

Abstract
Skeletal muscle insulin resistance, which increases the risk for developing various metabolic diseases, including type 2 diabetes, is a common metabolic disorder in obesity and aging. If potential treatments are to be developed to treat insulin resistance, then it is important to fully understand insulin signaling and glucose metabolism. While recent large-scale “omics” studies have revealed the acetylome to be comparable in size to the phosphorylome, the acetylation of insulin signaling proteins and its functional relevance to insulin-stimulated glucose transport and glucose metabolism is not fully understood. In this Mini Review we discuss the acetylation status of proteins involved in the insulin signaling pathway and review their potential effect on, and relevance to, insulin action in skeletal muscle.
Keywords: acetyltransferase, deacetylase, insulin signaling, p300/CBP, SIRT1/HDAC
INTRODUCTION
First described by Allfrey and co-workers some 60 years ago as a possible means of regulating RNA synthesis, reversible lysine acetylation (Ac-Lys) is well conserved across organisms, having been described in prokaryotes and eukaryotes (Choudhary et al., 2014; Kim and Yang, 2011; Verdin and Ott, 2014). Along with other post-translational modifications (PTMs; e.g., phosphorylation, methylation, ubiquitylation), acetylation is one mechanism by which the cell refines and enacts a complex control strategy in response to varying cellular and physiological processes (Choudhary et al., 2014; Verdin and Ott, 2014). Indeed, regulatory enzymes that control acetylation have been implicated in cancer, cardiovascular disease, neurodegeneration, and type 2 diabetes (T2D) (Choudhary et al., 2014; Verdin and Ott, 2014). Lysine acetylation involves the transfer of an acetyl group from acetyl-CoA to the ɛ-amino group in the target lysine. Acetyltransferases (KATs) and deacetylases (DACs) regulate this enzymatic process. The twenty identified KATs are split into three major families: MOZ YBF2/SAS3 and TIP60 protein 1 (MYST), general control of amino acid synthesis protein 5 [GCN5] related N-acetyltransferase (GNAT), and CREB binding protein/E1A binding protein p300 (CBP/p300) (Berndsen and Denu, 2008; Choudhary et al., 2014; Verdin and Ott, 2014). The eighteen DACs are categorized in two major families; Zn2+-dependent and NAD+-dependent (Choudhary et al., 2014; Verdin and Ott, 2014).
Despite the discovery of acetylation 60 years ago and identification of the first DACs and KATs some 20 years ago, only within the last decade has the extent to which acetylation influences cellular function begun to be appreciated (Choudhary et al., 2014; Philip et al., 2014; Verdin and Ott, 2014). A driving force behind this transition has been mass spectrometry (MS)-based proteomic approaches, and the advent of ‘acetylomics’. A detailed discussion on MS studies of acetylation and potential relevant physiology, including in skeletal muscle, can be found elsewhere (Choudhary et al., 2014; Philip et al., 2014; Verdin and Ott, 2014). For example, a recent study using high-resolution MS in 16 rat tissues mapped 15,474 acetyl specific modification sites on 4,541 proteins, revealing the acetylome to be at least comparable in size to the phosphorylome (Lundby et al., 2012). Of particular interest to this Mini Review, pathway enrichment analysis in human and rat skeletal muscle identified numerous acetylated proteins related to the insulin signaling pathway and glucose metabolism (Lundby et al., 2012). And because cellular NAD+ and acetyl CoA concentrations change in response to perturbations in nutrient and metabolic flux, which in turn alters the activity of KATs and NAD+-dependent DACs, reversible acetylation provides a natural link between nutrient availability and regulation of insulin action. Indeed, brief caloric restriction (CR) in humans and rodents potently enhances skeletal muscle insulin sensitivity (Kelley et al., 1993; Wang et al., 2001), with clinical studies demonstrating that the majority of the improvement in insulin action during weight loss, or after gastric bypass, occurs during the first week of intervention, and before substantial weight loss occurs (Assali et al., 2001; Gumbs et al., 2005; Kelley et al., 1993). The opposite is also true, as short-term hypercaloric feeding rapidly induces skeletal muscle insulin resistance (Wang et al., 2001). Conceptually, this presents an intriguing physiological model, such that changes in food intake are rapidly “translated” into a metabolic adaptation (namely insulin action) at the level of skeletal muscle. An obvious follow up question being, how is a nutritional cue, at the level of food intake, “sensed” and translated into altered insulin sensitivity at the level of skeletal muscle? Accordingly, the primary focus of this review is on the potential contribution of acetylation to regulation of insulin signaling and glucose metabolism in skeletal muscle.
THE BASICS OF INSULIN SIGNALING
Canonical insulin signaling is characterized by the step-wise progression of kinase phosphorylation, with select phosphatases negatively regulating these kinases. Our focus here is on insulin signaling for glucose transport and metabolism through Akt-PI3K (phosphoinositide [PI]-3 kinase), which we discuss briefly below. A detailed perspective on the nuances of canonical and non-canonical insulin signaling, can be found elsewhere (Cohen, 2006; Leto and Saltiel, 2012; Taniguchi et al., 2006).
Regarding insulin signaling for glucose transport, insulin binds the extracellular pocket of the transmembrane insulin receptor (IR), leading to intrinsic autophosphorylation on its cytoplasmic domain (Cohen, 2006; Leto and Saltiel, 2012; Taniguchi et al., 2006). The phosphorylated cytoplasmic domain of IR recruits and then phosphorylates its downstream target, IR substrate (IRS) 1 and/or 2 (Cohen, 2006; Leto and Saltiel, 2012; Taniguchi et al., 2006). Upon IRS phosphorylation, the PI3K p85/p110 heterodimer is recruited and binds IRS at the plasma membrane, where it phosphorylates the plasma membrane lipid PI-(4,5)-bisphosphate (PIP2), producing PI-(3,4,5)-bisphosphate (PIP3). PIP3 binds 3-PI-dependent protein kinase-1 (PDK1) and Akt (also known as protein kinase B), allowing PDK1 to phosphorylate Akt at Thr308. Subsequently, Akt is phosphorylated at Ser473 by mammalian target of rapamycin complex 2 (mTORC2), with phosphorylation on these two sites being necessary for full Akt activation (Cohen, 2006; Leto and Saltiel, 2012; Taniguchi et al., 2006). Activated Akt phosphorylates numerous downstream substrates leading to physiological changes in glucose, lipid, and protein metabolism within the skeletal muscle (Leto and Saltiel, 2012; Taniguchi et al., 2006). Regarding glucose metabolism, Akt-substrate of 160 kDa (AS160) and glycogen synthase kinase-3β (GSK3β), are two key Akt substrates that facilitate glucose uptake and storage, respectively (Cohen, 2006; Leto and Saltiel, 2012; Taniguchi et al., 2006). Specifically, Akt phosphorylation of AS160 relieves its inhibitory effect on GLUT4 allowing its translocation to the plasma membrane, which facilitates glucose diffusion into the cell (Stöckli et al., 2011). GSK3β phosphorylation relieves its inhibition of glycogen synthase (GS), thereby promoting glycogenesis (Cohen, 2006).
EFFECTS OF ACETYLATION ON INSULIN SIGNALING
Below we take a step-wise look at the insulin signaling pathway, discuss known and proposed effects of acetylation on insulin signaling, and postulate the implications for this on insulin-stimulated glucose transport. A list of known acetylation sites in insulin signaling proteins is provided in Table 1.
Table 1.
Acetylation sites in the insulin signaling pathway
| Protein | Lys-Ac Sites | Ref | ||
|---|---|---|---|---|
|
| ||||
| Human | Mouse | Rat | ||
| Insulin Signaling | ||||
|
| ||||
| IR | -- | -- | -- | -- |
| IRS1 | K52, K61 | -- | K315 | 1, 11 |
| IRS2 | -- | K118, K289, K292, K412 | -- | 2, 4, 6 |
| p85α | K530 | -- | K633 | 1 |
| p55α | K106, K114 | -- | -- | 1 |
| p55γ | K308 | 2 | ||
| p110β | K159, K173 | -- | -- | 1 |
| p110γ | K1000 | -- | -- | 1 |
| PDK1 | K304 | K307, K495, K534 | K307 | 1, 2, 7 |
| mTORC2 | K25, K228, K629 | K229, K628 | K25, K228 | 1, 2, 4 |
| Rictor | (9)K, K213, K582, K1312 | K1311 | -- | 1, 8 |
| Akt1 | K14, K20, K420, K426 | -- | -- | 1, 7 |
| Akt2 | K378 | -- | -- | 1, 5 |
| AS160 | K477 | -- | -- | 2, 5 |
| GLUT4 | -- | -- | -- | -- |
| GSK3β | -- | -- | -- | -- |
| ACLY | (16)K | (12)K | (17)K | 1. 9 |
|
| ||||
| Negative Regulators | ||||
|
| ||||
| PTEN | (7)K, K125, K128 | K125, K128 | K266 | 1, 4, 10 |
| PTP1B | K131, K141 | -- | -- | 1 |
| SKIP | -- | -- | -- | -- |
| SHIP2 | K272 | K902 | -- | 1, 2 |
| TRB3 | -- | -- | -- | -- |
IR, Insulin receptor; IRS1/2, Insulin receptor substrate-1/2; p85, p50, p55, p110, PI3K subunits; PDK1,3-Phosphoinositide dependent protein kianse 1, mTORC2, Mammalian target of rapamycin complex 2; Rictor, Raptor independent companion of mTOR; Akt1, v-akt murine thymom viral oncogene homolog 1; Akt2, v-akt murine thymom viral oncogene homolog 2; AS160, Akt substrate of 160 kDa; Glut4, Insulin-responsive glucose transporter type 4; GSK3β, Glycogen synthase kinase 3 beta; ACLY, ATP-Citrate Lyase; PTEN, Phosphatase and tenisin homolog; SKIP, Inositol polyphosphate-5-phosphatase K; SHIP2, Inositol polyphosphate phosphatase-like 1; TRBP3, Tribbles Pseudokinase 3; --, Not available. Note: Numbers inside paranthese preceeding, K (Lys), denote the number of Lys-Ac sites identified in each species. (1), www.phosphosite.org; (2), Lundby et al, 2012; (3), Choudhary et al 2009; (4), Wang et al 2010; (5), Chen et al 2012; (6), Zhang 2007; (7), Sundaresan 2011; (8), Glidden et al 2012; (9), Lin et al 2013; (10), Okomura et al 2006; (11), Zhao, 2010.
IR and insulin-like growth factor 1 receptor (IGF1R)
Significant functional and structural similarities exist between IR and IGF1R (Leto and Saltiel, 2012; Taniguchi et al., 2006). In recent global acetylation analyses, Lys1088 in IGF1R was acetylated in a human myeloid leukemia cell line, MV4-11 (Choudhary et al., 2009). Interestingly, Lys1088 is in the kinase domain of IGFR1, a region that is highly conserved between IGF1R and IR, suggesting that this site may be important for regulating the function of these proteins (Choudhary et al., 2009; Pirola et al., 2012). Supporting this, in C2C12 myotubes (which is an immortalized muscle-like cell line) inhibition of histone deacetylase 2 (HDAC2) activity increased pIRTyr1146, pAktSer473, glucose uptake and glycogen synthesis (Sun and Zhou, 2008), although whether this was due to changes in acetylation of IR was not assessed. Thus, whether acetylation affects the binding of insulin to the receptor or the activity and downstream transduction of IR/IGF1 signaling remains to be determined. It is important to note that Lys1088 was not identified in human skeletal muscle (Choudhary et al., 2009), which perhaps questions the relevance of this modification to IR or IGF1R function in skeletal muscle.
IRS proteins
Three Ac-Lys sites on IRS1 and four on IRS2 have been identified (Table 1) (Zhang, 2007; Zhao et al., 2010). Interestingly, two Ac-Lys sites (Lys52 and Lys61) in IRS1 lie within the pleckstrin homology (PH) domain, which is required for recognizing and binding PIP3. This suggests that acetylation may affect how IRS1 recognizes phosphorylated domains in other proteins, and therefore insulin signaling through IRS1. In support of this, in co-immunopreciptiation experiments in liver lysates from Balb/cJJ and ob/ob mice, HDAC2 pulled down with IRS1, although this did not occur in lysates from C57BL/6J mice (Kaiser and James, 2004). Further supporting a role for acetylation in regulating IRS1 function, treatment of the MCF-7 breast cancer cell line (which has high IRS1 expression) with the DAC inhibitor, trichostatin A (TSA), increased both IRS1 acetylation and insulin-stimulated IRS1 tyrosine phosphorylation (pIRS1Tyr) (Kaiser and James, 2004). In contrast, phorbol myristic acid (PMA), a protein kinase C (PKC) activator, increased HDAC2-IRS1 interaction and reduced insulin-stimulated pIRS1Tyr (Kaiser and James, 2004). Lastly, in H4IIE cells, SIRT1 inhibitors decreased IRS2 phosphorylation, impaired the pAktSer473 insulin signaling response, and subsequent mutation of putative Lys118/289/292/412 residues in IRS2 reduced insulin-stimulated pIRS2Tyr (Zhang, 2007). Additionally, SIRT2 has been shown up regulated in insulin resistant C2C12 myotubes, however its effect on IRS1/2 acetylation state was not directly shown (Arora and Dey, 2014). Putting these studies together, it appears that acetylation of IRS1 and IRS2 has opposing effects; IRS1 acetylation promotes its tyrosine phosphorylation, whilst acetylation of IRS2 inhibits it. The utility of these opposing effects are not readily apparent, it presents an interesting paradigm in which two insulin receptor substrates require differential modulation of their acetylation marks to affect downstream changes.
PI3K
Class 1A PI3K, a central signaling node for insulin-stimulated glucose transport, is a heterodimeric protein that is composed of a regulatory subunit (p85α, p85β, p50α, or p55α) and a catalytic subunit (p110α or p110β) (Taniguchi et al., 2006). MS analysis in non-muscle tissue has identified multiple Ac-Lys residues within the regulatory (p85α and p55α) and catalytic (p110β and p100γ) subunits (Table 1). For p85α, acetylation sites have been identified in the C-terminal SH2 and the inter-SH2 (which is between the N- and C-terminal SH2 domains) domains. These regions are important for docking to tyrosine-phosphorylated proteins, such as IRS proteins, and also for binding p110. For p110, there are no reported acetylation sites on p110α, whilst for p110β there are two Ac-Lys sites in close proximity to the p85 binding domain and the PI3K ras-binding domain. However, whether or not these acetylation marks effect heterodimer formation or PI3K function remains to be determined. In addition, to date, in skeletal muscle only the p55α subunit has been identified as having an Ac-Lys (Lundby et al., 2012), so clearly more detailed work is needed.
PDK1 and Akt acetylation
PDK1 and its downstream target, Akt, are serine-theronine kinases that recognize the PI3K generated second messenger, PIP3, through their pleckstrin homology (PH) domain. Multiple acetylation sites have been identified in the kinase and PH domain of PDK1 and Akt (Table 1). For example, in cardiac myocytes, p300 and p300/CBP-associated factor (PCAF) acetylate lysine residues in the PH domains of Akt (Lys14 and Lys20) and PDK1 (Lys495 and Lys534), negatively regulating their interaction with PIP3 (Sundaresan et al., 2011). Additionally, in MS analysis, Lys378 within the kinase domain of Akt2, was acetylated. In contrast, SIRT1-mediated deacetylation of these residues promoted their activation through enhanced binding of Akt and PDK1 to PIP3 (Sundaresan et al., 2011) Interestingly, in skeletal muscle, there was a decrease in Ac-Akt after insulin stimulation in fasted mice (but not non-fasted mice), and concomitant greater activation of Akt (Sundaresan et al., 2011). While effects on insulin-stimulated glucose uptake were not assessed in this study, we (White et al., 2013; 2014) and others (Banks et al., 2008) find that overexpression of SIRT1 in skeletal muscle does not enhance Akt activation (as measured by pAktSer473 and pAktThr308) or insulin sensitivity. In contrast, SIRT1 is required for the ability of caloric restriction to enhance skeletal muscle insulin action, through a mechanism upstream of PDK1-Akt, at the level of PI3K (Schenk et al., 2011). Although Ac-Akt was not measured in these studies, overall they do suggest that the contribution of SIRT1 to PDK1-Akt function in skeletal muscle is less definitive than in cardiac tissue.
Interestingly, in non-skeletal muscle cell lines that have normal insulin-regulated Akt activation, SIRT2, but not SIRT1, bound strongly to Akt and appeared to negatively regulate insulin-stimulated Akt activation (Ramakrishnan et al., 2014). Supporting a role for SIRT2 in negatively regulating Akt function, SIRT2 expression is increased in insulin resistant C2C12 myotubes and pharmacological- or knockdown-based inhibition of SIRT2 increases insulin signaling, Akt phosphorylation, and glucose uptake (Arora and Dey, 2014). While SIRT2 was not specifically shown to deacetylate Akt in these studies, inhibition of SIRT2 in a hepatocellular carcinoma cell line increased Ac-Akt (Chen et al., 2013).
Negative regulators of insulin signaling
Phosphatase and tensin homolog (PTEN) negatively regulates insulin signaling by dephosphorylating PIP3 (Tamgueny and Stokoe, 2007). Multiple Ac-Lys sites are present in the phosphatase domain of PTEN (Table 1), and growth factor stimulation in HEK293 cells suggest that PCAF-mediated acetylation in this domain at Lys125 and Lys128 inhibits its ability to dephosphorylate PIP3 (Okumura et al., 2006). In addition, PTEN acetylation is increased in SIRT1-deficient embryonic stem cell (ES), suggesting that PCAF and SIRT1 reciprocally regulate PTEN acetylation and PIP3 accumulation (Chae and Broxmeyer, 2011).
Protein tyrosine phosphotase 1B (PTP1B) negatively regulates insulin signaling by dephosphorylating the IR (Klaman et al., 2000; Salmeen et al., 2000). Two acetylation sites are present in the phosphatase domain of PTP1B (Table 1), although whether these activate or inhibit its function is not known. Lastly, examination of potential acetylation sites in other negative regulators of insulin signaling such as SKIP2, SHIP and TRB3 (Leto and Saltiel, 2012; Taniguchi et al., 2006), reveals that only SHIP2 has a known Ac-Lys site (Table 1); its function, however, is not known.
mTORC2
mTOR is a serine-threonine kinase that plays a central role in glucose metabolism and insulin signaling (Leto and Saltiel, 2012; Shimobayashi and Hall, 2014; Taniguchi et al., 2006). mTORC2, which contains the rapamycin-insensitive complex, Rictor, is required for Ser473 phosphorylation and full activation of Akt (Leto and Saltiel, 2012; Taniguchi et al., 2006). Rictor is a scaffolding protein within the mTORC2 complex that is required for proper function of mTORC2 activity, and its response to upstream insulin signaling (Sarbassov et al., 2005; Shimobayashi and Hall, 2014). Rictor contains a central region (amino acids between 1040–1140) that confers stability to mTORC2 and this region is highly acetylated with nine Ac-Lys residues (Table 1); lysine mutants in this region of Rictor exhibit impaired pAktSer473 (Glidden et al., 2012). Additionally, in HEK293 cells p300 overexpression or NAD+-dependent DAC inhibitors increased acetylation of Rictor, implicating these proteins in the regulation of Rictor and mTORC2 function.
GLUCOSE METABOLISM
In skeletal muscle, a post-prandial increase in circulating insulin normally results in increased glucose uptake into the myocyte, of which approximately one-third enters glycolysis and two-thirds is converted to glycogen (DeFronzo and Tripathy, 2009). Thus, coordination and matching of insulin-stimulated glucose uptake with this glucose ‘disposal’ is essential. In line with our discussions above on insulin signaling, recent MS-based analysis have found that essentially all enzymes in glycolysis and glycogen synthesis contain at least one, and in some cases as many as twenty nine, Ac-Lys sites (Table 2) (Choudhary et al., 2009; Lundby et al., 2012; Wang et al., 2010). While the precise role of acetylation to the function of these enzymes, especially those that are highly acetylated, remains to be fully elucidated, it is not unreasonable to infer that they likely contribute to their enzymatic activity. We discuss some examples of this below.
Table 2.
Acetylation and glucose metabolism
| Protein | Lys-Ac Sites | Ref | ||
|---|---|---|---|---|
|
| ||||
| Human | Mouse | Rat | ||
| Glycolysis | ||||
|
| ||||
| HK | (3)K | (4)K | (18)K | 1, 2, 3 |
| G6P | (12)K | (8)K | (20)K | 1, 2, 3, 4, 5 |
| PFK | (6)K | -- | (13)K | 1, 2, 3, 4, 5 |
| ALDO | (16)K | (7)K | (14)K | 1, 2, 3, 4, 5 |
| TPI | (12)K | (12)K | (14)K | 1, 2, 3, 4, 5 |
| GAPDH | (18)K | (14)K | (15)K | 1, 2, 3, 4, 5 |
| PGK | (29)K | (19)K | (22)K | 1, 2, 3, 4, 5 |
| PGlyM | (3)K | (1)K | (8)K | 1, 2, 3, 4, 5 |
| ENO | (2)K | (4)K | (17)K | 1, 2, 3, 4, 5 |
| PK | (23)K | (3)K | (12)K | 1, 2, 3, 4, 5 |
|
| ||||
| Glycogen | ||||
|
| ||||
| GCK | K296 | K296 | -- | 1, 2 |
| PGluM | (3)K | (18)K | (14)K | 1, 2, 3, 4 |
| UGP | (5)K | (13)K | (12)K | 1, 2, 4 |
| GS | (3)K | -- | (2)K | 1, 2, 4 |
| GP | (4)K | (13)K, K470, K796 | -- | 1, 2, 3, 4, 5, 6 |
| GLYG | -- | -- | K27, K209, K229 | 1, 2 |
HK, Hexokinase; G6P, Glucose-6-phosphate isomerase; PFK, Phosphofructose-kinase; ALDO, Aldolase; TPI, Triosephosphate isomerase; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; PGK, phosphoglycerate kinase; PGlyM, phosphoglycerate mutase; ENO, Enolase; PK, Pyruvate Kianse; GCK, glucokinase; GS, glycogen synthase; PGluM, phosphoglucose mutase; UGP, UTP-glucose-1-phosphate uridyltransferase; GS, Glycogen synthase; GP, Glycogen phosphorylase; GLYG, Glycogenin; H, Human; M, Mouse; R, Rat; --, Not available. Note: Numbers inside paranthese preceeding, K (Lys), denote the number of Lys-Ac sites identified in each species. (1) www.phosphosite.org; (2), Lundby et al 2012. (3) Choudhary et al 2009. (4) Guan et al 2010. (5) Chen et al 2012; (6), Zhang et al 2012.
Gycolysis
Recently, Lundby et al. (2012) investigated the functional significance of Ac-Lys in the glycolytic enzymes, aldolase B (ALDOB) and glycerol-3-phosphate dehydrogenase (GPD1). Lysine to glutamine mutation (which mimics constitutive acetylation) ablated enzymatic activity of both enzymes. This finding is intriguing as it presents acetylation as a mechanism to slow down glycolysis. In contrast, in Salmonella enterica acetylation of glycolytic enzymes stimulated glycolysis (Wang et al., 2010). While differences between these studies likely lie in, among other things, the complexity of the cell type studied, the underlying message is that reversible acetylation of glycolytic enzymes is sufficient to modulate their activity.
Glycogen synthesis
Glycogen phosphorylase (GP) breaks down glycogen by catalyzing the reaction of glycogen to glucose-1-phosphate (Wang et al., 2010). GP is classically thought to be regulated by phosphorylation through the opposing actions of phosphorylase kinase (PK) and protein phosphatase 1 (PP1). While PP1 acetylation sites have been identified (Choudhary et al., 2009; Lundby et al., 2012), the interplay of phosphorylation and acetylation on the activity of PP1 or its targets is unclear. Recently, GP Lys470 and Lys796 acetylation were found to negatively regulate glycogen catabolism by promoting PP1-GP interaction through the substrate targeting subunit GL, which leads to GP dephosphorylation and inhibition of its activity; however, the KAT responsible was not identified (Zhang et al., 2012). Additionally, pGSK3βSer9 was reduced in GH3 cells treated with resveratrol, a SIRT1 innibitor, although the mechanism underlying this effect is unknown (Monteserin-Garcia et al., 2013). Taken together, these results infer that insulin activation leads to acetylation of GP and deacetylation of GSK3β, thereby facilitating glycogen storage.
COORDINATING AND MATCHING CYTOSOLIC PROTEIN ACETYLATION TO GLUCOSE DISPOSAL
From our discussions above, it is clear that there are many cytosolic proteins that undergo reversible acetylation. Given that acetyl CoA is primarily generated in the mitochondria, a key question is of course, how is acetyl CoA generated in the cytosol to meet requirements for acetylation of proteins found within the cytosol? Citrate generated from the tricarboxylic acid (TCA) cycle is shuttled from the mitochondria into the cytosol when citrate begins to accumulate, where it is cleaved in the cytosol by ATP citrate lyase (ACLY) to produce acetyl CoA (Elshourbagy et al., 1992). Thus, production of cytosolic acetyl CoA would increase under conditions of increased glucose flux, such as with insulin stimulation. Interestingly, ACLY is directly phosphorylated by Akt, which increases its activity (Berwick et al., 2002; Migita et al., 2008; Pierce et al., 1982). Moreover, PCAF-mediated acetylation of ACLY at Lys540/546/554 stabilizes the protein (Lin et al., 2013). Putting these findings together, regulation of ACLY by Akt and acetylation presents an elegant model of matching insulin-stimulated Akt activation and glucose uptake to generation of cytosolic acetyl CoA and subsequent acetylation of cytosolic proteins.
ACETYLATION AS A METABOLIC RHEOSTAT: TYING THE PIECES TOGETHER
Traditionally, insulin signaling to glucose transport has been thought to progress via stepwise phosphorylation. The advent of the ‘acetylome’, however, provides an exciting additional level of control; such that perturbations in glucose flux are directly linked to changes in the activity (or binding) of insulin signaling proteins and enzymes central to glucose disposal or protein function, through changes in cytosolic acetyl CoA and NAD+ levels. By extension this presents a fascinating, but perhaps not surprising model in which acetylation allows for glucose availability to “talk” to proteins in a manner that allows prompt, flexible modulation of enzyme activity and pathway flux. The end result being a transient increase in glucose disposal after insulin stimulation that is in time down-regulated once the cell is sufficiently “full”.
While it is easy to ascribe a simple “on” or “off” tag to the effects of acetylation on function, acetylation does not appear binary, but rather a mechanism by which the cell finely tunes its response to insulin. One might think of phosphorylation as the key that turns the engine on or off, whilst acetylation is the accelerator and brake that by extension controls speed (or flux) across a wide spectrum. This is perhaps supported by the fact that many of the proteins in glycolysis are heavily acetylated, thereby providing stepwise control of glucose uptake in conjunction with insulin signaling.
We present a theoretical overview of this in Fig. 1. The translocation of GLUT4 to the plasma membrane is central to insulin-stimulated glucose transport. Under physiological conditions, the number of GLUT4 vesicles recruited to the membrane is a function of the efficiency of insulin signaling. Moreover, maintenance of the interstitial-to-intracellular glucose gradient is essential to maintain glucose transport through GLUT4 after insulin stimulation. In skeletal muscle, this gradient is maintained by conversion of glucose to glucose-6-phosphate (G6P), by hexokinase II (HKII). Once inside the muscle cell, glucose is disposed of oxidatively or non-oxidatively, via glycolysis or glycogen synthesis, respectively, with approximately two-thirds of glucose being shuttled toward glycogen. Thus, if one ascribes that acetylation of glycolytic enzymes results in a general slowing of reaction rates, then there would be an accumulation of intracellular G6P and reduced glucose transport. If concurrently, however, acetylation of glycogen-related enzymes favors glycogen synthesis, then G6P will be incorporated into glycogen, and a picture of flexible coordination of glucose disposal toward glycogen synthesis is revealed. Parallel changes in the cytosolic availability of acetyl CoA and NAD+, as a function of glucose flux, would allow for rapid temporal control of the flow of glucose, as well as simultaneous modulation of the acetylation status of insulin signaling proteins. The end result being coordinated control of insulin signaling, with glucose uptake and glucose disposal by acetylation. Certainly, the fact that insulin stimulation leads to activation of ACLY adds credence to this line of thinking.
Fig. 1.
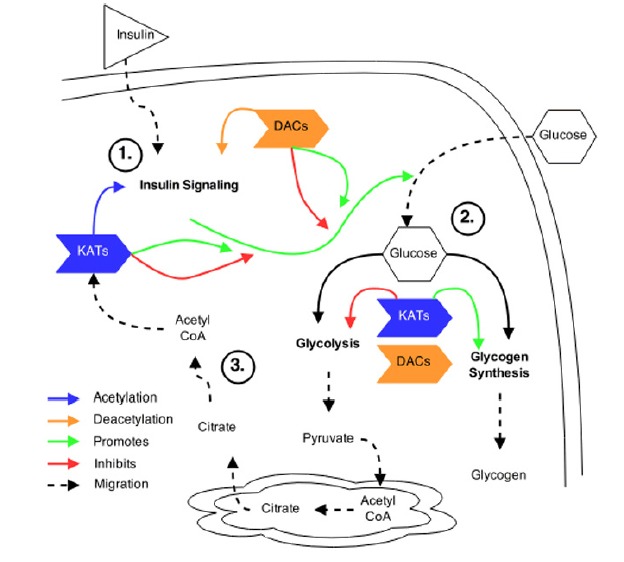
Theoretical overview of acetylation in insulin signaling and glucose metabolism. (A) KATs and DACs have been shown to regulate the acetylation patterns of various proteins within the insulin signaling pathway. However, the exact pattern of acetylation and the effect acetylation plays in response to insulin stimulation is not known. (B) Acetylation within glucose metabolism has been shown to regulate glycolytic enzymes and promote glycogen synthesis. However, the dynamics of acetylation patterns within these pathways, and the KATs and DACs that regulate them, following insulin stimulation, are not known. (C) Insulin stimulation may activate ACLY within the cytosol that produces acetyl CoA, from Citrate, required for subsequent acetylation of target proteins.
In this Minireview our goal has been to extrapolate the importance of acetylation to the regulation of insulin-stimulated glucose uptake and glucose metabolism, with a primary focus on skeletal muscle. Our reason for focusing on skeletal muscle is two-fold: skeletal muscle is the primary site for glucose disposal in the post-prandial state, and impairments in this disposal is a key predictor for the development of many metabolic diseases, including type 2 diabetes (DeFronzo and Tripathy, 2009). Thus, if we can understand “if and how” acetylation affects insulin signaling and glucose metabolism in muscle, then more effective treatment can be developed. Overall, however, there are clearly more questions than answers. One major issue in extrapolating to skeletal muscle is that many of the to-date identified Ac-Lys residues in the insulin signaling pathway (but not glucose metabolism) were not identified in human skeletal muscle (Choudhary et al., 2009; Lundby et al., 2012). Part of the reason for this could be that the muscle tissue was not insulin-stimulated, which might limit overall coverage. As such, it will be very interesting in future studies to compare the acetylome of basal vs. insulin-stimulated muscle, our hypothesis being that more proteins will be revealed to be acetylated after insulin stimulation. As part of this, determining if different patterns of acetylation exist between insulin sensitive compared to insulin resistant muscle, and whether this is altered by interventions that enhance insulin action, such as caloric restriction or exercise, will be highly insightful. For instance, in rat skeletal muscle, the acetylation of proteins is higher in aged/insulin resistant vs. young/insulin sensitive rats, whilst exercise normalizes acetylation in aged muscle to a level seen in young (Koltai et al., 2010); whether or not any of these proteins are related to insulin signaling or glucose metabolism is unknown.
Another pressing question is, does acetylation of insulin signaling proteins in skeletal muscle modulate their function? In addition, very little data exists regarding the specific KATs and DACs that regulate Ac-Lys residues of the enzymes and proteins of insulin signaling and glucose metabolism, let alone the acetylome as a whole. Furthermore, the discovery of various other lysine acylations, such as malonylation, succinylation, proprionylation, provide an additional link between nutrient flux and protein function (Choudhary et al., 2014). Studies using pharmacological or gene-based approaches, in parallel with ‘omics analysis and measurement of insulin-stimulated glucose uptake will be most helpful in addressing these gaps in knowledge.
In conclusion, in this review we examined the exciting topic of acetylation as an additional regulatory pathway in insulin signaling and glucose metabolism. Clearly ‘omics analysis has unlocked the door to the acetylome, and as a result, it is tempting to extrapolate functional outcomes (in our case, insulin action) from changes in protein and enzyme acetylation. Without doubt, however, many questions remain to be answered before the door is fully opened, and we look forward to exciting discoveries in the coming years.
Acknowledgments
This work was supported in part by National Institutes of Health grants, R01 AG043120, R24 HD050837 and P30 AR058878.
REFERENCES
- Arora A., Dey C.S. SIRT2 negatively regulates insulin resistance in C2C12 skeletal muscle cells. Biochim. Biophys. Acta. 2014;1842:1372–1378. doi: 10.1016/j.bbadis.2014.04.027. [DOI] [PubMed] [Google Scholar]
- Assali A.R., Ganor A., Beigel Y., Shafer Z., Hershcovici T., Fainaru M. Insulin resistance in obesity: body-weight or energy balance? J. Endocrinol. 2001;171:293–298. doi: 10.1677/joe.0.1710293. [DOI] [PubMed] [Google Scholar]
- Banks A.S., Kon N., Knight C., Matsumoto M., Gutiérrez-Juárez R., Rossetti L., Gu W., Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndsen C.E., Denu J.M. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 2008;18:682–689. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berwick D.C., Hers I., Heesom K.J., Moule S.K., Tavare J.M. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 2002;277:33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- Chae H.D., Broxmeyer H.E. SIRT1 Deficiency Downregulates PTEN/JNK/FOXO1 Pathway to Block Reactive Oxygen Species-Induced Apoptosis in Mouse Embryonic Stem Cells. Stem Cells and Development. 2011;20:1277–1285. doi: 10.1089/scd.2010.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Chan A.W.H., To K.F., Chen W., Zhang Z., Ren J., Song C., Cheung Y.-S., Lai P.B.S., Cheng S.-H., et al. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3β/β-catenin signaling. Hepatology. 2013;57:2287–2298. doi: 10.1002/hep.26278. [DOI] [PubMed] [Google Scholar]
- Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Choudhary C., Weinert B.T., Nishida Y., Verdin E., Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- Cohen P. The twentieth century struggle to decipher insulin signalling. Nat. Rev. Mol. Cell Biol. 2006;7:867–873. doi: 10.1038/nrm2043. [DOI] [PubMed] [Google Scholar]
- DeFronzo R.A., Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S157–63. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshourbagy N.A., Near J.C., Kmetz P.J., Wells T.N., Groot P.H., Saxty B.A., Hughes S.A., Franklin M., Gloger I.S. Cloning and expression of a human ATP–citrate lyase cDNA. Eur. J. Biochem. 1992;204:491–499. doi: 10.1111/j.1432-1033.1992.tb16659.x. [DOI] [PubMed] [Google Scholar]
- Glidden E.J., Gray L.G., Vemuru S., Li D., Harris T.E., Mayo M.W. Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J. Biol. Chem. 2012;287:581–588. doi: 10.1074/jbc.M111.304337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbs A.A., Modlin I.M., Ballantyne G.H. Changes in insulin resistance following bariatric surgery: role of caloric restriction and weight loss. Obes. Surg. 2005;15:462–473. doi: 10.1381/0960892053723367. [DOI] [PubMed] [Google Scholar]
- Kaiser C., James S.R. Acetylation of insulin receptor substrate-1 is permissive for tyrosine phosphorylation. BMC Biol. 2004;2:23. doi: 10.1186/1741-7007-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D.E., Wing R., Buonocore C., Sturis J., Polonsky K., Fitzsimmons M. Relative effects of calorie restriction and weight loss in noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1993;77:1287–1293. doi: 10.1210/jcem.77.5.8077323. [DOI] [PubMed] [Google Scholar]
- Kim G.W., Yang X.J. Comprehensive lysine acetylomes emerging from bacteria to humans. Trends Biochem. Sci. 2011;36:211–220. doi: 10.1016/j.tibs.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Klaman L.D., Boss O., Peroni O.D., Kim J.K., Martino J.L., Zabolotny J.M., Moghal N., Lubkin M., Kim Y.B., Sharpe A.H., et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltai E., Szabo Z., Atalay M., Boldogh I., Naito H., Goto S., Nyakas C., Radak Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech. Ageing Dev. 2010;131:21–28. doi: 10.1016/j.mad.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto D., Saltiel A.R. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- Lin R., Tao R., Gao X., Li T., Zhou X., Guan K.L., Xiong Y., Lei Q.-Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell. 2013;51:506–518. doi: 10.1016/j.molcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A., Lage K., Weinert B.T., Bekker-Jensen D.B., Secher A., Skovgaard T., Kelstrup C.D., Dmytriyev A., Choudhary C., Lundby C., et al. Proteomic Analysis of Lysine Acetylation Sites in Rat Tissues Reveals Organ Specificity and Subcellular Patterns. Cell Rep. 2012;2:419–431. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migita T., Narita T., Nomura K., Miyagi E., Inazuka F., Matsuura M., Ushijima M., Mashima T., Seimiya H., Satoh Y., et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547–8554. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- Monteserin-Garcia J., Al-Massadi O., Seoane L.M., Alvarez C.V., Shan B., Stalla J., Paez-Pereda M., Casanueva F.F., Stalla G.K., Theodoropoulou M. Sirt1 inhibits the transcription factor CREB to regulate pituitary growth hormone synthesis. FASEB J. 2013;27:1561–1571. doi: 10.1096/fj.12-220129. [DOI] [PubMed] [Google Scholar]
- Okumura K., Mendoza M., Bachoo R.M., DePinho R.A., Cavenee W.K., Furnari F.B. PCAF modulates PTEN activity. J. Biol. Chem. 2006;281:26562–26568. doi: 10.1074/jbc.M605391200. [DOI] [PubMed] [Google Scholar]
- Philip A., Rowland T., Perez-Shindler J., Schenk S. Understanding the acetylome: Translating targeted proteomics into meaningful physiology. American Journal of Physiology, Cell Physiology. 2014 doi: 10.1152/ajpcell.00399.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce M.W., Palmer J.L., Keutmann H.T., Hall T.A., Avruch J. The insulin-directed phosphorylation site on ATP-citrate lyase is identical with the site phosphorylated by the cAMP-dependent protein kinase in vitro. J. Biol. Chem. 1982;257:10681–10686. [PubMed] [Google Scholar]
- Pirola L., Zerzaihi O., Vidal H., Solari F. Protein acetylation mechanisms in the regulation of insulin and insulin-like growth factor 1 signalling. Mol. Cell. Endocrinol. 2012;362:1–10. doi: 10.1016/j.mce.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan G., Davaakhuu G., Kaplun L., Chung W.C., Rana A., Atfi A., Miele L., Tzivion G. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J. Biol. Chem. 2014;289:6054–6066. doi: 10.1074/jbc.M113.537266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeen A., Andersen J.N., Myers M.P., Tonks N.K., Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol. Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schenk S., McCurdy C.E., Philp A., Chen M.Z., Holliday M.J., Bandyopadhyay G.K., Osborn O., Baar K., Olefsky J.M. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J. Clin. Invest. 2011;121:4281–4288. doi: 10.1172/JCI58554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M., Hall M.N. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- Stöckli J., Fazakerley D.J., James D.E. GLUT4 exocytosis. J. Cell Sci. 2011;124:4147–4159. doi: 10.1242/jcs.097063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Zhou J. Trichostatin A improves insulin stimulated glucose utilization and insulin signaling transduction through the repression of HDAC2. Biochem. Pharmacol. 2008;76:120–127. doi: 10.1016/j.bcp.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Sundaresan N.R., Pillai V.B., Wolfgeher D., Samant S., Vasudevan P., Parekh V., Raghuraman H., Cunningham J.M., Gupta M., Gupta M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011;4:ra46. doi: 10.1126/scisignal.2001465. [DOI] [PubMed] [Google Scholar]
- Tamguney T., Stokoe D. New insights into PTEN. J. Cell Sci. 2007;120:4071–4079. doi: 10.1242/jcs.015230. [DOI] [PubMed] [Google Scholar]
- Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Verdin E., Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2014 doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- Wang J., Obici S., Morgan K., Barzilai N., Feng Z., Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001;50:2786–2791. doi: 10.2337/diabetes.50.12.2786. [DOI] [PubMed] [Google Scholar]
- Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A.T., McCurdy C.E., Philp A., Hamilton D.L., Johnson C.D., Schenk S. Skeletal muscle-specific overexpression of SIRT1 does not enhance whole-body energy expenditure or insulin sensitivity in young mice. Diabetologia. 2013;56:1629–1637. doi: 10.1007/s00125-013-2912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A.T., Philp A., Fridolfsson H.N., Schilling J.M., Murphy A.N., Hamilton D.L., McCurdy C.E., Patel H.H., Schenk S. High-fat diet-induced impairment of skeletal muscle insulin sensitivity is not prevented by SIRT1 overexpression. Am. J. Physiol. Endocrinol. Metab. 2014;307:E764–72. doi: 10.1152/ajpendo.00001.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J. Biol. Chem. 2007;282:34356–34364. doi: 10.1074/jbc.M706644200. [DOI] [PubMed] [Google Scholar]
- Zhang T., Wang S., Lin Y., Xu W., Ye D., Xiong Y., Zhao S., Guan K.-L. Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab. 2012;15:75–87. doi: 10.1016/j.cmet.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]