limited.
Novel in vivo and in vitro analysis of APOE‐modulated Aβ‐induced neuroinflammation
Introduction
Because of the paucity of research and critical importance of the topic, we have begun to dissect the effects of the h‐apoE isoforms on Aβ‐induced neuroinflammation. We present data testing whether h‐apoE modulates the NIP in the new EFAD‐Tg mouse model, and the effect of APOE on oAβ‐induced TNFα secretion in vitro.
Methods
mRNA analysis of EFAD mice
EFAD mice are the result of crossing 5xFAD mice (APP K670N/M671L + I716V + V717I and PS1 M146L + L286V) and APOE‐TR mice (Youmans et al. 2012). All experiments follow the University of Illinois at Chicago (UIC) Institutional Animal Care and Use Committee protocols. EFAD mice were housed in the College of Medicine Research Building (COMRB) animal facility (IACUC 14‐088). The COMRB is the campus barrier facility that oversees the procurement, care, and maintenance of animals used in the teaching and research programs of the UIC. This oversight responsibility includes ensuring that the UIC program meets federal regulations, the requirements of the American Association for the Accreditation of Laboratory Animal Care (AAALAC), and currently accepted standards for providing adequate veterinary care and proper animal husbandry (the PHS assurance number is A3460‐01). The COMRB staff continually monitors temperature, light timers, cage‐washing procedures, sterilizer operation, automatic watering system, and animal health. EFAD mice were housed two to five mice per cage and maintained on a standard 12‐h light–dark cycle with access to food and water ad libitum. Six and 8‐month‐old male E3FAD and E4FAD mice were anesthetized with sodium pentobarbital (50 mg/kg) and transcardially perfused with ice‐cold phosphate‐buffered saline containing protease inhibitors (set 3; Calbiochem, San Diego, CA, USA). One milliliter of RNAlater solution (Ambion, Austin, TX, USA) was added to 20 mg of cortical brain tissue dissected from the right hemi‐brains of EFAD mice, samples were incubated at 4°C overnight and stored at −80°C to ensure minimal mRNA degradation. Quantitative mRNA analysis was performed using a custom‐designed QuantiGene 2.0 Multiplex assay system (Affymetrix, Santa Clara, CA, USA) for the markers in Table 1, according to the manufacturer's protocol, by an investigator blinded to APOE genotype and age. Briefly, samples were thawed, RNAlater removed, and cortices lysed in the QuantiGene homogenizing solution. Subsequently, magnetic beads containing target‐specific probes were added, followed by sequential hybridization and signal amplification. Signal intensity was measured using a Luminex array system (Bio‐Rad Bio‐Plex; Bio‐Rad, Hercules, CA, USA). Mean fluorescence intensity values were adjusted by blank value subtraction, and standardized to the housekeeping gene GUSB. Data were analyzed with the MasterPlex®EX software (Hitachi‐Solution America, San Bruno, CA, USA).
An important consideration for the analysis of mRNA expression data is the reference gene utilized for normalization. For this manuscript, the expression of three reference genes was assessed: ACTB, RPLN0, and GUSB. ACTB levels were at the upper end of the mean fluorescence intensity limit of detection at the sample dilutions required to measure the genes in Table 1, and therefore ACTB was not suitable as a reference gene. The levels of RPLN0 and GUSB were not significantly different by APOE genotype (APOE3 compared to APOE4) or age (6 months compared to 8 months) in EFAD mice when assessed using two‐way anova followed by Bonferroni post hoc analysis. Through our analysis of mRNA expression in cortical tissue from EFAD mice, similar results are obtained when normalizing with either RPLN0 or GUSB; however, baseline RPLN0 expression is more varied (data not shown). Furthermore, GUSB is reported as one of the three most stable reference genes in the cerebral CX of rats following traumatic brain injury (Cook et al. 2009). Thus, GUSB was utilized as the reference gene for this study. An important caveat is that GUSB expression has been described as the eighth most stable reference in feline brain (Kessler et al. 2009), to fluctuate in some experimental conditions (Cook et al. 2009; Gubern et al. 2009; Pernot et al. 2010; Swijsen et al. 2012), and to decrease with age (from post‐natal days 7–6 months) in the cerebellum of mice (Boda et al. 2009). An ongoing aspect of our work is dissecting in larger sample sets the expression of reference genes by brain region, APOE genotype, and Aβ accumulation (age) in EFAD mice.
Mixed glial cultures
Mixed glial cultures (95% astrocytes, 5% microglia) were isolated from APOE‐TR and APOE‐KO mice as described (White et al. 2005; Nwabuisi‐Heath et al. 2012). Glial cultures (24‐well plates) were incubated with oAβ42 (0–10 μM) (see Stine et al. 2003; White et al. 2005 for details) or LPS (0‐100 ng/mL), +/− LPSRS, IAXO101, IAXO103, IAXO202 (Innaxon, Tewkesbury, UK), MW181 (Watterson et al. 2013, Bachstetter et al. 2014), or vehicle control, at the concentrations indicated in the figures (N.B. MW181 was added 30 min prior to the addition of LPS or oAβ). Twenty four hours later, TNFα levels were measured in the conditioned media by ELISA (Invitrogen, Carlsbad, CA, USA).
Statistical analysis
Data were analyzed by one‐way anova followed by Tukey's post hoc analysis or two‐way anova followed by Bonferroni post hoc analysis (Fig. 2b) using GraphPad Prism version 6 for Macintosh. Heat map complete‐linkage clustering between gene expression profiles was performed in R, using Pearson's correlation as the distance measure. IL‐2 and IFNγ values from the E3FAD‐CX at 8 months were removed because of low quality and were replaced with mean expression for the gene during clustering (represented by black spaces in the heat map).
Results
EFAD‐Tg mice: APOE modulates neuroinflammation
A lack of tractable h‐APOE/FAD‐Tg models has hindered research efforts to determine the effect of APOE on AD‐relevant pathways (Tai et al. 2011). To identify the effects of APOE genotype‐specific modulated neuroinflammation induced by Aβ pathology, we utilized EFAD‐Tg mice (Youmans et al. 2012). EFAD mice are an AD model expressing h‐APOE under the regulated control of the endogenous m‐apoE promoter (APOE‐TR) (Sullivan et al. 1997,1998), and over‐expressing h‐Aβ42 via the 5xFAD‐Tg mice (Oakley et al. 2006). EFAD mice have enabled research on the role of APOE in AD‐like pathology (Youmans et al. 2012; Kunzler et al. 2013; Tai et al. 2013) and for preclinical assessment of potential AD therapeutics (Tai et al. 2014a). Briefly, we have demonstrated that in E4FAD mice, compared to E3FAD mice, there are reduced post‐synaptic protein levels, increased extracellular and soluble Aβ levels (including oAβ), reduced levels of soluble Aβ bound to apoE (apoE/Aβ complex), and reduced apoE lipoprotein association/lipidation at 6 months in the CX and hippocampus. Furthermore, microglial activation is increased in the vicinity of plaques in the CX of 6‐month‐old E4FAD mice compared to E3FAD mice (Rodriguez et al. 2014). Although immunohistochemistry analysis is important for analyzing the local environment and gliosis, biochemical analysis is critical to begin defining a NIP. Therefore, the markers in Table 1 were measured at the mRNA level in the CX of EFAD mice at 6 and 8 months using multiplex analysis.
Cluster analysis of mRNA data for EFAD mice at 6 and 8 months demonstrates that APOE has a pronounced impact on factors selected as representative of the inflammatory activation states and associated receptors (Table 1 and Fig. 2a). A large cluster of genes (C1) was identified with lower expression in the E3FAD‐CX at 6 months, including select markers in the pro‐inflammatory (TNFα, IL‐1β, IL‐2, IL‐6, and IFNγ) and anti‐inflammatory (TGFβ1 and IL‐10) classifications. The expression of genes in C1 increases in the E3FAD‐CX from 6 to 8 months, including pro‐inflammatory markers (e.g., TNFα, IL‐6), a subcluster (C1.1.2) of select anti‐inflammatory markers (suppressor of cytokine signalling (SOCS) 2, SOCS3, IL‐10, SPHK1/1), immunoregulatory markers (MRC1), and inflammatory receptors (CCR2), potentially as an adaptive response not observed in E4FAD mice. In the E4FAD‐CX, a subset of genes in C1 (C1.2) is consistently high at both 6 and 8 months, which encompasses markers in all NIP classifications (e.g., TNFα, IL‐1β, IL‐10, and TLR2). In addition, the expression of genes in a different subcluster of C1, C1.1.1 also increases from 6 to 8 months in the E4FAD‐CX mice, including repair (FIZZ1) and immunoregulatory (CCL2 GAL3R) markers, potentially as an APOE4‐specific adaptive response. Importantly, a second large cluster (C2) was identified with higher expression in E3FAD‐CX and lower expression in the E4FAD‐CX at 6 months. The majority of C2 markers are in the anti‐inflammatory (CXCL1, TGFβ3, and SOCS1) and repair (chitinase‐like 3 (YM1), arginase 1 (ARG1), colony stimulating factor 3 (CSF3)) categories and also include the IL‐4Rα subunit (see ‘EFAD‐Tg mice: TLR4 associated cytokines are higher and IL‐4R lower with APOE4' below). The expression of C2 markers was decreased from 6 to 8 months in both E3FAD and E4FAD mice.
EFAD‐Tg mice: TLR4‐associated cytokines are higher and IL‐4R lower with APOE4
To further dissect these data, inflammatory receptor expression was specifically compared, expressed relative to E3FAD‐CX at 6 months (Fig. 2b). Three significant results are observed. First, from 6 to 8 months, expression of CD33 decreases and TREM2 increased in E3FAD and E4FAD mice. These data are confounding, as genetic evidence indicates that TREM2 is protective (Guerreiro et al. 2013; Jonsson et al. 2013) while CD33 increases AD risk (Hollingworth et al. 2011; Naj et al. 2011; Green et al. 2014) and therefore might be expected to decrease and increase, respectively, with increasing Aβ pathology. Thus, our data suggest that the changes in TREM2 and CD33 in EFAD mice are an adaptive response to high Aβ levels regardless of APOE genotype. Second, TLR4 expression increases from 6 to 8 months in EFAD mice for both APOE3 and APOE4. Interestingly, TLR4‐dependent neuroinflammation has been shown to decrease neuronal viability in vivo (Lehnardt et al. 2003; Lehnardt 2010). Third, IL‐4Rα expression is 50% lower in 6‐month‐old E4FAD mice compared to E3FAD mice. In the periphery, TLR4 and IL‐4R are described as opposite ends of the inflammatory spectrum, with TLR4 linked to a pro‐inflammatory response and IL‐4R to an anti‐inflammatory/repair response, at least for select markers (Chhor et al. 2013; Cherry et al. 2014). Therefore, the expression of inflammatory markers linked to TLR4 and IL‐4R signaling was plotted at 6 months, expressed relative to E3FAD‐CX (Fig. 2c). Data demonstrate that select markers of TLR4 signaling are greater (e.g., TNFα, IL‐1β, and IL‐1RN), and markers of IL‐4R signaling lower (e.g., SOCS1, YM1, and ARG1), in E4FAD mice at 6 months. Thus, even though TLR4 expression is lower in E4FAD mice at 6 months, TLR4‐associated cytokines are higher. As described above, age (6–8 months) significantly impacts the expression of the inflammatory genes in EFAD mice, which in general results in greater increases in TLR4 and decreases in IL‐4R related genes for E3FAD mice compared to E4FAD mice; however there are exceptions. Thus, a comparison of the same genes at 8 months (Fig. 2d) demonstrates that select TLR4‐related genes are non‐significantly (TNFα) and significantly higher (IL‐10, IL‐1RN) in E4FAD mice compared to E3FAD mice. For IL‐4R‐related genes at 8 months, SOCS1 and YM1 remained lower in E4FAD mice compared to E3FAD mice, whereas FIZZ1 was higher in E4FAD mice, data that contrast with the results at 6 months. These data re‐emphasize that the APOE‐modulated NIP can vary as AD pathology progresses.
In summary, these data support the differential modulation of TLR4 and IL‐4R pathways as underling the unique NIP at early stages of Aβ pathology in E4FAD mice at 6 months.
TLR4/LPS‐dependent TNFα secretion in mixed glial cultures isolated from APOE‐TR and APOE‐KO mice: APOE‐KO >APOE4 > APOE3 > APOE2
The overall goal of the in vitro experiments was to dissect the effect of APOE on oAβ‐induced TNFα secretion. Mixed cultures were selected to account for the paracrine‐like signaling that can occur between astrocytes and microglia, and therefore to mimic more closely in vivo neuroinflammation. However, as TLR4/LPS‐induced TNFα secretion follows opposing rank orders in astrocytes (APOE2 ≥ APOE3 ≥ APOE4) and microglia (APOE4 > APOE3 > APOE2) (Fig. 1a), initially the effect of APOE on LPS‐induced TNFα secretion was determined in mixed glial cultures (95% astrocytes, 5% microglia) isolated from APOE‐TR and APOE‐KO mice. To monitor neuroinflammation in vitro, TNFα (pro‐inflammatory) secretion was measured in the media by ELISA, as overall data support a detrimental role for TNFα in AD (reviewed in Tobinick 2009). In the mixed glial cultures from APOE‐TR and APOE‐KO mice, LPS (100 ng/mL, 24‐h incubation)‐induced TNFα levels from highest to lowest are: APOE‐KO > APOE4 > APOE3 > APOE2 (Fig. 3a). Indeed, compared with APOE3, TNFα levels are 50% higher with APOE4 and 50% lower with APOE2. These data are consistent with microglial rather than astrocytic APOE genotype‐specific TNFα secretion (Maezawa et al. 2006a), potentially because of paracrine‐like signaling between the two glial cell types.
Next, whether TLR4 antagonism could inhibit LPS‐induced TNFα secretion was determined. LPS activates TLR4 through four proteins, LPS‐binding protein (LBP), CD14, the myeloid differentiation protein‐2 (MD‐2), and TLR4. Essentially, LBP interacts with and transfers LPS to CD14, which in turn transfers LPS to MD‐2 (Ulevitch & Tobias 1999). An activated [TLR4.MD‐2.LPS]2 complex was then formed that induces TLR4 signaling. LPS from Rhodobacter sphaeroides (LPS‐RS) inhibits the MD2–TLR4 interaction and was therefore a TLR4 antagonist. Consistent with TLR4 activation, LPS‐RS (10 μg/mL) inhibits LPS (100 ng/mL)‐induced TNFα secretion in mixed glial cultures isolated from all three strains of APOE‐TR mice and APOE‐KO mice (Fig. 3a).
oAβ‐induced TNFα secretion in mixed glial cultures isolated from APOE‐TR and APOE‐KO mice: APOE‐KO > APOE4 > APOE3 > APOE2 and blocked by TLR4 antagonism
As described (see Aβ‐dependent activation of neuroinflammation), dissecting APOE modulation of oAβ‐induced neuroinflammation in vitro remains an important question. Indeed, unlike TLR4/LPS‐induced activation of microglia and astrocytes, the effect and potential pathways underlying APOE modulation of oAβ‐induced acute cytokine secretion in vitro remain unknown, which confounds the development of future in vitro experiments to mimic long‐term neuroinflammation. oAβ (10 μM, 24‐h incubation)‐induced TNFα secretion follows the identical rank order as for LPS in the isolated mixed glial cultures: APOE‐KO > APOE4 > APOE3 > APOE2 (Fig. 3b). For example, TNFα levels are 80% higher with APOE4 compared to APOE3. As APOE modulated both oAβ‐ and TLR4/LPS‐induced neuroinflammation, we explored whether TLR4 antagonism also blocks oAβ‐induced TNFα secretion. LPS‐RS (10 μg/mL) antagonizes oAβ‐induced (10 μM) TNFα secretion, resulting in an ~ 80% reduction in TNFα levels in glia isolated from all three strains of APOE‐TR mice and APOE‐KO mice (Fig. 3b). To dissect whether antagonists of additional TLR4 signaling complex components also inhibit oAβ‐induced TNFα secretion, novel antagonists of the CD14‐MD2 interaction, IAXO101 and IAXO103, were utilized. At an equivalent concentration (5 μg/mL), IAXO101 and IAXO103 are more potent than LPS‐RS, whereas the inactive IAXO202 control did not suppress oAβ‐induced TNFα secretion in mixed glial cultures from APOE4‐TR mice (Fig. 3c).
LPS‐ and oAβ‐induced TNFα secretion in mixed glial cultures isolated from APOE3‐ and APOE4‐TR mice: Prevented by p38α inhibition
A potential down‐stream signaling pathway of TLR4 is p38α, and p38α inhibitors suppress LPS‐ and Aβ42‐induced neuroinflammation in vitro and in vivo (Watterson et al. 2013; Bachstetter et al. 2014). As LPS‐ and oAβ‐induced TNFα secretion are both inhibited by TLR4 antagonists, mixed glial cultures from APOE3‐TR and APOE4‐TR mice were used to define the role of p38α in both LPS‐ and oAβ‐induced TNFα secretion. In this study, MW181, a p38α inhibitor (Watterson et al. 2013), dose‐dependently inhibits LPS‐ and oAβ‐induced TNFα secretion for both APOE3 and APOE4 (Fig. 3d and e).
In summary, in vitro data indicate that apoE4 represents a loss of positive function rather than a toxic gain of function for oAβ‐induced TNFα secretion (i.e., TNFα levels are higher in mixed glia isolated from APOE‐KO compared to APOE4 mice, see APOE4 is likely a loss of positive function for Aβ‐induced neuroinflammation), and APOE genotype‐specific modulation of TNFα secretion follows the same pattern of APOE‐induced AD risk: APOE4 > APOE3 > APOE2. An additional observation is that LPS (100 ng/mL) induces greater TNFα secretion than oAβ (10 μM). LPS is a high‐affinity agonist for TLR4, and therefore at low concentrations induces robust functional effects in vivo, including TNFα secretion and neuronal dysfunction in acute studies (Maezawa et al. 2006b; Pocivavsek et al. 2009a,b; Vitek et al. 2009; Keene et al. 2011; Zhu et al. 2012; Gale et al. 2014). As a chronic condition with progressive cognitive decline over time, changes in the NIP, e.g., TNFα levels, are likely less pronounced but more sustained in AD than with acute LPS treatment. Therefore, the lower TNFα levels with oAβ compared to LPS, potentially because of lower TLR4 agonist activity, may allow for the future development of long‐term mixed glial cultures designed to mimic the AD‐NIP. In addition, TLR4 antagonism and p38α inhibition may be particularly beneficial for APOE4 carriers, as LPS‐ and oAβ‐induced TNFα secretion is higher for APOE4 than APOE3, and TLR4 antagonists and p38α inhibitors can decrease TNFα levels to baseline for both APOE genotypes.
Discussion
Using the EFAD mice as an AD‐Tg mouse model that combines the regulated expression of the h‐apoE isoforms with the over‐production of Aβ42, we have begun to address the effect of APOE on the complex AD‐NIP in vivo. One important finding was that not all markers within pro‐inflammatory, anti‐inflammatory, repair, and immunoregulatory classifications were equally modulated by APOE genotype. As an example, anti‐inflammatory/repair markers were both lower (IL‐10, FIZZ1, SOCS2, SOCS3) and higher (ARG, SOCS1) in E3FAD mice at 6 months when assessed by cluster analysis comparing 6‐ and 8‐month‐old E3FAD and E4FAD mice (Fig. 2a). Our current work is focused on further dissecting the NIP, including the signaling pathways that may underlie the differential expression of markers within each class.
Our novel data also demonstrate consistently higher levels of genes considered pro‐inflammatory (e.g., TNFα, IL‐1β) in E4FAD mice compared to E3FAD mice, regardless of age. These markers were low in 6‐month‐old E3FAD mice and increased by 8 months. The dramatic differences in neuroinflammation between 6 and 8 months in EFAD mice are likely related to the significant increases in APOE‐modulated Aβ pathology that occurs between these time points (manuscript in preparation). Critically, there were lower levels of an anti‐inflammatory and repair gene cluster in E4FAD mice compared to E3FAD at 6 months. Through further in vivo and in vitro analysis, an important finding emerged, that the cytokine TNFα, and the receptor pathways TLR4‐p38α and IL‐4R might play a key role in APOE‐modulated Aβ‐induced neuroinflammation.
TNFα as a target for APOE‐modulated Aβ‐induced neuroinflammation
Targeting TNFα may be particularly efficacious for APOE4 carriers, as Aβ‐induced TNFα levels were higher with APOE4 than APOE3 in vitro and in vivo. TNFα has been demonstrated to directly inhibit long‐term potentiation and synaptic plasticity, to impair insulin signaling in neurons and induce neuronal toxicity (reviewed in Tobinick 2009). Furthermore, TNFα levels are up to 25‐fold higher in CSF from AD patients compared to controls, and correlate with dementia (Tobinick 2009). Based on these lines of evidence, anti‐TNFα therapies appear to be promising candidates for AD treatment. Entanercept is the therapeutic that is furthest advanced clinically. The drug binds to and neutralizes TNFα, and has been reported to improve cognition in mild‐ to late‐AD patients in a 6‐month, open‐label pilot study, a result that is supported in select case reports (Tobinick 2009). Although the cellular target of this therapy remains unclear, and may involve cells in the immediate vicinity of the blood–cerebrospinal fluid barrier, these results are encouraging and support TNFα as a potential therapeutic target.
TLR4 as a target for APOE‐modulated Aβ‐induced neuroinflammation
Targeting the TLR4‐p38α pathway may provide a therapeutic opportunity for APOE‐modulated Aβ‐induced neuroinflammation. Indeed, select markers associated with TLR4 were higher in E4FAD mice compared to E3FAD mice at 6 months, TLR4 expression was increased from 6 to 8 months in both E3FAD and E4FAD mice, and TLR4 antagonists inhibited APOE‐modulated oAβ‐induced TNFα secretion in vitro. Data from AD patients, FAD‐Tg mice, and in vitro studies conflict on the effect of TLR4 antagonism for AD risk, inflammation, and Aβ levels, which can be broadly grouped into beneficial or detrimental. Evidence for beneficial effects of TLR4 antagonism include: (i) decreased AD risk in individuals expressing a blunted TLR4 signaling polymorphism (Minoretti et al. 2006); (ii) expression of inactive TLR4 in FAD‐Tg mice reduces neuroinflammation (Jin et al. 2008); (iii) inactivation of microglial TLR4 signaling prevents Aβ‐mediated neurotoxicity in vitro (Walter et al. 2007); (iv) chronic LPS treatment increases Aβ deposition in FAD‐Tg mice (Sheng et al. 2003; Lee et al. 2008); and (v) TLR4 knockdown in neurons prevents Aβ‐induced neurotoxicity (Tang et al. 2008). Evidence for detrimental effects of TLR4 antagonism include: (i) increased AD risk in individuals with a TLR4 mutation that reduces TLR4 expression (Wang et al. 2011); (ii) increased levels of both soluble and insoluble Aβ with genetic inactivation of TLR4 signaling in vivo (Tahara et al. 2006; Song et al. 2011); and (iii) that LPS (DiCarlo et al. 2001; Quinn et al. 2003; Herber et al. 2004; Malm et al. 2005; Herber et al. 2007) or a partial TLR4 agonist, monophosphoryl lipid A (MPL), (Michaud et al. 2013) decreases Aβ levels in FAD‐Tg mice.
As reviewed in section Neuroinflammatory receptors in AD, an important consideration is the contribution of TLR4 to Aβ phagocytosis and inflammatory mediator production. One perspective is that when Aβ levels are high over a short‐to‐mid time frame, TLR4 agonists will further exacerbate pro‐inflammatory phenotype and synaptic dysfunction, and therefore TLR4 antagonists are the appropriate strategy. If with longer periods of high Aβ levels there is a general suppression of all glial neuroinflammation, i.e., both pro‐inflammatory and anti‐inflammatory/repair, and Aβ phagocytosis is suppressed, then TLR4 agonists may reactivate aspects of glial activation (both astrocytes and microglia) and induce beneficial effects, e.g., phagocytosis and an adaptive IL‐4R response (see APOE modulation of the Aβ‐induced NIP: IL‐4R signaling).
An additional consideration for dissecting the role of TLR4 in AD is the cell‐ and species‐specific expression. Although microglia express functional TLR4, data are conflicting on the expression and activity of TLR4 in isolated astrocyte cultures (Bsibsi et al. 2002; Lehnardt et al. 2002; Sola et al. 2002; Bowman et al. 2003; Falsig et al. 2004; Carpentier et al. 2005). One confounding factor is the purity of the in vitro cultures, as contaminating microglia may be responsible for the TLR4 agonist‐induced pro‐inflammatory cytokine secretion directly, and/or through activating astrocytes via paracrine‐like signaling. A further issue is whether human and murine astrocytes respond differentially to TLR4 activation. Recent data demonstrate that isolated human and murine astrocytes display differential innate immune activation pathways (Tarassishin et al. 2014). TLR4/LPS activation of isolated murine, but not human astrocytes induces TNFα secretion because of a lack of CD14 expression in human astrocytes. Further cell‐type‐ and species‐specific analysis of TLR4 agonist‐induced signaling pathway activation, including the contribution of paracrine‐like signaling between glial cell types, will yield critical information for dissecting the translational significance of these findings.
Data for p38α appear more straightforward as a therapeutic, as p38α inhibition with MW181 reduces brain pro‐inflammatory cytokine production after LPS administration and attenuates synaptic dysfunction in an AD‐relevant mouse model (Watterson et al. 2013; Bachstetter et al. 2014). Pre‐clinical therapeutic studies are needed to clarify the role of the TLR4‐p38α pathway on AD‐like pathology, including addressing confounding factors that influence overall efficacy of TLR4 agonists or antagonists such as length of treatment, time of treatment, and the incorporation of h‐APOE.
APOE modulation of the Aβ‐induced NIP: IL‐4R signaling
Differential activation of the IL‐4R pathway may also significantly underlie APOE genotype‐specific Aβ‐induced neuroinflammation, as markers associated with the IL‐4R pathway were lower in E4FAD mice compared to E3FAD mice at 6 months, and IL‐4R expression decreased in E3FAD mice from 6 to 8 months. TLR4 and IL‐4R exert opposing functions in peripheral inflammation. In peripheral macrophages, TLR4 activation induces a pro‐inflammatory response leading to tissue damage, whereas IL‐4R activation induces a response associated with cellular proliferation and repair (Mantovani et al. 2004; Benoit et al. 2008). Importantly, evidence indicates that TLR4 and IL‐4R also exert opposing effects on neuroinflammation. Peripheral LPS injection results in a TLR4‐mediated pro‐inflammatory response in the CNS, countered by increased microglial IL‐4R expression in young mice (Fenn et al. 2012). Furthermore, the adaptive IL‐4R response is diminished in old mice (Fenn et al. 2012), data that are significant as age is the greatest non‐genetic AD risk factor. The products of IL‐4R signaling play a number of important functions that may be relevant for the CNS. For example, IL‐4/IL‐4R signaling decreases NO levels through increasing Arg1, which competitively inhibits iNOS for arginine metabolism (Colton et al. 2004, 2006), and also increases the production of polyamines, proline, and ornithines that increase matrix deposition and promote a repair function in the CNS (Munder 2009). However, whether the increased levels of Arg1 are beneficial or detrimental in AD pathology remains unclear, particularly as Arg1 levels are reportedly higher in AD patients compared to controls (Colton et al. 2004, 2006). In addition, in vitro, IL‐4/IL‐4R signaling suppresses LPS‐induced TNFα secretion by astrocytes, and conditioned media from IL‐4 activated astrocytes and microglia promotes neuronal viability (Gadani et al. 2012). In terms of modulating Aβ levels, IL‐4 has been demonstrated to both decrease (Gadani et al. 2012) and increase Aβ levels (Chakrabarty et al. 2012) in FAD‐Tg mice, conflicting data that is subject to the same issues as TLR4.
The data presented here indicate that in response to Aβ, lower IL‐4R expression and/or signaling may impart a lower anti‐inflammatory and repair response with APOE4. However, as described, this is a relatively simplistic description of the data and it is clear that there is a more complex interplay between and across NIP markers. For example, although IL‐4R expression was decreased from 6 to 8 months in E3FAD mice, SOCS2 and SOCS3 expression was increased, despite the fact that both cytokines are produced by IL‐4R signaling in the periphery (Ratthe et al. 2007).
Interpretation and future directions
How might APOE modulate the neuroinflammatory phenotype?
APOE4 is likely a loss of positive function for Aβ‐induced neuroinflammation
For virtually every function linked to APOE in AD, there is the unresolved question of whether apoE4 or all the h‐apoE isoforms represent a toxic gain of function or loss of positive function. To dissect this issue requires a comparison among mice or cells that express the APOE genotypes and APOE‐KO. Overall, if apoE4 is a toxic gain of function for a detrimental neuroinflammatory response, e.g., LPS‐induced TNFα secretion, then the expected result would be: APOE4 > APOE‐KO, i.e., APOE4 is detrimental compared to a lack of APOE. If apoE4 is a loss of positive function then the expected result would be: APOE‐KO ≥ APOE4 i.e., APOE4 is neutral or beneficial compared to no APOE. Our in vitro data suggest that for neuroinflammation, apoE4 is a relative loss of positive function compared to apoE3. Indeed, in mixed glial cultures isolated from APOE‐TR mice and APOE‐KO mice, both oAβ and LPS‐induced TNFα secretion follows the order from highest to lowest: APOE‐KO > APOE4 > APOE3 > APOE2, consistent with LPS ICV injection data in APOE‐TR and APOE‐KO mice (Zhu et al. 2012). These data support that apoE4 is anti‐inflammatory compared to the absence of apoE, but less anti‐inflammatory than apoE3. However, the overall effect of APOE4 on any given CNS‐specific measure will be the product of multiple overlapping mechanisms. Thus, caution remains critical in determining whether APOE gene inactivation or therapies that correct the loss of positive function related to apoE4, are the appropriate therapeutic response.
Proposed hypothesis of APOE‐modulated Aβ‐induced neuroinflammation
How the apoE isoforms modulate neuroinflammation is likely not through a single mechanism, but the combination of multiple pro‐ and anti‐inflammatory processes. One potential pathway is apoE isoform‐specific effects on non‐inflammatory functions, such as the increased Aβ levels that occur with APOE4 in vivo. Although modulation of Aβ levels will contribute to the APOE‐modulated NIP, TLR4/LPS‐induced neuroinflammation is also apoE isoform‐specific, highlighting a pathway(s) independent of Aβ levels. Based on published and the data presented herein, we propose the hypothesis that apoE modulates inflammation through receptor signaling pathways. Specifically, impaired apoE4 function compromises apoE receptor activation, modulating Aβ‐induced effects on inflammatory receptor signaling, specifically causing amplification of detrimental (TLR4‐p38α), and suppression of beneficial (IL‐4R‐nuclear receptor) pathways, resulting in an adverse NIP that causes neuronal dysfunction (Fig. 4).
ApoE isoform‐specific functional effects could be mediated through structural differences including lipidation, and/or apoE levels. As the protein component of apolipoproteins, apoE function is, in part, modulated by lipidation (Fisher & Ryan 1999; Dergunov et al. 2000; LaDu et al. 2006; Tai et al. 2014a,b). Recent data support the long‐held belief that apoE4‐containing CNS lipoproteins are less lipidated than apoE3‐containing CNS lipoproteins (Youmans et al. 2012; Boehm‐Cagan & Michaelson 2014; Tai et al. 2014a,b), reducing the stability of apoE4 (Hirsch‐Reinshagen et al. 2004; Wahrle et al. 2004) and resulting in reduced apoE4 levels (Riddell et al. 2008; Vitek et al. 2009; Sullivan et al. 2011; Cruchaga et al. 2012; Youmans et al. 2012; Martinez‐Morillo et al. 2014). In addition, cellular recycling of apoE4 is impaired (Chen et al. 2010) contributing to the lower apoE4 levels.
The different structural properties and lower levels of apoE4, compared to apoE3, may result in effects on apoE receptor activation, including lower apoE receptor‐binding and agonist activity. Thus, an important consideration is whether the apoE isoforms differentially bind to apoE receptors. In the periphery, apoE2 binds with lower affinity to LDLR and LRP1 compared to apoE3 and apoE4 (Weisgraber et al. 1982; Mahley et al. 2009; Hauser et al. 2011). These data potentially conflict with our in vitro data that apoE2 suppresses both LPS and oAβ‐induced TNFα secretion to a greater extent than apoE3 and apoE4, as well as our stated hypothesis. Therefore, it is important to determine whether the apoE isoforms differentially bind to the LDLR family using CNS‐relevant apoE‐containing lipoproteins and cell types. For example, apoE binds to heparin sulfate proteoglycans, to facilitate receptor dependent and independent (Mahley & Ji 1999) uptake of apoE. These processes are likely apoE isoform specific and dependent on the cell type (Weisgraber et al. 1982; Mahley et al. 2009; Hauser et al. 2011). Indeed, extracellular matrix in the CNS is extensive and complex, includes a high content of heparin sulfate proteoglycans, and its composition varies with cell type. Thus, it is difficult to predict receptor‐binding affinity for apoE receptors in the CNS based on results primarily using non‐CNS apoE‐containing lipoproteins/lipid particles and fibroblast cells (Weisgraber et al. 1982; Mahley et al. 2009; Hauser et al. 2011). In addition to agonist activity, the apoE isoforms may differentially modulate apoE receptor levels. In neurons, apoE4 impairs apoE receptor recycling, resulting in lower apoE receptor levels (Chen et al. 2010). This will likely have a paracrine‐like effect on glia via changes in neuronal derived factors. Importantly, similar processes may occur in glia, thus the apoE isoforms may directly modulate apoE receptor signaling pathway interactions with Aβ‐induced neuroinflammatory receptors.
TLR4 and IL‐4R are two inflammatory receptors that may be modulated by apoE receptor signaling, as there is significant potential signaling pathway overlap particularly with the apoE receptors LRP1 and LDLR (Fig. 4). In vitro, apoE signaling (apoE3 > apoE4) via the apoE receptors may suppress Aβ‐induced activation of the TLR4‐p38α pathway. In vitro, cytokines associated with TLR4 activation are higher in E4FAD compared to E3FAD mice, at 6 months (Fig. 2c), oAβ‐induced TNFα secretion is APOE genotype‐specific (apoE4 > apoE3) (Fig. 3b), and LPS‐ and Aβ‐induced TNFα secretion is inhibited by TLR4 antagonists and p38α inhibitors (Fig. 3d and e; Bachstetter et al. 2014). For the IL‐4R, apoE may modulate pathways that converge on nuclear receptor signaling (Fig. 4). IL‐4R signaling is linked to the JAK‐STAT‐nuclear receptor pathway (e.g., PPARγ, RXR, LXR) in the periphery, and nuclear receptor agonists suppress pro‐inflammatory and increase anti‐inflammatory/repair markers in FAD‐Tg mice (Xu et al. 2005; Lefterov et al. 2007; Zelcer et al. 2007; Cherry et al. 2014; Skerrett et al. 2014; Spillmann et al. 2014). Specific to apoE, our data demonstrate that in E4FAD mice, markers of IL‐4R signaling (Fig. 3a) and the expression of nuclear receptors (PPARγ, RXR, LXR) were lower compared to E3FAD mice (Tai et al. 2014a). Furthermore, chronic neuroinflammation caused by Aβ and age may exacerbate the apoE4 loss of positive function that results in the higher suppression of IL‐4R signaling compared to apoE3. Indeed, increasing Aβ pathology leads to lower expression of nuclear receptors, an effect greater in E4FAD compared to E3FAD mice (Tai et al. 2014a), IL‐4R expression was decreased from 6 to 8 months in E3FAD mice, and the IL‐4R response was lower in old versus young mice (Fenn et al. 2012). Thus, activating apoE receptors, e.g., by apoE mimetic peptides (Vitek et al. 2012), and/or targeting the inflammatory receptor pathways modulated by apoE and Aβ may represent promising therapeutic strategies. The apoE isoform‐specific modulation of cellular apoE receptor binding/activation (e.g., LRP1, LDLR) and effects on downstream signaling pathways remain unclear (Phillips 2014). As presented in Fig. 4, this hypothesis is designed to spark debate and further research in this largely overlooked and complicated field (Michaelson 2014).
Future perspective
The complex nature of chronic neuroinflammation has resulted in conflicting data surrounding whether any specific inflammatory mediator or receptor is beneficial or detrimental for AD progression, and incorporating APOE into the equation amplifies this complexity. We have begun to address the role of APOE in Aβ‐induced neuroinflammation, and have identified TLR4 and IL‐4R responses as critical pathways. However, further research into this topic is sorely needed based on the proposed central role of neuroinflammation in AD and the 12‐fold increased risk for AD with APOE4 (Verghese et al. 2011).
We propose a number of future research directions to enable a deeper understanding of APOE‐modulated neuroinflammation in AD. The first, and most critical, is to identify the APOE‐specific NIP in Aβ‐relevant models and in AD patients across disease progression. An additional aspect of this approach is to assess the extent that in vitro and in vivo models recapitulate the NIP during aging and AD progression in humans. Furthermore, developing an AD‐relevant in vitro model of chronic neuroinflammation that incorporates astrocyte–microglia–neuron interactions and, as technology evolves, other inflammatory cell types such as components of the neurovascular unit (e.g., pericytes, BECs), is vital for mechanistic studies. This approach will delineate whether apoE isoforms modulate predisposition, amplification, and/or the adaptive response as chronic neuroinflammation progresses. Furthermore, for individual or groups of markers, it is important to determine conceptually whether APOE4 accelerates or imparts a differential multiphasic NIP compared to APOE3. In addition, dissecting how and through which pathways apoE modulates neuroinflammation and the functional consequences for neuronal function may ultimately lead to APOE genotype‐specific therapeutics.
Acknowledgments and conflict of interest disclosure
This work was supported by NIH/NIA P01AG030128 (MJL), NIH/NIA R21AG048498 (MJL), NIH/NINDS R01NS064247 (LVE), University of Illinois at Chicago (UIC), Center for Clinical and Translational Science Grant UL1RR029879 (MJL), a UIC OVCR grant and NIH UL1TR000050 (University of Illinois CTSA that supports the UIC Center for Research Informatics).
All experiments were conducted in compliance with the ARRIVE guidelines. The authors have no conflict of interest to declare.
References
- ADAPT‐FS‐Research‐Group (2014) Follow‐up evaluation of cognitive function in the randomized Alzheimer's Disease Anti‐inflammatory Prevention Trial and its Follow‐up Study. Alzheimers Dement. doi: 10.1016/j.jalz.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ADAPT‐Research‐Group (2013) Results of a follow‐up study to the randomized Alzheimer's disease Anti‐inflammatory Prevention Trial (ADAPT). Alzheimers Dement. 9, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A., Barres B. A. and Bennett M. L. (2013) Microglia: scapegoat, saboteur, or something else? Science 339, 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Barger S., Barnum S. et al (2000) Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo O. G., Bolick D. T., Roche J. K., Pinkerton R. F., Lima A. A., Vitek M. P., Warren C. A., Oria R. B. and Guerrant R. L. (2014) Apolipoprotein E plays a key role against cryptosporidial infection in transgenic undernourished mice. PLoS ONE 9, e89562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter A. D. and Van Eldik L. J. (2010) The p38 MAP kinase family as regulators of proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. 1, 199–211. [PMC free article] [PubMed] [Google Scholar]
- Bachstetter A. D., Watterson D. M. and Van Eldik L. J. (2014) Target engagement analysis and link to pharmacodynamic endpoint for a novel class of CNS‐penetrant and efficacious p38alpha MAPK inhibitors. J. Neuroimmune Pharmacol. 9, 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks W. A. and Robinson S. M. (2010) Minimal penetration of lipopolysaccharide across the murine blood‐brain barrier. Brain Behav. Immun. 24, 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum A. N., Cunha C., Sidhu H., Alkam T., Scolnick J., Rosario E. R. and Ethell D. W. (2014) Women with the Alzheimer's risk marker ApoE4 lose Aβ‐specific CD4(+) T cells 10‐20 years before men. Transl. Psychiatry 4, e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinson H. and Michaelson D. M. (2009) ApoE4‐dependent Aβ‐mediated neurodegeneration is associated with inflammatory activation in the hippocampus but not the septum. J. Neural. Transm. 116, 1427–1434. [DOI] [PubMed] [Google Scholar]
- Bell R. D., Winkler E. A., Singh I. et al (2012) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit M., Desnues B. and Mege J. L. (2008) Macrophage polarization in bacterial infections. J. Immunol. 181, 3733–3739. [DOI] [PubMed] [Google Scholar]
- Bettoni I., Comelli F., Rossini C., Granucci F., Giagnoni G., Peri F. and Costa B. (2008) Glial TLR4 receptor as new target to treat neuropathic pain: efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia 56, 1312–1319. [DOI] [PubMed] [Google Scholar]
- Boda E., Pini A., Hoxha E., Parolisi R. and Tempia F. (2009) Selection of reference genes for quantitative real‐time RT‐PCR studies in mouse brain. J. Mol. Neurosci. 37, 238–253. [DOI] [PubMed] [Google Scholar]
- Boehm‐Cagan A. and Michaelson D. M. (2014) Reversal of apoE4‐driven brain pathology and behavioral deficits by bexarotene. J. Neurosci. 34, 7293–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bont N., Netea M. G., Demacker P. N., Verschueren I., Kullberg B. J., van Dijk K. W., van der Meer J. W. and Stalenhoef A. F. (1999) Apolipoprotein E knock‐out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J. Lipid Res. 40, 680–685. [PubMed] [Google Scholar]
- Bowman C. C., Rasley A., Tranguch S. L. and Marriott I. (2003) Cultured astrocytes express toll‐like receptors for bacterial products. Glia 43, 281–291. [DOI] [PubMed] [Google Scholar]
- Bradshaw E. M., Chibnik L. B., Keenan B. T. et al (2013) CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitner J. C., Baker L. D., Montine T. J. et al (2011) Extended results of the Alzheimer's disease anti‐inflammatory prevention trial. Alzheimers Dement. 7, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britschgi M., Takeda‐Uchimura Y., Rockenstein E., Johns H., Masliah E. and Wyss‐Coray T. (2012) Deficiency of terminal complement pathway inhibitor promotes neuronal tau pathology and degeneration in mice. J. Neuroinflammation 9, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers N., Van Cauwenberghe C., Engelborghs S. et al (2012) Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry 17, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. M., Wright E., Colton C. A., Sullivan P. M., Laskowitz D. T. and Vitek M. P. (2002) Apolipoprotein E isoform mediated regulation of nitric oxide release. Free Radic. Biol. Med. 32, 1071–1075. [DOI] [PubMed] [Google Scholar]
- Bsibsi M., Ravid R., Gveric D. and van Noort J. M. (2002) Broad expression of Toll‐like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61, 1013–1021. [DOI] [PubMed] [Google Scholar]
- Bu G. (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan M. M., Hutchinson M., Watkins L. R. and Yin H. (2010) Toll‐like receptor 4 in CNS pathologies. J. Neurochem. 114, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchart J. and Holmes C. (2012) Systemic and central immunity in Alzheimer's disease: therapeutic implications. CNS Neurosci. Ther. 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O., Jedrychowski M. P., Moore C. S. et al (2014) Identification of a unique TGF‐beta‐dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron B. and Landreth G. E. (2010) Inflammation, microglia, and Alzheimer's disease. Neurobiol. Dis. 37, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier P. A., Begolka W. S., Olson J. K., Elhofy A., Karpus W. J. and Miller S. D. (2005) Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 49, 360–374. [DOI] [PubMed] [Google Scholar]
- Cash J. G., Kuhel D. G., Basford J. E., Jaeschke A., Chatterjee T. K., Weintraub N. L. and Hui D. Y. (2012) Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J. Biol. Chem. 287, 27876–27884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P., Tianbai L., Herring A., Ceballos‐Diaz C., Das P. and Golde T. E. (2012) Hippocampal expression of murine IL‐4 results in exacerbation of amyloid deposition. Mol. Neurodegener. 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Durakoglugil M. S., Xian X. and Herz J. (2010) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl Acad. Sci. USA 107, 12011–12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J. D., Olschowka J. A. and O'Banion M. K. (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhor V., Le Charpentier T., Lebon S. et al (2013) Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 32, 70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouraki V. and Seshadri S. (2014) Genetics of Alzheimer's disease. Adv. Genet. 87, 245–294. [DOI] [PubMed] [Google Scholar]
- Colton C. A., Xu Q., Burke J. R., Bae S. Y., Wakefield J. K., Nair A., Strittmatter W. J. and Vitek M. P. (2004) Disrupted spermine homeostasis: a novel mechanism in polyglutamine‐mediated aggregation and cell death. J. Neurosci. 24, 7118–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton C. A., Mott R. T., Sharpe H., Xu Q., Van Nostrand W. E. and Vitek M. P. (2006) Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J. Neuroinflammation 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook N. L., Vink R., Donkin J. J. and van den Heuvel C. (2009) Validation of reference genes for normalization of real‐time quantitative RT‐PCR data in traumatic brain injury. J. Neurosci. Res. 87, 34–41. [DOI] [PubMed] [Google Scholar]
- Crehan H., Holton P., Wray S., Pocock J., Guerreiro R. and Hardy J. (2012) Complement receptor 1 (CR1) and Alzheimer's disease. Immunobiology 217, 244–250. [DOI] [PubMed] [Google Scholar]
- Cruchaga C., Kauwe J. S., Nowotny P. et al (2012) Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer's disease. Hum. Mol. Genet. 21, 4558–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudaback E., Yang Y., Montine T. J. and Keene C. D. (2015) APOE genotype‐dependent modulation of astrocyte chemokine CCL3 production. Glia 63, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergunov A. D., Smirnova E. A., Merched A., Visvikis S., Siest G., Yakushkin V. V. and Tsibulsky V. (2000) Conformation of apolipoprotein E both in free and in lipid‐bound form may determine the avidity of triglyceride‐rich lipoproteins to the LDL receptor: structural and kinetic study. Biochim. Biophys. Acta 1484, 14–28. [DOI] [PubMed] [Google Scholar]
- DiCarlo G., Wilcock D., Henderson D., Gordon M. and Morgan D. (2001) Intrahippocampal LPS injections reduce Aβ load in APP+PS1 transgenic mice. Neurobiol. Aging 22, 1007–1012. [DOI] [PubMed] [Google Scholar]
- Dorey E., Chang N., Liu Q. Y., Yang Z. and Zhang W. (2014) Apolipoprotein E, amyloid‐beta, and neuroinflammation in Alzheimer's disease. Neurosci. Bull. 30, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drye L. T. and Zandi P. P. (2012) Role of APOE and age at enrollment in the Alzheimer's disease anti‐inflammatory prevention trial (ADAPT). Dement. Geriatr. Cogn. Dis. Extra 2, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P., Archer H. A., Gerhard A. et al (2008) Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK11195‐PET and [11C]PIB‐PET study. Neurobiol. Dis. 32, 412–419. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P. and van Gool W. A. (2004) Neuroinflammatory perspectives on the two faces of Alzheimer's disease. J. Neural. Transm. 111, 281–294. [DOI] [PubMed] [Google Scholar]
- Ennaciri J. and Girard D. (2009) IL‐4R(alpha), a new member that associates with Syk kinase: implication in IL‐4‐induced human neutrophil functions. J. Immunol. 183, 5261–5269. [DOI] [PubMed] [Google Scholar]
- Falsig J., Latta M. and Leist M. (2004) Defined inflammatory states in astrocyte cultures: correlation with susceptibility towards CD95‐driven apoptosis. J. Neurochem. 88, 181–193. [DOI] [PubMed] [Google Scholar]
- Farlow M. R., Lahiri D. K., Poirier J., Davignon J., Schneider L. and Hui S. L. (1998) Treatment outcome of tacrine therapy depends on apolipoprotein genotype and gender of the subjects with Alzheimer's disease. Neurology 50, 669–677. [DOI] [PubMed] [Google Scholar]
- Fenn A. M., Henry C. J., Huang Y., Dugan A. and Godbout J. P. (2012) Lipopolysaccharide‐induced interleukin (IL)‐4 receptor‐alpha expression and corresponding sensitivity to the M2 promoting effects of IL‐4 are impaired in microglia of aged mice. Brain Behav. Immun. 26, 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti M. T. and Cuello A. C. (2011) Does a pro‐inflammatory process precede Alzheimer's disease and mild cognitive impairment? Curr. Alzheimer Res. 8, 164–174. [DOI] [PubMed] [Google Scholar]
- Fisher C. A. and Ryan R. O. (1999) Lipid binding‐induced conformational changes in the N‐terminal domain of human apolipoprotein E. J. Lipid Res. 40, 93–99. [PubMed] [Google Scholar]
- Ford J. W. and McVicar D. W. (2009) TREM and TREM‐like receptors in inflammation and disease. Curr. Opin. Immunol. 21, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller J. P., Stavenhagen J. B. and Teeling J. L. (2014) New roles for Fc receptors in neurodegeneration‐the impact on immunotherapy for Alzheimer's disease. Front. Neurosci. 8, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadani S. P., Cronk J. C., Norris G. T. and Kipnis J. (2012) IL‐4 in the brain: a cytokine to remember. J. Immunol. 189, 4213–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale S. C., Gao L., Mikacenic C. et al (2014) APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 134, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. (2003) Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35. [DOI] [PubMed] [Google Scholar]
- Gorelick P. B. (2010) Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann. N. Y. Acad. Sci. 1207, 155–162. [DOI] [PubMed] [Google Scholar]
- Green A. E., Gray J. R., Deyoung C. G., Mhyre T. R., Padilla R., Dibattista A. M. and William Rebeck G. (2014) A combined effect of two Alzheimer's risk genes on medial temporal activity during executive attention in young adults. Neuropsychologia 56, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin W. S. and Mrak R. E. (2002) Interleukin‐1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer's disease. J. Leukoc. Biol. 72, 233–238. [PMC free article] [PubMed] [Google Scholar]
- Griffin W. S. T., Stanley L. C., Ling C., White L., MacLeod V., Perrot L. J., White C. L. and Araoz C. (1989) Brain interleukin 1 and S‐100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl Acad. Sci. USA 86, 7611–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubern C., Hurtado O., Rodriguez R., Morales J. R., Romera V. G., Moro M. A., Lizasoain I., Serena J. and Mallolas J. (2009) Validation of housekeeping genes for quantitative real‐time PCR in in‐vivo and in‐vitro models of cerebral ischaemia. BMC Mol. Biol. 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R., Wojtas A., Bras J. et al (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., LaDu M. J. and Van Eldik L. J. (2004) A dual role for apolipoprotein e in neuroinflammation: anti‐ and pro‐inflammatory activity. J. Mol. Neurosci. 23, 205–212. [DOI] [PubMed] [Google Scholar]
- Haass C. and Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Hanamsagar R., Hanke M. L. and Kielian T. (2012) Toll‐like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 33, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. (2006) Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J. Alzheimers Dis. 9, 151–153. [DOI] [PubMed] [Google Scholar]
- Hardy J., Bogdanovic N., Winblad B., Portelius E., Andreasen N., Cedazo‐Minguez A. and Zetterberg H. (2014) Pathways to Alzheimer's disease. J. Intern. Med. 275, 296–303. [DOI] [PubMed] [Google Scholar]
- Hashioka S., Monji A., Ueda T., Kanba S. and Nakanishi H. (2005) Amyloid‐beta fibril formation is not necessarily required for microglial activation by the peptides. Neurochem. Int. 47, 369–376. [DOI] [PubMed] [Google Scholar]
- Hauser P. S., Narayanaswami V. and Ryan R. O. (2011) Apolipoprotein E: from lipid transport to neurobiology. Prog. Lipid Res. 50, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden E. Y. and Teplow D. B. (2013) Amyloid beta‐protein oligomers and Alzheimer's disease. Alzheimers Res. Ther. 5, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy E. J., Parker A. E. and O'Neill L. A. (2010) Targeting toll‐like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307. [DOI] [PubMed] [Google Scholar]
- Herber D. L., Roth L. M., Wilson D., Wilson N., Mason J. E., Morgan D. and Gordon M. N. (2004) Time‐dependent reduction in Aβ levels after intracranial LPS administration in APP transgenic mice. Exp. Neurol. 190, 245–253. [DOI] [PubMed] [Google Scholar]
- Herber D. L., Mercer M., Roth L. M., Symmonds K., Maloney J., Wilson N., Freeman M. J., Morgan D. and Gordon M. N. (2007) Microglial activation is required for Aβ clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J. Neuroimmune Pharmacol. 2, 222–231. [DOI] [PubMed] [Google Scholar]
- Hickman S. E. and El Khoury J. (2014) TREM2 and the neuroimmunology of Alzheimer's disease. Biochem. Pharmacol. 88, 495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S. E., Allison E. K. and El Khoury J. (2008) Microglial dysfunction and defective beta‐amyloid clearance pathways in aging Alzheimer's disease mice. J. Neurosci. 28, 8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L. C., Means T. K. and El Khoury J. (2013) The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch‐Reinshagen V., Zhou S., Burgess B. L. et al (2004) Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279, 41197–41207. [DOI] [PubMed] [Google Scholar]
- Hollingworth P., Harold D., Sims R. et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman D. M., Herz J. and Bu G. (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect Med. 2, a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S., Quintero‐Monzon O., Ostaszewski B. L., Podlisny D. R., Cavanaugh W. T., Yang T., Holtzman D. M., Cirrito J. R. and Selkoe D. J. (2011) Dynamic analysis of amyloid beta‐protein in behaving mice reveals opposing changes in ISF versus parenchymal Aβ during age‐related plaque formation. J. Neurosci. 31, 15861–15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., LaDu M. J. and Van Eldik L. J. (1998) Apolipoprotein E attenuates beta‐amyloid‐induced astrocyte activation. J. Neurochem. 71, 1626–1634. [DOI] [PubMed] [Google Scholar]
- Hudry E., Dashkoff J., Roe A. D. et al (2013) Gene transfer of human apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 5, 212ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo B. P., Solfrizzi V. and Panza F. (2010) Are NSAIDs useful to treat Alzheimer's disease or mild cognitive impairment? Front. Aging Neurosci. 2, doi: 10.3389/fnagi.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T., Yu J. T., Hu N., Tan M. S., Zhu X. C. and Tan L. (2014) CD33 in Alzheimer's disease. Mol. Neurobiol. 49, 529–535. [DOI] [PubMed] [Google Scholar]
- Jin J. J., Kim H. D., Maxwell J. A., Li L. and Fukuchi K. (2008) Toll‐like receptor 4‐dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J. Neuroinflammation 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M., Shepardson N., Yang T., Chen G., Walsh D. and Selkoe D. J. (2011) Soluble amyloid beta‐protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl Acad. Sci. USA 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jofre‐Monseny L., Loboda A., Wagner A. E., Huebbe P., Boesch‐Saadatmandi C., Jozkowicz A., Minihane A. M., Dulak J. and Rimbach G. (2007) Effects of apoE genotype on macrophage inflammation and heme oxygenase‐1 expression. Biochem. Biophys. Res. Commun. 357, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T., Stefansson H., Steinberg S. et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E. and Hardy J. (2014) Antiamyloid therapy for Alzheimer's disease–are we on the right road? N. Engl. J. Med. 370, 377–378. [DOI] [PubMed] [Google Scholar]
- Keene C. D., Cudaback E., Li X., Montine K. S. and Montine T. J. (2011) Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer's disease. Curr. Opin. Neurobiol. 21, 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M. E., Clay M. A., Mistry M. J., Hsieh‐Li H. M. and Harmony J. A. (1994) Apolipoprotein E inhibition of proliferation of mitogen‐activated T lymphocytes: production of interleukin 2 with reduced biological activity. Cell. Immunol. 159, 124–139. [DOI] [PubMed] [Google Scholar]
- Kessler Y., Helfer‐Hungerbuehler A. K., Cattori V. et al (2009) Quantitative TaqMan real‐time PCR assays for gene expression normalisation in feline tissues. BMC Mol. Biol. 10, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein W. L. (2002) Aβ toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 41, 345–352. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy G., Kelley J., Yerra L., Smith J. K. and Chi D. S. (1999) Human endothelium as a source of multifunctional cytokines: molecular regulation and possible role in human disease. J. Interferon Cytokine Res. 19, 91–104. [DOI] [PubMed] [Google Scholar]
- Kunzler J., Youmans K. L., Yu C., Ladu M. J. and Tai L. (2013) APOE modulates the effect of estrogen therapy on Aβ accumulation EFAD‐Tg mice. Neurosci. Lett. 560, 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y. M., Emmerling M. R., Vigo‐Pelfrey C., Kasunic T. C., Kirkpatrick J. B., Murdoch G. H., Ball M. J. and Roher A. E. (1996) Water‐soluble Aβ (N‐40, N‐42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 271, 4077–4081. [DOI] [PubMed] [Google Scholar]
- LaDu M. J., Reardon C., Van Eldik L., Fagan A. M., Bu G., Holtzman D. and Getz G. S. (2000) Lipoproteins in the central nervous system. Ann. N. Y. Acad. Sci. 903, 167–175. [DOI] [PubMed] [Google Scholar]
- LaDu M. J., Shah J. A., Reardon C. A., Getz G. S., Bu G., Hu J., Guo L. and Van Eldik L. J. (2001) Apolipoprotein E and apolipoprotein E receptors modulate A beta‐induced glial neuroinflammatory responses. Neurochem. Int. 39, 427–434. [DOI] [PubMed] [Google Scholar]
- LaDu M. J., Stine W. B., Jr , Narita M., Getz G. S., Reardon C. A. and Bu G. (2006) Self‐assembly of HEK cell‐secreted ApoE particles resembles ApoE enrichment of lipoproteins as a ligand for the LDL receptor‐related protein. Biochemistry 45, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J. C., Ibrahim‐Verbaas C. A., Harold D. et al (2013) Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambracht‐Washington D. and Rosenberg R. N. (2013) Anti‐amyloid beta to tau ‐ based immunization: developments in immu‐notherapy for Alzheimer disease. Immunotargets Ther. 2013, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreth G. E. (2009) Microglia in central nervous system diseases. J. Neuroimmune Pharmacol. 4, 369–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreth G. E. and Reed‐Geaghan E. G. (2009) Toll‐like receptors in Alzheimer's disease. Curr. Top. Microbiol. Immunol. 336, 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O. and Marr R. A. (2013) Of mice and men: neurogenesis, cognition and Alzheimer's disease. Front. Aging Neurosci. 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. W., Lee Y. K., Yuk D. Y., Choi D. Y., Ban S. B., Oh K. W. and Hong J. T. (2008) Neuro‐inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta‐amyloid generation. J. Neuroinflammation 5, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterov I., Bookout A., Wang Z., Staufenbiel M., Mangelsdorf D. and Koldamova R. (2007) Expression profiling in APP23 mouse brain: inhibition of Aβ amyloidosis and inflammation in response to LXR agonist treatment. Mol. Neurodegener. 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S. (2010) Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll‐like receptor‐mediated neuronal injury. Glia 58, 253–263. [DOI] [PubMed] [Google Scholar]
- Lehnardt S., Lachance C., Patrizi S., Lefebvre S., Follett P. L., Jensen F. E., Rosenberg P. A., Volpe J. J. and Vartanian T. (2002) The toll‐like receptor TLR4 is necessary for lipopolysaccharide‐induced oligodendrocyte injury in the CNS. J. Neurosci. 22, 2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S., Massillon L., Follett P., Jensen F. E., Ratan R., Rosenberg P. A., Volpe J. J. and Vartanian T. (2003) Activation of innate immunity in the CNS triggers neurodegeneration through a Toll‐like receptor 4‐dependent pathway. Proc. Natl Acad. Sci. USA 100, 8514–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon C. G., Tory R., Jia J., Sivak O. and Wasan K. M. (2008) Discovery and development of toll‐like receptor 4 (TLR4) antagonists: a new paradigm for treating sepsis and other diseases. Pharm. Res. 25, 1751–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoutsakos J. M., Muthen B. O., Breitner J. C. and Lyketsos C. G. (2012) Effects of non‐steroidal anti‐inflammatory drug treatments on cognitive decline vary by phase of pre‐clinical Alzheimer disease: findings from the randomized controlled Alzheimer's Disease Anti‐inflammatory Prevention Trial. Int. J. Geriatr. Psychiatry 27, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S. E., Sherman M. A., Grant M., Kuskowski M., Schneider J. A., Bennett D. A. and Ashe K. H. (2013) Brain amyloid‐beta oligomers in ageing and Alzheimer's disease. Brain 136, 1383–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Shen N., Zhang S. et al (2014) CD33 rs3865444 polymorphism contributes to Alzheimer's disease susceptibility in Chinese, European, and North American populations. Mol. Neurobiol. doi: 10.1007/s12035‐014‐8880‐9. [DOI] [PubMed] [Google Scholar]
- Lindberg C., Selenica M. L., Westlind‐Danielsson A. and Schultzberg M. (2005) Beta‐amyloid protein structure determines the nature of cytokine release from rat microglia. J. Mol. Neurosci. 27, 1–12. [DOI] [PubMed] [Google Scholar]
- Linton M. F., Gish R., Hubl S. T., Butler E., Esquivel C., Bry W. I., Boyles J. K., Wardell M. R. and Young S. G. (1991) Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J. Clin. Invest. 88, 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C., Kanekiyo T., Xu H. and Bu G. (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. H., Wang Y. R., Xiang Y., Zhou H. D., Giunta B., Manucat‐Tan N. B., Tan J., Zhou X. F. and Wang Y. J. (2014) Clearance of amyloid‐beta in Alzheimer's disease: shifting the action site from center to periphery. Mol. Neurobiol. 51, 1–7. [DOI] [PubMed] [Google Scholar]
- Lue L. F., Kuo Y. M., Roher A. E. et al (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L. F., Schmitz C. and Walker D. G. (2014) What happens to microglial TREM2 in Alzheimer's disease: immunoregulatory turned into immunopathogenic? Neuroscience doi: 10.1016/j.neuroscience.2014.09.050. [DOI] [PubMed] [Google Scholar]
- Lyketsos C. G., Breitner J. C., Green R. C., Martin B. K., Meinert C., Piantadosi S. and Sabbagh M. (2007) Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68, 1800–1808. [DOI] [PubMed] [Google Scholar]
- Ma L., Dong F., Zaid M., Kumar A. and Zha X. (2012) ABCA1 protein enhances Toll‐like receptor 4 (TLR4)‐stimulated interleukin‐10 (IL‐10) secretion through protein kinase A (PKA) activation. J. Biol. Chem. 287, 40502–40512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I., Maeda N., Montine T. J. and Montine K. S. (2006a) Apolipoprotein E‐specific innate immune response in astrocytes from targeted replacement mice. J. Neuroinflammation 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I., Nivison M., Montine K. S., Maeda N. and Montine T. J. (2006b) Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 20, 797–799. [DOI] [PubMed] [Google Scholar]
- Maezawa I., Zaja‐Milatovic S., Milatovic D., Stephen C., Sokal I., Maeda N., Montine T. J. and Montine K. S. (2006c) Apolipoprotein E isoform‐dependent dendritic recovery of hippocampal neurons following activation of innate immunity. J. Neuroinflammation 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley R. W. and Ji Z. S. (1999) Remnant lipoprotein metabolism: key pathways involving cell‐surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 40, 1–16. [PubMed] [Google Scholar]
- Mahley R. W., Weisgraber K. H. and Huang Y. (2009) Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. J. Lipid Res. 50(Suppl), S183–S188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm T. M., Koistinaho M., Parepalo M., Vatanen T., Ooka A., Karlsson S. and Koistinaho J. (2005) Bone‐marrow‐derived cells contribute to the recruitment of microglial cells in response to beta‐amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol. Dis. 18, 134–142. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Sozzani S., Locati M., Allavena P. and Sica A. (2002) Macrophage polarization: tumor‐associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A. and Locati M. (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686. [DOI] [PubMed] [Google Scholar]
- Martin B. K., Meinert C. L. and Breitner J. C. (2002) Double placebo design in a prevention trial for Alzheimer's disease. Control. Clin. Trials 23, 93–99. [DOI] [PubMed] [Google Scholar]
- Martin B. K., Szekely C., Brandt J., Piantadosi S., Breitner J. C., Craft S., Evans D., Green R. and Mullan M. (2008) Cognitive function over time in the Alzheimer's Disease Anti‐inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol. 65, 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Morillo E., Hansson O., Atagi Y., Bu G., Minthon L., Diamandis E. P. and Nielsen H. M. (2014) Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer's disease patients and controls. Acta Neuropathol. 127, 633–643. [DOI] [PubMed] [Google Scholar]
- McGeer P. L. and McGeer E. G. (2013) The amyloid cascade‐inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 126, 479–497. [DOI] [PubMed] [Google Scholar]
- McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I. and Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866. [DOI] [PubMed] [Google Scholar]
- Medeiros R. and LaFerla F. M. (2013) Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 239, 133–138. [DOI] [PubMed] [Google Scholar]
- Meinert C. L., McCaffrey L. D. and Breitner J. C. (2009) Alzheimer's Disease Anti‐Inflammatory Prevention Trial: design, methods, and baseline results. Alzheimers Dement. 5, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz‐Rios M. A., Toral‐Rios D., Franco‐Bocanegra D., Villeda‐Hernandez J. and Campos‐Pena V. (2013) Inflammatory process in Alzheimer's disease. Front. Integr. Neurosci. 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson D. M. (2014) ApoE4: the most prevalent yet understudied risk factor for Alzheimer's disease. Alzheimers Dement. 10, 861–868. [DOI] [PubMed] [Google Scholar]
- Michaud J. P., Halle M., Lampron A. et al (2013) Toll‐like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease‐related pathology. Proc. Natl Acad. Sci. USA 110, 1941–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoretti P., Gazzaruso C., Vito C. D., Emanuele E., Bianchi M., Coen E., Reino M. and Geroldi D. (2006) Effect of the functional toll‐like receptor 4 Asp299Gly polymorphism on susceptibility to late‐onset Alzheimer's disease. Neurosci. Lett. 391, 147–149. [DOI] [PubMed] [Google Scholar]
- Mistry M. J., Clay M. A., Kelly M. E., Steiner M. A. and Harmony J. A. (1995) Apolipoprotein E restricts interleukin‐dependent T lymphocyte proliferation at the G1A/G1B boundary. Cell. Immunol. 160, 14–23. [DOI] [PubMed] [Google Scholar]
- Mohamed A. and Posse de Chaves E. (2011) Aβ internalization by neurons and glia. Int. J. Alzheimers Dis. 2011, 127984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D. M. and Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munder M. (2009) Arginase: an emerging key player in the mammalian immune system. Br. J. Pharmacol. 158, 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj A. C., Jun G., Beecham G. W. et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat. Genet. 43, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzzo D., Picone P., Caruana L., Vasto S., Barera A., Caruso C. and Di Carlo M. (2014) Inflammatory mediators as biomarkers in brain disorders. Inflammation 37, 639–648. [DOI] [PubMed] [Google Scholar]
- Nwabuisi‐Heath E., LaDu M. J. and Yu C. (2012) Simultaneous analysis of dendritic spine density, morphology and excitatory glutamate receptors during neuron maturation in vitro by quantitative immunocytochemistry. J. Neurosci. Methods 207, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H., Cole S. L., Logan S. et al (2006) Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E., Griffioen K. J., Lathia J. D., Tang S. C., Mattson M. P. and Arumugam T. V. (2009) Toll‐like receptors in neurodegeneration. Brain Res. Rev. 59, 278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E., Griffioen K. J. and Mattson M. P. (2011) Toll‐like receptor signaling in neural plasticity and disease. Trends Neurosci. 34, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opal S. M., Laterre P. F., Francois B. et al (2013) Effect of eritoran, an antagonist of MD2‐TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309, 1154–1162. [DOI] [PubMed] [Google Scholar]
- Parvathy S., Rajadas J., Ryan H., Vaziri S., Anderson L. and Murphy G. M., Jr (2009) Aβ peptide conformation determines uptake and interleukin‐1alpha expression by primary microglial cells. Neurobiol. Aging 30, 1792–1804. [DOI] [PubMed] [Google Scholar]
- Peri F. and Piazza M. (2012) Therapeutic targeting of innate immunity with Toll‐like receptor 4 (TLR4) antagonists. Biotechnol. Adv. 30, 251–260. [DOI] [PubMed] [Google Scholar]
- Perlmutter L. S., Barron E. and Chui H. C. (1990) Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci. Lett. 119, 32–36. [DOI] [PubMed] [Google Scholar]
- Pernot F., Dorandeu F., Beaup C. and Peinnequin A. (2010) Selection of reference genes for real‐time quantitative reverse transcription‐polymerase chain reaction in hippocampal structure in a murine model of temporal lobe epilepsy with focal seizures. J. Neurosci. Res. 88, 1000–1008. [DOI] [PubMed] [Google Scholar]
- Persson M., Brantefjord M., Hansson E. and Ronnback L. (2005) Lipopolysaccharide increases microglial GLT‐1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF‐alpha. Glia 51, 111–120. [DOI] [PubMed] [Google Scholar]
- Phillips M. C. (2014) Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 66, 616–623. [DOI] [PubMed] [Google Scholar]
- Pocivavsek A., Burns M. P. and Rebeck G. W. (2009a) Low‐density lipoprotein receptors regulate microglial inflammation through c‐Jun N‐terminal kinase. Glia 57, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A., Mikhailenko I., Strickland D. K. and Rebeck G. W. (2009b) Microglial low‐density lipoprotein receptor‐related protein 1 modulates c‐Jun N‐terminal kinase activation. J. Neuroimmunol. 214, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M. and Priller J. (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312. [DOI] [PubMed] [Google Scholar]
- Qiu W. Q., Mwamburi M., Besser L. M. et al (2013) Angiotensin converting enzyme inhibitors and the reduced risk of Alzheimer's disease in the absence of apolipoprotein E4 allele. J. Alzheimers Dis. 37, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J., Montine T., Morrow J., Woodward W. R., Kulhanek D. and Eckenstein F. (2003) Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer's disease. J. Neuroimmunol. 137, 32–41. [DOI] [PubMed] [Google Scholar]
- Ratthe C., Pelletier M., Chiasson S. and Girard D. (2007) Molecular mechanisms involved in interleukin‐4‐induced human neutrophils: expression and regulation of suppressor of cytokine signaling. J. Leukoc. Biol. 81, 1287–1296. [DOI] [PubMed] [Google Scholar]
- Reyes T. M., Fabry Z. and Coe C. L. (1999) Brain endothelial cell production of a neuroprotective cytokine, interleukin‐6, in response to noxious stimuli. Brain Res. 851, 215–220. [DOI] [PubMed] [Google Scholar]
- Riddell D. R., Zhou H., Atchison K. et al (2008) Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. 28, 11445–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risner M. E., Saunders A. M., Altman J. F., Ormandy G. C., Craft S., Foley I. M., Zvartau‐Hind M. E., Hosford D. A. and Roses A. D. (2006) Efficacy of rosiglitazone in a genetically defined population with mild‐to‐moderate Alzheimer's disease. Pharmacogenomics J 6, 246–254. [DOI] [PubMed] [Google Scholar]
- Rodriguez G. A., Tai L. M., LaDu M. J. and Rebeck G. W. (2014) Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Abeta deposition. J. Neuroinflammation 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselaar S. E. and Daugherty A. (1998) Apolipoprotein E‐deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J. Lipid Res. 39, 1740–1743. [PubMed] [Google Scholar]
- Rosenberg G. A. (2002) Matrix metalloproteinases in neuroinflammation. Glia 39, 279–291. [DOI] [PubMed] [Google Scholar]
- Ross J., Sharma S., Winston J. et al (2013) CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: a 12‐week, double‐blind, placebo‐controlled study. Curr. Alzheimer Res. 10, 742–753. [DOI] [PubMed] [Google Scholar]
- Rubio‐Perez J. M. and Morillas‐Ruiz J. M. (2012) A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal 2012, 756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo I., Barlati S. and Bosetti F. (2011) Effects of neuroinflammation on the regenerative capacity of brain stem cells. J. Neurochem. 116, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi F., Fassina L., Venturini L., Inguscio M., Guerriero F., Rolfo E. and Ricevuti G. (2011) Alzheimer's disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun. Rev. 11, 149–153. [DOI] [PubMed] [Google Scholar]
- Sebollela A., Mustata G. M., Luo K. et al (2014) Elucidating molecular mass and shape of a neurotoxic Aβ oligomer. ACS Chem. Neurosci. 5, 1238–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (2011) Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Shen Y., Yang L. and Li R. (2013) What does complement do in Alzheimer's disease? Old molecules with new insights. Transl. Neurodegener. 2, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng J. G., Bora S. H., Xu G., Borchelt D. R., Price D. L. and Koliatsos V. E. (2003) Lipopolysaccharide‐induced‐neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 14, 133–145. [DOI] [PubMed] [Google Scholar]
- Shimizu E., Kawahara K., Kajizono M., Sawada M. and Nakayama H. (2008) IL‐4‐induced selective clearance of oligomeric beta‐amyloid peptide(1‐42) by rat primary type 2 microglia. J. Immunol. 181, 6503–6513. [DOI] [PubMed] [Google Scholar]
- Simard A. R., Soulet D., Gowing G., Julien J. P. and Rivest S. (2006) Bone marrow‐derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49, 489–502. [DOI] [PubMed] [Google Scholar]
- Skerrett R., Malm T. and Landreth G. (2014) Nuclear receptors in neurodegenerative diseases. Neurobiol. Dis. 72(Pt A), 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares H. D., Potter W. Z., Pickering E. et al (2012) Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Arch. Neurol. 69, 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola C., Casal C., Tusell J. M. and Serratosa J. (2002) Astrocytes enhance lipopolysaccharide‐induced nitric oxide production by microglial cells. Eur. J. Neurosci. 16, 1275–1283. [DOI] [PubMed] [Google Scholar]
- Solomon S. D., McMurray J. J., Pfeffer M. A. et al (2005) Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 352, 1071–1080. [DOI] [PubMed] [Google Scholar]
- Sondag C. M., Dhawan G. and Combs C. K. (2009) Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflammation 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M., Jin J., Lim J. E. et al (2011) TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. J. Neuroinflammation 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Jack C. R., Jr , Black S. E. et al (2011) Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 7, 367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R., Salloway S., Brooks D. J. et al (2012) Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 11, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillmann F., Van Linthout S., Miteva K., Lorenz M., Stangl V., Schultheiss H. P. and Tschope C. (2014) LXR agonism improves TNF‐alpha‐induced endothelial dysfunction in the absence of its cholesterol‐modulating effects. Atherosclerosis 232, 1–9. [DOI] [PubMed] [Google Scholar]
- Stine W. B., Jr , Dahlgren K. N., Krafft G. K. and LaDu M. J. (2003) In vitro characterization of conditions for amyloid‐beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622. [DOI] [PubMed] [Google Scholar]
- Sullivan P. M., Mezdour H., Aratani Y., Knouff C., Najib J., Reddick R. L., Quarfordt S. H. and Maeda N. (1997) Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet‐induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 272, 17972–17980. [DOI] [PubMed] [Google Scholar]
- Sullivan P. M., Mezdour H., Quarfordt S. H. and Maeda N. (1998) Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J. Clin. Invest. 102, 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P. M., Han B., Liu F., Mace B. E., Ervin J. F., Wu S., Koger D., Paul S. and Bales K. R. (2011) Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol. Aging 32, 791–801. [DOI] [PubMed] [Google Scholar]
- Swijsen A., Nelissen K., Janssen D., Rigo J. M. and Hoogland G. (2012) Validation of reference genes for quantitative real‐time PCR studies in the dentate gyrus after experimental febrile seizures. BMC Res. Notes 5, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara K., Kim H. D., Jin J. J., Maxwell J. A., Li L. and Fukuchi K. (2006) Role of toll‐like receptor signalling in Aβ uptake and clearance. Brain 129, 3006–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai L. M., Youmans K. L., Jungbauer L., Yu C. and Ladu M. J. (2011) Introducing human APOE into Aβ transgenic mouse models. Int. J. Alzheimers Dis. 2011, 810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai L. M., Bilousova T., Jungbauer L. et al (2013) Levels of soluble apolipoprotein E/amyloid‐beta (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 288, 5914–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai L. M., Koster K. P., Luo J., Lee S. H., Wang Y. T., Collins N. C., Ben Aissa M., Thatcher G. R. and LaDu M. J. (2014a) Amyloid‐beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 289, 30538–30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai L. M., Mehra S., Shete V., Estus S., Rebeck G. W., Bu G. and Ladu M. J. (2014b) Soluble apoE/Aβ complex: mechanism and therapeutic target for APOE4‐induced AD risk. Mol. Neurodegener. 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S. C., Lathia J. D., Selvaraj P. K. et al (2008) Toll‐like receptor‐4 mediates neuronal apoptosis induced by amyloid beta‐peptide and the membrane lipid peroxidation product 4‐hydroxynonenal. Exp. Neurol. 213, 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassishin L., Suh H. S. and Lee S. C. (2014) LPS and IL‐1 differentially activate mouse and human astrocytes: role of CD14. Glia 62, 999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplow D. B. (2013) On the subject of rigor in the study of amyloid beta‐protein assembly. Alzheimers Res. Ther. 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal D. R., Capetillo‐Zarate E., Del Tredici K. and Braak H. (2006) The development of amyloid beta protein deposits in the aged brain. Sci. Aging Knowledge Environ. 2006, re1. [DOI] [PubMed] [Google Scholar]
- Thal D. R., Griffin W. S. and Braak H. (2008) Parenchymal and vascular Aβ‐deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J. Cell Mol. Med. 12, 1848–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilleux S. and Hermans E. (2007) Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 85, 2059–2070. [DOI] [PubMed] [Google Scholar]
- Tobinick E. (2009) Tumour necrosis factor modulation for treatment of Alzheimer's disease: rationale and current evidence. CNS Drugs 23, 713–725. [DOI] [PubMed] [Google Scholar]
- Tomic J. L., Pensalfini A., Head E. and Glabe C. G. (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 35, 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulevitch R. J. and Tobias P. S. (1999) Recognition of gram‐negative bacteria and endotoxin by the innate immune system. Curr. Opin. Immunol. 11, 19–22. [DOI] [PubMed] [Google Scholar]
- Vadeboncoeur N., Segura M., Al‐Numani D., Vanier G. and Gottschalk M. (2003) Pro‐inflammatory cytokine and chemokine release by human brain microvascular endothelial cells stimulated by Streptococcus suis serotype 2. FEMS Immunol. Med. Microbiol. 35, 49–58. [DOI] [PubMed] [Google Scholar]
- Van Eldik L. J., Thompson W. L., Ralay Ranaivo H., Behanna H. A. and Martin Watterson D. (2007) Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function‐based and target‐based discovery approaches. Int. Rev. Neurobiol. 82, 277–296. [DOI] [PubMed] [Google Scholar]
- Verghese P. B., Castellano J. M. and Holtzman D. M. (2011) Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 10, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek M. P., Brown C. M. and Colton C. A. (2009) APOE genotype‐specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek M. P., Christensen D. J., Wilcock D., Davis J., Van Nostrand W. E., Li F. Q. and Colton C. A. (2012) APOE‐mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer's disease transgenics. Neurodegener. Dis. 10, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries H. E., Blom‐Roosemalen M. C., van Oosten M., de Boer A. G., van Berkel T. J., Breimer D. D. and Kuiper J. (1996) The influence of cytokines on the integrity of the blood‐brain barrier in vitro. J. Neuroimmunol. 64, 37–43. [DOI] [PubMed] [Google Scholar]
- de Vries H. E., Kuiper J., de Boer A. G., Van Berkel T. J. and Breimer D. D. (1997) The blood‐brain barrier in neuroinflammatory diseases. Pharmacol. Rev. 49, 143–155. [PubMed] [Google Scholar]
- Wahrle S. E., Jiang H., Parsadanian M., Legleiter J., Han X., Fryer J. D., Kowalewski T. and Holtzman D. M. (2004) ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte‐secreted apoE. J. Biol. Chem. 279, 40987–40993. [DOI] [PubMed] [Google Scholar]
- Walter S., Letiembre M., Liu Y. et al (2007) Role of the toll‐like receptor 4 in neuroinflammation in Alzheimer's disease. Cell. Physiol. Biochem. 20, 947–956. [DOI] [PubMed] [Google Scholar]
- Wang J., Dickson D. W., Trojanowski J. Q. and Lee V. M. (1999) The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 158, 328–337. [DOI] [PubMed] [Google Scholar]
- Wang L. Z., Yu J. T., Miao D., Wu Z. C., Zong Y., Wen C. Q. and Tan L. (2011) Genetic association of TLR4/11367 polymorphism with late‐onset Alzheimer's disease in a Han Chinese population. Brain Res. 1381, 202–207. [DOI] [PubMed] [Google Scholar]
- Watterson D. M., Grum‐Tokars V. L., Roy S. M. et al (2013) Development of novel chemical probes to address CNS protein kinase involvement in synaptic dysfunction. PLoS ONE 8, e66226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisgraber K. H., Innerarity T. L. and Mahley R. W. (1982) Abnormal lipoprotein receptor‐binding activity of the human E apoprotein due to cysteine‐arginine interchange at a single site. J. Biol. Chem. 257, 2518–2521. [PubMed] [Google Scholar]
- White J. A., Manelli A. M., Holmberg K. H., Van Eldik L. J. and LaDu M. J. (2005) Differential effects of oligomeric and fibrillar amyloid‐beta1‐42 on astrocyte‐mediated inflammation. Neurobiol. Dis. 18, 459–465. [DOI] [PubMed] [Google Scholar]
- Wilcock D. M. and Griffin W. S. (2013) Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflammation 10, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M. A., Itzhaki R. F., Faragher E. B., James M. W., Ryder S. D., Irving W. L. and Trent H. C. V. S. G. (2002) Apolipoprotein E‐epsilon 4 protects against severe liver disease caused by hepatitis C virus. Hepatology 36, 456–463. [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray T. and Rogers J. (2012) Inflammation in Alzheimer disease‐a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect Med. 2, a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing B., Bachstetter A. D. and Van Eldik L. J. (2011) Microglial p38alpha MAPK is critical for LPS‐induced neuron degeneration, through a mechanism involving TNFalpha. Mol. Neurodegener. 6, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Storer P. D., Chavis J. A., Racke M. K. and Drew P. D. (2005) Agonists for the peroxisome proliferator‐activated receptor‐alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J. Neurosci. Res. 81, 403–411. [DOI] [PubMed] [Google Scholar]
- Xu Q., Bernardo A., Walker D., Kanegawa T., Mahley R. W. and Huang Y. (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Cudaback E., Jorstad N. L. et al (2013) APOE3, but not APOE4, bone marrow transplantation mitigates behavioral and pathological changes in a mouse model of Alzheimer disease. Am. J. Pathol. 183, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans K. L., Tai L. M., Nwabuisi‐Heath E. et al (2012) APOE4‐specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem. 287, 41774–41786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. and Ye R. D. (2014) Microglial Aβ receptors in Alzheimer's disease. Cell. Mol. Neurobiol. 35, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. T., Miao D., Cui W. Z., Ou J. R., Tian Y., Wu Z. C., Zhang W. and Tan L. (2012) Common variants in toll‐like receptor 4 confer susceptibility to Alzheimer's disease in a Han Chinese population. Curr. Alzheimer Res. 9, 458–466. [DOI] [PubMed] [Google Scholar]
- Zelcer N., Khanlou N., Clare R., Jiang Q., Reed‐Geaghan E. G., Landreth G. E., Vinters H. V. and Tontonoz P. (2007) Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc. Natl Acad. Sci. USA 104, 10601–10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Lin S., Bales K. R. et al (2009) Macrophage‐mediated degradation of beta‐amyloid via an apolipoprotein E isoform‐dependent mechanism. J. Neurosci. 29, 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Nwabuisi‐Heath E., Dumanis S. B., Tai L. M., Yu C., Rebeck G. W. and LaDu M. J. (2012) APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60, 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]