

Abstract
PRMT3 catalyzes the asymmetric dimethylation of arginine residues of various proteins. It is essential for maturation of ribosomes, may have a role in lipogenesis, and is implicated in several diseases. A potent, selective, and cell- active PRMT3 inhibitor would be a valuable tool for further investigating PRMT3 biology. Here we report the discovery of the first PRMT3 chemical probe, SGC707, by structure-based optimization of the allosteric PRMT3 inhibitors we reported previously, and thorough characterization of this probe in biochemical, biophysical, and cellular assays. SGC707 is a potent PRMT3 inhibitor (IC50 = 31 ± 2 nm, KD = 53 ± 2 nm) with outstanding selectivity (selective against 31 other methyltransferases and more than 250 non-epigenetic targets). The mechanism of action studies and crystal structure of the PRMT3-SGC707 complex confirm the allosteric inhibition mode. Importantly, SGC707 engages PRMT3 and potently inhibits its methyltransferase activity in cells. It is also bioavailable and suitable for animal studies. This well- characterized chemical probe is an excellent tool to further study the role of PRMT3 in health and disease.
Keywords: allosteric inhibition, chemical probes, enzyme inhibitors, histone methylation, X-ray diffraction
Protein arginine methyltransferase (PRMT) 3 is a type I PRMT that catalyzes mono- and asymmetric dimethylation of arginine residues. It was shown to be a cytosolic protein methylating 40S ribosomal protein S2 (rpS2) which is critical for proper maturation of 80S ribosome.[1] Recently it was shown that in cells treated with palmitic acid or T0901317 (a liver X receptor a (LXRa) agonist) PRMT3 co-localizes with LXRa in the cell nucleus, regulating hepatic lipogenesis.[2] However, this effect appears to be independent of PRMT3 methyltransferase activity. PRMT3 as well as PRMT1 meth- ylate the recombinant mammalian nuclear poly(A)-binding protein (PABPN1)[3] and have been implicated in oculophar- yngeal muscular dystrophy, which is caused by polyalanine expansion in PABPN1.[3c, 4] PRMT3 has been shown to be overexpressed in human myocardial tissues from patients with coronary heart disease.[5] It also methylates a histone peptide (H4 1–24) in vitro.[6] Histone H4R3 is a modification site associated with increased transcription of a number of genes, including those under control of estrogen and andro- gen receptors.[7] Interaction of PRMT3 with the tumor suppressor DAL-1/4.1B and consequent inhibition of its methyltransferase activity suggest a possible role of PRMT3 in tumor growth and possible epigenetic regulation of gene expression.[8]
Well-characterized chemical probes[9] that display high potency and selectivity, and robust on-target activities for protein(s) of interest in cells would be extremely valuable for elucidating biological functions of the protein(s) and testing therapeutic hypotheses. However, selective and cell-active PRMT inhibitors have not been reported to date.[10] We recently discov- ered a novel allosteric site for PRMT3 and the first allosteric inhibitor of PRMT3 (Compound 1; Figure 1).[11] We later reported structure–activity rela- tionship studies of this inhibitor, leading to the iden- tification of a sub-micromolar PRMT3 inhibitor.[12]
Figure 1.
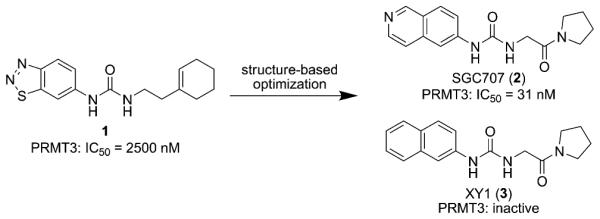
Discovery of SGC707 and its inactive control XY1.
To discover the first chemical probe of PRMT3, we further optimized both the left-hand side (LHS) and right-hand side (RHS) moieties of this scaffold through structure-guided design and synthesis. In particular, we conducted a scaffold hopping exercise (see the Sup- porting Information (SI) for details) for replacing the LHS benzothiadiazole moiety, which resulted in the selection of several replacement candidates, including an isoquinoline moiety. We also explored various substituents at the isoquinoline ring. For the RHS moiety, we extensively investigated various amides, ketones, and bicyclic heterocycles. In total, more than 100 analogues were designed, synthesized, and tested. From this study, we discovered SGC707 (Figure 1), a potent, low molecular weight (MW = 298) PRMT3 inhibitor with an IC50 value of 31 ± 2 nm (n = 3) by scintillation proximity assay (SPA) and 66 nm (n = 2) by LC-MS detection assay at balanced conditions (sub- strate and cofactor concentrations equal to their Km values; Figures 2 A and S1, SI). We also designed and synthesized an inactive control, XY1 (Figure 1), for chemical biology studies based on the crystal structure of the PRMT3-SGC707 complex. XY1, a close analogue of SGC707, was completely inactive against PRMT3 at concentrations as high as 100 mm (Figure 2 A). Binding of SGC707 was confirmed by iso- thermal titration calorimetry (ITC) with a KD value of 53 ± 2 nm (n = 3) (Figure S2, SI) and surface plasmon resonance (SPR) with a KD value of 85 ± 1 nm (n = 3) (kon of 1.17±0.05 × 105M−1S−1 and koff of 0.99±0.03 × 10−2 S−1; Figure 2B). SGC707 exhibited a reasonably long residence time, a significant improvement over compound 1 (kon=5.1±0.57 × 104M−1 s−1 and koff=0.76±0.09 s−1).[11] We confirmed that SGC707 had a longer residence time (9.7 min) than compound 1 (6.4 min) using jump dilution experiments (Figure S3, SI). Remarkably, SGC707 was selective for PRMT3 over 31 protein-, DNA-, and RNA-methyltransferases (Figure 2C) as well as a broad range of non-epigenetic targets including more than 250 kinases, G protein-coupled receptors (GPCRs), ion channels, and transporters (Table S1, SI). It showed modest inhibition at 10000 nm for only six (5HT2B 69%, BRSK1 56%, DLK1 60%, MSK2 55%, PKG2 58%, and PRKX 61%) out of more than 250 targets. Its binding affinity (Ki) for 5HT2B was determined to be higher than 15000 nm with a maximum inhibition of about 50% at 32000 nm (Figure S4, SI). In mechanism of action studies (Figure 2D and E), SGC707 displayed a noncompetitive inhibition pattern with respect to both the cofactor S-(5′-adenosyl)-l-methionine (SAM) and peptide substrate, suggesting it is an allosteric inhibitor of PRMT3.
Figure 2.

SGC707 is a potent, selective, and non-competitive inhibitor of PRMT3. A) IC50 determination for SGC707 (●) and XY1 (○) was performed at balanced condition (Km of both substrates). B) SPR studies confirmed that SGC707 had a high binding affinity to PRMT3 (KD = 85 ± 1 nm (n = 3)). C) Effect of SGC707 on the activity of 27 protein methyltransferases as well as DNMT1, DNMT3A-3L, DNMT3B-3L, and BCDIN3D (an RNA-methyltransfer- ase) were assessed at 1 (red), 5 (green), and 20 μm (purple) of compound and no inhibition was observed. No change in IC50 values at varying D) SAM or E) peptide concentrations was consistent with a noncompetitive pattern, confirming the allosteric mode of inhibition.
To further confirm that SGC707 binds to the allosteric site of PRMT3, we solved the crystal structure of the PRMT3-SGC707 complex (PDB code: 4RYL; Table S2, SI). As observed with parent compounds,[11] SGC707 occupies a cavity located in the β-barrel of PRMT3, at the base of the dimerization arm, more than 15 Å away from the site of methyl transfer (Figure 3). The isoquinoline group forms a hydrogen bond at the bottom of the cavity with T466, the urea group forms hydrogen bonds with E422 and R396 side chains, and the pyrrolidine amide is buttressed against the ahelix of the other PRMT3 subunit. This α-helix is a dynamic secondary element that is conserved in class I PRMTs and is critical for catalytic activity.[13] The N-terminal section of this helix, that wraps around the cofactor in the active conformation of the enzyme (PDB code: 2FYT), is unresolved in inhibitor-bound structures, and we believe that SGC707 induces conformational constraints on the α-helix that prevents formation of a catalytically competent state. We attribute the increased potency of SGC707 over parent compounds to the presence of the isoquinoline ring that optimally occupies the PRMT3 cavity, and the pyrrolidine amide that could form direct or water-mediated contacts with the side chain of K392.[12] Our inactive control XY1, which contains a naphthyl group replacing the isoquinoline group, lacks the key hydrogen bond with T466. This hydrogen bond is absolutely desolvated, thus contributing strongly to the PRMT3–SGC707 interaction. There is no other hydrogenbond acceptor in the vicinity of the T466 side chain, which, in the absence of an inhibitor, forms a hydrogen bond with a water molecule (PDB code: 2FYT). The naphthyl ring of XY1 could act as a weak hydrogen-bond acceptor, but this should come with a substantial enthalpic penalty. The more than 1000-fold potency loss of XY1 compared with SGC707 supports this analysis. It is unclear whether other factors such as electronic effects also contributed to the potency loss of XY1 compared with SGC707.
Figure 3.

Structure of the PRMT3–SGC707 complex. SGC707 (magenta) binds an allosteric site at the interface of the two PRMT3 subunits that is distant from the site of methyl transfer (red star). The allosteric site comprises a PRMT3-specific pocket in one subunit (yellow/orange) and a portion of the “activation α-helix” (light blue) of the other subunit.
To demonstrate the target engagement of SGC707 in cells, we used an InCELL Hunter Assay, which measures intracellular binding of SGC707 to the methyltransferase domain of PRMT3 in cell lines expressing the methyltransferase domain of PRMT3 tagged with a short fragment of β-galactosidase (enhanced ProLabel or ePL). Binding of a compound to PRMT3(211-531)-ePL increases the fusion protein half-life. SGC707 stabilized PRMT3 in both HEK293 and A549 cells with EC50 values of 1.3 μm and 1.6 μm, respectively (Figure 4A). We have previously shown that PRMT3 can methylate histone peptides in vitro.[11] To determine if SGC707 can inhibit the PRMT3 catalytic activity in cells we investigated its effect on H4R3 asymmetric dimethylation. Methylated arginine residues have relatively slow turnover[14] thus we overexpressed human Flag-tagged PRMT3 and looked at the methylation of both endogenous H4 and exogenously introduced GFP-tagged H4. Overexpressed PRMT3 increased the endogenous H4R3me2a from the baseline levels and SGC707 was able to reduce this increase (Figure 4 B and C) with an IC50 of 225 nm. The asymmetric dimethylation of exogenous H4R3 was also inhibited (IC50 = 91 nm) indicating potent cellular effect of SGC707 (Figure 4 B and D). The dependency on the transfected PRMT3 catalytic activity was deter- mined by using the catalytically dead PRMT3 mutant (E335Q) that did not affect endogenous or exogenous H4R3me2a levels and therefore was used to establish the baseline levels of the mark. SGC707 at 1 mm was almost as effective at reducing the H4R3me2a mark as the catalytically dead PRMT3 mutant E335Q (Figure 4B) indicating full PRMT3 inhibition at this concentration. Taken together, these results clearly indicate that SGC707 can engage PRMT3 and effectively inhibit its catalytic activity in cells and that overexpressed PRMT3 can methylate histone H4 in cells. No toxicity was observed under the conditions of the 24 h assays. Compound treatment for 72 h resulted in some toxicity, but only at very high concentrations, 50 and 100 mm (Figure S5, SI).
Figure 4.

SGC707 binds to PRMT3 in cells and reduces PRMT3-dependent H4R3me2a. A) SGC707 stabilized PRMT3 in both HEK293 and A549 cells with EC50 values of 1.3 μm and 1.6 μm in PRMT3 InCELL Hunter Assays. RLU, relative light units. B) Western blot analysis of H4R3me2a levels. HEK293 cells were co-transfected with FLAG-tagged PRMT3 (WT) or its catalytically dead mutant E335Q (Mut) and treated with different concentrations of SGC707, as indicated. The color panels below endogenous and exogenous H4R3me2a and H4 panels are overlays of the above panels in which the methylated H4R3 signal is in green and total H4 is in red thus both signals generating yellow. C) Quantitation of SGC707 effect on the endogenous H4R3me2a from the top panels in (B). D) Quantitation of SGC707 effect on the exogenous H4R3me2a from the bottom panels in (B). The graphs represent nonlinear fits of H4R3me2a signal intensities normalized to intensities of GFP or H4 for exogenous and endogenous H4, respectively, and subtracted for the baseline signal from mutant PRMT3. The results are mean ±SEM of three replicates.
Lastly, we assessed in vivo pharmacokinetic (PK) properties of SGC707. Intraperitoneal injection of SGC707 at 30 mg kg−1 gave good plasma exposure in CD-1 male mice over 6 h with the peak plasma level of 38000 nm (Figure S6, SI). The plasma level of SGC707 at 6 h post injection was 208 nm, more than 2-fold higher than its IC50 value in the cellular assay and the half-life of SGC707 was about 1 h. This dose was well tolerated by the test animals. These results suggest that SGC707 is suitable for animal studies in addition to cell-based studies.
In summary, we discovered SGC707, a first-in-class PRMT3 chemical probe which is a potent and cell-active allosteric inhibitor of PRMT3. SGC707 is remarkably selec- tive for PRMT3 over 31 other methyltransferases and more than 250 kinases, GPCRs, ion channels, and transporters. In multiple cell-based assays, SGC707 engages PRMT3 and potently inhibits its methyltransferase activity. SGC707 is bioavailable and suitable for animal studies. In addition, we discovered XY1, which is a very close analogue of SGC707, but is completely inactive. Thus, SGC707 and XY1 are a pair of excellent tools for the biomedical community to further elucidate biological functions and disease associations of PRMT3.
Supplementary Material
Footnotes
This research was supported by the grant R01GM103893 (to J.J.) from the U.S. National Institutes of Health. The S.G.C. is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, Genome Canada through Ontario Genomics Institute Grant OGI- 055, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Well- come Trust Grant 092809/Z/10/Z. H.Ü.K. was supported by a postdoctoral fellowship from the SGC. We thank Jacob Stuckey and Dr. Ashutosh Tripathy for assisting with ITC studies, Dr. Anqi Ma for checking NMR spectra, and Drs. Yuan Mi, Bin Xiang, and Justin Gu for valuable discussion.
Supporting information for this article (including detailed synthetic procedures and compound characterization as well as methods for scaffold hopping, crystallization, structure determination, bio- chemical assays, SPR, ITC, PRMT3 InCELL Hunter assay, cellular PRMT3 assay, cell viability assay, and mouse PK studies) is available on the WWW under http://dx.doi.org/10.1002/anie.201412154.
References
- [1].a) Tang J, Gary JD, Clarke S, Herschman HR. J. Biol. Chem. 1998;273:16935–16945. doi: 10.1074/jbc.273.27.16935. [DOI] [PubMed] [Google Scholar]; b) Bachand F, Silver PA. EMBO J. 2004;23:2641–2650. doi: 10.1038/sj.emboj.7600265. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Di Lorenzo A, Bedford MT. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Swiercz R, Person MD, Bedford MT. Biochem. J. 2005;386:85–91. doi: 10.1042/BJ20041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kim DI, Park MJ, Lim SK, Park JI, Yoon KC, Han HJ, Gustafsson JA, Lim JH, Park SH. Diabetes. 2015;64:60–71. doi: 10.2337/db13-1394. [DOI] [PubMed] [Google Scholar]
- [3].a) Fronz K, Otto S, Kolbel K, Kuhn U, Friedrich H, Schierhorn A, Beck-Sickinger AG, Ostareck-Lederer A, Wahle E. J. Biol. Chem. 2008;283:20408–20420. doi: 10.1074/jbc.M802329200. [DOI] [PubMed] [Google Scholar]; b) Smith JJ, Rucknagel KP, Schierhorn A, Tang J, Nemeth A, Linder M, Herschman HR, Wahle E. J. Biol. Chem. 1999;274:13229–13234. doi: 10.1074/jbc.274.19.13229. [DOI] [PubMed] [Google Scholar]; c) Tavanez JP, Bengoechea R, Berciano MT, Lafarga M, Carmo-Fonseca M, Enguita FJ. PloS One. 2009;4:e6418. doi: 10.1371/journal.pone.0006418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O, Blumen S, Korczyn AD, Heutink P, Mathieu J, Duranceau A, Codere F, Fardeau M, Rouleau GA. Nat. Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- [5].Chen X, Niroomand F, Liu Z, Zankl A, Katus HA, Jahn L, Tiefenbacher CP. Basic Res. Cardiol. 2006;101:346–353. doi: 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- [6].Allali-Hassani A, Wasney GA, Siarheyeva A, Hajian T, Arrowsmith CH, Vedadi M. J. Biomol. Screening. 2012;17:71–84. doi: 10.1177/1087057111422256. [DOI] [PubMed] [Google Scholar]
- [7].a) Herrmann F, Pably P, Eckerich C, Bedford MT, Fackelmayer FO. J. Cell Sci. 2009;122:667–677. doi: 10.1242/jcs.039933. [DOI] [PubMed] [Google Scholar]; b) Obianyo O, Causey CP, Jones JE, Thompson PR. ACS Chem. Biol. 2011;6:1127–1135. doi: 10.1021/cb2001473. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wagner S, Weber S, Kleinschmidt MA, Nagata K, Bauer UM. J. Biol. Chem. 2006;281:27242–27250. doi: 10.1074/jbc.M605172200. [DOI] [PubMed] [Google Scholar]
- [8].Singh V, Miranda TB, Jiang W, Frankel A, Roemer ME, Robb VA, Gutmann DH, Herschman HR, Clarke S, Newsham IF. Oncogene. 2004;23:7761–7771. doi: 10.1038/sj.onc.1208057. [DOI] [PubMed] [Google Scholar]
- [9].Frye SV. Nat. Chem. Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- [10].a) Finley A, Copeland RA. Chem. Biol. 2014;21:1196–1210. doi: 10.1016/j.chembiol.2014.07.024. [DOI] [PubMed] [Google Scholar]; b) Ü. Kaniskan H, Konze KD, Jin J. J. Med. Chem. 2014 DOI: 10.1021/jm501234a. [Google Scholar]
- [11].Siarheyeva A, Senisterra G, Allali-Hassani A, Dong A, Dobrovetsky E, Wasney GA, Chau I, Marcellus R, Hajian T, Liu F, Korboukh I, Smil D, Bolshan Y, Min J, Wu H, Zeng H, Loppnau P, Poda G, Griffin C, Aman A, Brown PJ, Jin J, Al-Awar R, Arrowsmith CH, Schapira M, Vedadi M. Structure. 2012;20:1425–1435. doi: 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- [12].Liu F, Li F, Ma A, Dobrovetsky E, Dong A, Gao C, Korboukh I, Liu J, Smil D, Brown PJ, Frye SV, Arrowsmith CH, Schapira M, Vedadi M, Jin J. J. Med. Chem. 2013;56:2110–2124. doi: 10.1021/jm3018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schapira M, Freitasa R. F. d. Med. Chem. Commun. 2014;5:1779–1788. [Google Scholar]
- [14].Bedford MT, Clarke SG. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.