

Abstract
EphA7 has been implicated in the regulation of apoptotic cell death in neural epithelial cells. In this report, we provide evidence that EphA7 interacts with caspase-8 to induce apoptotic cell signaling. First, a pull-down assay using biotinylated ephrinA5-Fc showed that EphA7 co-precipitated with wild type caspase-8 or catalytically inactive caspase-8 mutant. Second, co-transfection of EphA7 with caspase-8 significantly increased the number of cleaved caspase-3 positive apoptotic cells under an experimental condition where transfection of EphA7 or caspase-8 alone did not affect cell viability or apoptosis. EphA4 also had a causative role in inducing apoptotic cell death with caspase-8, whereas EphA8 did not. Third, caspase-8 catalytic activity was essential for the apoptotic signaling cascade, whereas tyrosine kinase activity of the EphA4 receptor was not. Interestingly, we found that kinase-inactive EphA4 was well co-localized at the plasma membrane with catalytically inactive caspase-8, suggesting that an interaction between these mutant proteins was more stable. Finally, we observed that the extracellular region of the EphA7 receptor was critical for interacting with caspase-8, whereas the cytoplasmic region of EphA7 was not. Therefore, we propose that Eph receptors physically associate with a transmembrane protein to form an apoptotic signaling complex and that this unidentified receptor-like protein acts as a biochemical linker between the Eph receptor and caspase-8.
Keywords: apoptosis, caspase-8, EphA
INTRODUCTION
Early apoptotic cell death in neuroepithelial cells in the developing brain has been implicated in regulating mammalian brain size (Kuan et al., 2000). One class of proteins implicated in apoptotic neuroepithelial cell death is Eph receptors and their ligands, ephrins, which are abundantly expressed during early brain development (Park, 2013). The first evidence that Eph/ephrin signaling triggers apoptosis came from an ectopic ephrin-A5 expression study in EphA7-expressing brain tissue, where massive apoptosis and a subsequent decrease in cortical size was observed as a result of Eph and ephrin co-expression (Depaepe et al., 2005). In addition, ephrin-A5 null mutant mouse embryos display neural tube closure defects (Holmberg et al., 2000). Although it was postulated that fusion of neural folds at the midline is mediated by an adhesive interaction between EphA7 and ephrin-A5, an alternative explanation is that a significant increase in the dorsal midline cell population may be responsible for the neural tube closure defects in ephrin-A5 null mutant embryos. Consistent with this hypothesis, recent studies using in vivo expression of ephrinA5-Fc or EphA8-Fc revealed that upregulation of Eph/ephrin signaling in the dorsal midline play a causative role in triggering massive apoptotic cell death (Kim et al., 2013; Park et al., 2013). These findings suggest that cell-cell contact in a brain region where Eph and ephrin are co-expressed triggers the pro-apoptotic signaling pathway downstream of the Eph-ephrin complex, a critical mechanism for modulating the size of the neuroepithelial cell population or remodeling brain tissue (Park, 2013). Although the pro-apoptotic pathway downstream of the Eph/ephrin complex has not been clearly elucidated, a recent study suggested that EphA receptors may cross-talk with cell death receptors such as tumor necrosis factor receptor 1 (TNFR1) (Lee et al., 2013).
Caspase-8 is a member of the conserved cysteine-aspartic acid protease (caspase) family with a central role in executing cell apoptosis in the cell death receptor downstream pathway (Kumar, 2007). Caspase-8 is synthesized as a pro-enzyme and contains a large N-terminal prodomain and a C-terminal catalytic domain composed of a large and small subunit separated by a small linker (Pop and Salvesen, 2009; Wang et al., 2005). Caspase-8 is an initiator caspase (Nicholson, 1999), implicated in cleaving inactive pro-forms of effector caspases, thereby activating them to trigger apoptosis (Ashkenazi and Herbst, 2008). The caspase-8 prodomain contains the so-called death effector domain (DED), which enables it to interact with other proteins to regulate its activation. The caspase-8-dependent extrinsic apoptotic pathway is triggered by various cell death receptors including TNFR1. Activation of TNFR1 upon binding to its cognate ligand leads to recruitment of the adaptor molecule TRADD through a death domain (DD) interaction, subsequently forming TNFR1 complex I along with other proteins such as RIP1, TRAF2, and cIAP1/2 (Hsu et al., 1996a; 1996b). After internalization of the TNFR1 complex I, TRADD enables recruitment of FADD via an interaction of the DD’s of the two adaptors, forming TNFR1 complex II (Schneider-Brachert et al., 2004). FADD further recruits procaspase-8 via an interaction between the DED’s of the two proteins, forming a death-inducing signaling complex (DISC) (Chang et al., 2003; Kischkel et al., 1995; Peter and Krammer, 2003). Oligomerization of procaspase-8 seems to be sufficient to trigger autocatalytic cleavage and activation (Boatright and Salvesen, 2003). The procaspase-8 prodomain contains two cleavage sites for autoproteolytic processing, so interdomain cleavage events lead to the formation of a heterodimeric enzyme consisting of two large and two small subunits (Johnson and Kornbluth, 2008). The fully active caspase-8 heterodimeric enzyme transduces pro-apoptotic signals by cleaving and activating downstream executioner caspases or the BH3-interacting domain death agonist. Null mutation of caspase-8 or FADD in mice leads to embryonic lethality at embryonic day 10.5 (E10.5) due to failure of yolk sac vascularization and hematopoiesis (Kang et al., 2004; Zhang et al., 1998). However, little is known about whether caspase-8 plays a role in inducing apoptosis in a specific brain region in response to extrinsic cues.
In this report, we found that caspase-8 was physically associated with EphA7 and that this distinct protein complex induced caspase-dependent apoptotic cell death. Although evidence suggests that this interaction may not occur directly, we propose that a Eph-ephrin signaling complex may constitute a novel DISC involving caspase-8.
MATERIALS AND METHODS
Expression constructs
EphA7 (GenBank accession no. BC026153), caspase-8 (GenBank accession no. BC049955), and TNFR1 (GenBank accession no. BC004599) cDNAs were obtained from Thermo Scientific (USA). EphA4, EphA8, or TNFR1 cDNA in the pcDNA3neo expression vector was stably transfected into HEK293 cells, as described previously (Gu et al., 2005). A 206 bp polymerase chain reaction (PCR) product was amplified using primers matching nt 1740–1772 (5′-GTTGCCACACTTGAGGAAGCTTCAGGTAAAATG-3′) and nt 1924–1947 (5′-GCTAGCTCAGCAATGAAAGTAGAGTTCTTC-3′) of EphA7 cDNA to construct EphA7-D1 (with deletion of mouse EphA7 aa 601-994). Next, the 206 bp PCR product was digested with HindIII/NheI and subcloned into the same enzyme sites of the expression vector: HindIII site at nt 1616 of the full-length EphA7 cDNA and the NheI site in pCMV-SPORT6. The same procedures were used to generate EphA7-D2 (with deletion of mouse EphA7 aa 636-994), except that we amplified a 317-bp PCR product using primers matching nt 1740-1772 (5′-GTTGCCACACTTGAGGAAGCTTCAGGTAAAATG-3′) and nt 2042–2057 (5′-GCTAGCTCATGCACCAATCACACGCTCAAT-3′) of EphA7 cDNA. We amplified a 816-bp PCR product using primers matching nt 271–289 (5′-CGGGATCCAGACAATGCC-3′) and nt 1063–1087 (5′-GGCAGCCTGAATGAAAAAGATCTT-3′) of the caspase-8 cDNA, and a 375-bp PCR product using primers matching nt 1067-1105 (5′-ATCTTTTTCATTCAGGCTGCCCAAGGAAGTAACTTCCAG-3′) and nt 1426–1442 (5′-GCGGCCGCTTAGGGAGGGAAGAAGAG-3′) of caspase-8 cDNA to construct caspase-8 C362A (substitution of cysteine to alanine at aa 362). The resulting two partially complementary PCR fragments were annealed and used as templates in another PCR performed with primers matching nt 271–289 and 1426–1442 of caspase-8 cDNA. Next, the 1191-bp PCR product was digested with XbaI/ HindIII and subcloned into the corresponding region of the full-length caspase-8 cDNA.
Cell culture, transfection, and immunofluorescence staining
HEK293 cells were cultured as described previously (Gu et al., 2005). Transient transfection was carried out using ViaFect (Promega, USA), according to the manufacturer’s instructions. The cells were plated at a density of 1.0 × 106 cells per 35-mm dish for immunostaining. The cells were briefly washed with PBS 24 h post-transfection, fixed in 4% paraformaldehyde/2% sucrose in PBS on ice for 30 min, rinsed with PBS three times, and blocked for 30 min at room temperature with 3% bovine serum albumin, 5% horse serum, and 0.1% Triton X-100 in PBS. Rabbit anti-cleaved caspase-3 antibody (1:750, Cell Signaling Technology, USA) was applied for overnight at 4°C for apoptotic cell staining, and visualized with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG. The cells were incubated with ephrinA5-Fc (1 μg/ml) on ice and processed for immunofluorescence staining as described previously to co-localize EphA4 and casapse-8 (Lee et al., 2013). EphA4 was detected by staining cell surface bound ephrinA5-Fc with FITC-conjugated goat anti-human IgG, whereas caspase-8 was detected using rhodamine-conjugated anti-rabbit IgG.
Immunoprecipitation and Western blot
Immunoprecipitation and Western blotting were performed as described previously (Shin et al., 2007). Briefly, cells were grown to 70% confluence on 10-cm dishes and transiently transfected with 7 μg plasmid DNA. The cells were incubated with biotinylated ephrinA5-Fc (1 μg/ml, R&D Systems, USA) for 1 h at 4°C 16 h post-transfection. Labeled cells were lysed with PLC lysis buffer, as described previously, and bound to streptavidin-agarose beads (Thermo Scientific, USA). The bound material was eluted by boiling the beads and resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Antibodies
Anti-EphA4, anti-EphA7, and anti-TNFR1 antibodies were purchased from Santa Cruz Biotechnology (USA). Anti-caspase-8 and anti-cleaved caspase-3 antibodies were purchased from Cell Signaling Technology. FITC-conjugated goat anti-human IgG, rhodamine-conjugated anti-rabbit IgG, and FITC-conjugated anti-rabbit IgG antibodies were purchased from Invitrogen (USA). Horseradish peroxidase-conjugated anti-rat IgG and anti-rabbit IgG antibodies were acquired from Zymed (USA).
RESULTS
The EphA7 receptor is physically associated with caspase-8
The EphA7-mediated signaling pathway has been implicated in triggering apoptotic cell death during early brain development (Depaepe et al., 2005; Kim et al., 2013; Park et al., 2013). To explore the potential mechanism underlying the apoptotic signaling pathway downstream of EphA7, we investigated whether EphA7 can form a complex with caspase-8 in intact cells. For this purpose, 293 cells were transiently transfected with the EphA7 expression vector. Endogenous expression of EphA7 was not detectable in vector-transfected cells whereas ectopic expression of EphA7 was abundantly induced by transient transfection of the expression vector (data not shown; Fig. 1B). In contrast, endogenous caspase-8 expression was highly detectable, even in the absence of additional transfection (Fig. 1C). When the EphA7 receptor complexes were precipitated with biotinylated ephrinA5-Fc, caspase-8 specifically co-precipitated in the EphA7-transfected cells but not in the vector-transfected cells (Fig. 1A). In addition, co-immunoprecipitation using anti-caspse-8 antibody revealed that EphA7 was also associated with caspase-8 in EphA7-transfected cells (Figs. 1D and 1E, lane 1). To further confirm specific complex formation between EphA7 and caspase-8, EphA7 and caspase-8 were transfected into 293 cells. However, EphA7 level decreased markedly in the co-transfected cells, likely due to the high level of caspse-8 expression (Figs. 1F and 1G, lane 2). Although the EphA7 expression level was low in the co-transfected cells, it was evident that EphA7 was co-precipitated with caspase-8 (Fig. 1D, lane 2).
Fig. 1.
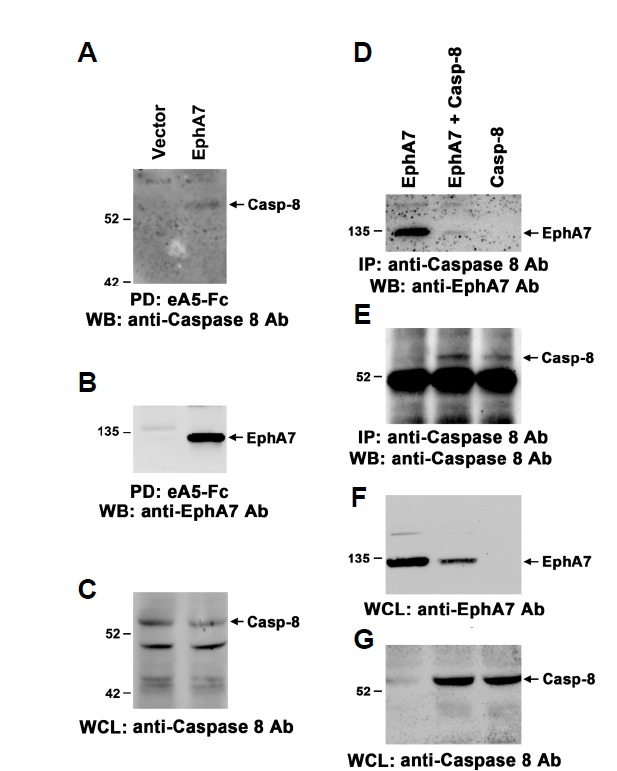
Physical association between EphA7 and caspase-8. HEK293 cells were transiently transfected with the indicated constructs, and the cells were treated with biotinylated ephrinA5-Fc on ice for 1 h 24 h post-transfection. Cell lysates were further incubated with streptavidin-agarose beads prior to pull-down (PD). Protein complexes were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to Western blot analysis using anti-caspase-8 (A) or EphA7 antibodies (B). Whole cell lysates (WCL) were also analyzed by Western blot using anti-caspase-8 antibody (C). (D-G) HEK293 cells were transiently transfected as above and the cell lysates were precipitated with anti-caspase-8 antibody 16 h post-transfection. Protein complexes were resolved by SDS-PAGE and subjected to Western blot analysis using the indicated antibodies.
To assess whether caspase-8 expression level affects that of EphA7 in co-transfected cells, 6 μg of EphA7 expression vector was transfected into 293 cells with varying amounts of the caspase-8 expression vector. As a result, EphA7 expression level was not significantly altered until the caspase-8 expression vector was added up to 0.25 μg (Supplementary Figs. 1A–1C, lanes 1–3). We observed that the level of caspase-8 that co-precipitated with EphA7 was significantly promoted in co-transfected cells compared with that in EphA7-transfected cells under these conditions (Supplementary Figs. 1A–1C, lanes 4–6). Interestingly, the elevated level of caspase-8 in the whole cell lysate was not observed in the cells co-transfected with EphA8, suggesting that this effect is due to EphA7 (data not shown). In addition, we generated a caspase8 mutant with deficient catalytic activity (cysteine at aa 362 was replaced by alanine) to test whether the EphA7 expression level was influenced by caspase-8 catalytic activity. Interestingly, EphA7 level was not significantly altered until caspase8 was added up to 5 μg under each co-transfection condition (Supplementary Figs. 1D–1F, lanes 1–3). Although EphA7 does not contain caspase-8 cleavage site, it needs to be further determined whether EphA7 is a direct substrate for caspase-8. More importantly, the level of caspase-8 co-precipitated with EphA7 increased proportionally according to the ectopic expression level of the inactive caspase-8 mutant. Interestingly, a low level of caspase-8 was detected in the protein complexes pull-downed by biotinylated ephrinA5–Fc when cells were transfected with caspase-8 alone, suggesting that the catalytically inactive caspase-8 mutant co-precipitated with other Eph receptors endogenously expressed in 293 cells (Supplementary Fig. 1D, lane 6). Taken together, these findings support our hypothesis that EphA7 physically forms a complex with caspase-8 and that this protein complex plays a role in triggering the apoptotic signaling pathway.
Co-expression of EphA7 with caspase-8 increases apoptotic cell death
As the transfection of 6 μg EphA7 mixed with 0.25 μg caspase-8 did not reduce EphA7 expression level, we further investigated whether EphA7 could cooperate with caspase-8 to promote apoptotic cell death under this transfection condition. As a control, cells transfected with vector alone barely displayed cleaved caspase-3 positive apoptotic cells (Figs. 2A and 2E, bar 1). Similarly, cells transfected with EphA7 or caspase-8 alone did not show a significant increase in the number of apoptotic cells (Figs. 2B, 2C, and 2E, bars 2 and 4). In contrast, co-transfection of EphA7 with caspase-8 resulted in an approximate 10-fold increase in the number of cleaved caspase-3 positive apoptotic cells (Figs. 2D and 2E, bar 3).
Fig. 2.
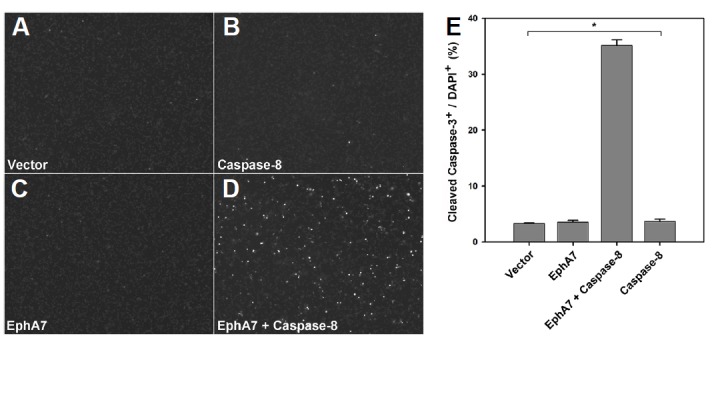
Stable complex formation between EphA7 and inactive caspase-8. (A–C) The EphA7 expression vector was transiently transfected into HEK293 cells with varying amounts of the caspase-8 expression vector. The cells were treated with biotinylated ephrinA5-Fc for pull-down 16 h post-transfection. Protein complexes were subjected to Western blot analysis using anti-caspase-8 (A) or anti-EphA7 antibodies (B). Whole cell lysates were also analyzed by Western blot using anti-caspase-8 antibody (C). (D–F) Experiments were performed essentially as in (A–C), except that the inactive caspase-8 mutant was used instead of wild-type caspase-8.
We further assessed whether other EphA receptors also synergistically cooperated with caspase-8 to enhance apoptotic cell death in 293 cells. As a positive control, co-transfection of TNFR-1 with caspase-8 was sufficient to induce massive apoptotic cell death in 293 cells (Figs. 3A, 3B, and 3G, bar 7). Interestingly, co-expression of EphA4 with caspase-8 also resulted in increasing apoptotic cell death to a similar degree as observed in EphA7/caspase-8 transfected cells (Figs. 3C–3E, and 3G, bars 2–5). However, EphA8 was not effective for inducing apoptotic cell death with caspase-8 (Figs. 3F and 3G, bar 6). EphA2 cooperated with caspase-8 to enhance apoptotic cell death to a similar degree as observed with EphA7 or EphA4 (data not shown). Taken together, our findings indicate that the Eph receptor/caspase-8 complex triggers the apoptotic signaling cascade and that this apoptotic protein complex is highly dependent on the type of Eph receptor.
Fig. 3.
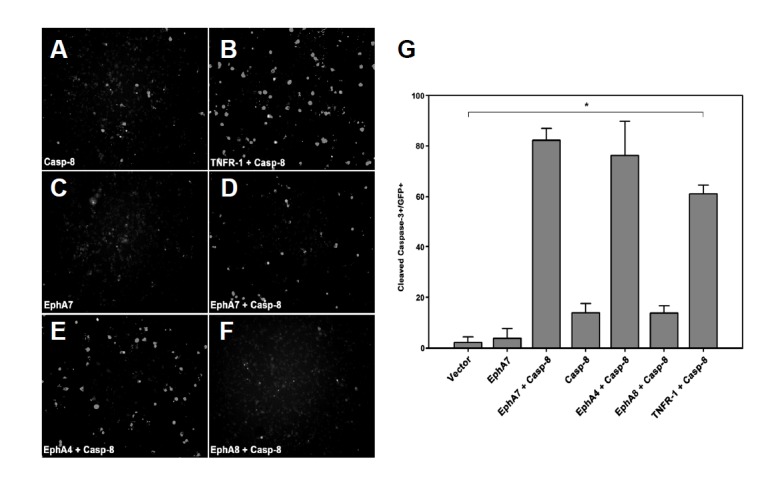
Apoptotic activation of caspase-8 is highly dependent on Eph receptor subtype. (A–F) HEK293 cells were transiently transfected with the indicated constructs as described in Fig. 3. Note that EphA8 does not promote apoptotic cell death even in the presence of caspase-8. (G) Cleaved caspase-3 stained apoptotic cells were counted as described in Fig. 3E but are presented as a percentage of total GFP+ cells. Caspase-8 was subcloned into a bicistronic pIRES-GFP vector. Data represent ± standard errors. *P < 0.001; single-factor analysis of variance.
Eph receptor tyrosine kinase activity is not required for inducing apoptotic signaling through caspase-8
Our results demonstrate that EphA4 or EphA7 physically interact with caspase-8 to induce the apoptotic signaling cascade. To further test whether caspase-8 catalytic activity is essential for triggering the apoptotic signaling cascade, the catalytically inactive caspase-8 mutant (C362A) was co-transfected into 293 cells with EphA4 or EphA7. Similar to the result for caspase-8 transfection alone, we did not observe a significant increase in apoptotic cell death in the transfected cells (Figs. 4D, 4G, and 4H, bars 4 and 7). We also tested whether EphA4 tyrosine kinase activity is essential for triggering apoptotic cell death with caspase-8. For this purpose, the tyrosine at EphA4 aa 653, an ATP binding residue, was replaced by phenylalanine. Strikingly, the tyrosine kinase inactive EphA4 mutant was as effective as wild-type EphA4 in enhancing apoptotic cell death with caspase-8 (Figs. 4F and 4H, bar 6). This finding suggests that Eph receptor tyrosine kinase activity is dispensable for its specific interaction with caspase-8 to trigger the apoptotic signaling cascade.
Fig. 4.
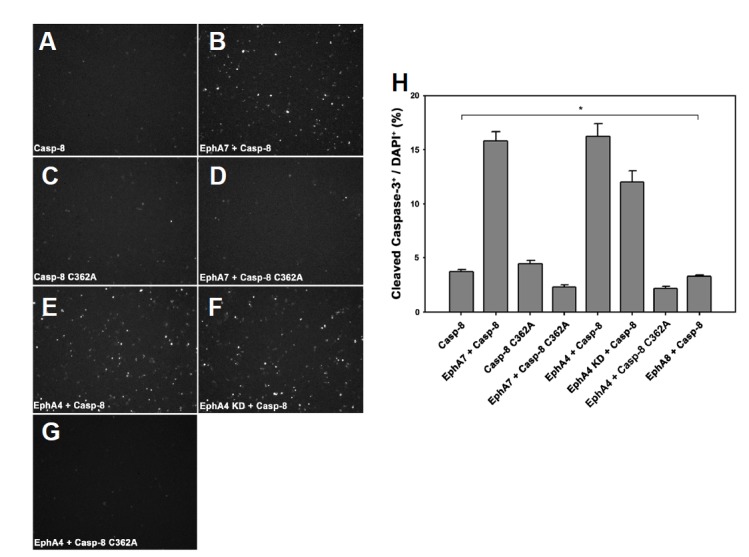
EphA4 autokinase activity is dispensable for caspase8-dependent apoptotic signaling. (A–G) HEK293 cells were transiently transfected with the indicated constructs. The cells were subjected to immunocytochemical staining using anti-cleaved caspase3 antibody 16 h post-transfection, as described in Fig. 3. (H) Data represent means ± standard errors. * P < 0.001; single-factor analysis of variance.
Although the kinase-inactive EphA4 mutant showed a similar result to that of wild type EphA4 for triggering apoptotic signaling, it was not efficiently internalized into the cells, and it more efficiently co-precipitated with the catalytically inactive caspase-8 mutant (data not shown). This result suggests that kinase-inactive EphA4 more stably complexes with catalytically inactive caspase-8 in cells. To confirm this possibility, we performed immunofluorescence staining using anti-caspase8 antibody and ephrinA5-Fc, which is bound to EphA4 and is visualized with Fc-specific secondary antibodies. As a result, the kinase-inactive EphA4 mutant was primarily localized to the cell surface, whereas wild-type caspase-8 was found in the cytoplasm (Figs. 5A–5C). The kinase-inactive EphA4 mutant was barely co-localized with wild-type caspase-8 on the cell surface, possibly due to their transient interaction on the plasma membrane. In contrast, when kinase-inactive EphA4 was co-expressed with catalytically inactive caspase-8, the two proteins co-localized well on the plasma membrane, suggesting that the interaction between the two proteins was prolonged due to the inactive mutation (Figs. 5D–5F). Taken together, our results indicate that the EphA4 or EphA7 receptor transiently associates with caspase-8 to trigger apoptotic cell death and that Eph receptor tyrosine kinase activity is not essential for this apoptotic process.
Fig. 5.
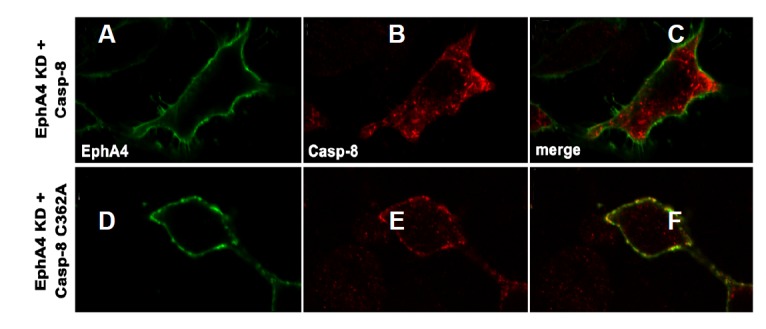
Co-localization of the kinase-inactive EphA4 mutant with the catalytically inactive caspase-8 mutant on the cell surface. (A–C) HEK293 cells were transfected with both kinase-inactive EphA4 mutant and wild-type caspase-8 expression vectors. The cells were treated with preclustered ephrinA5-Fc on ice for 1 h at 22 h post-transfection. EphrinA5-Fc (1 μg) was incubated for clustering with goat anti-human IgG antibody for 30 min on ice. The cells were washed, fixed, and subjected to immunocytochemical stain-using goat anti-human IgG antibody (conjugated with FITC) and anti-caspase-8 antibody (pre-incubated with rhodamine-conjugated secondary antibody). (D–F) Experiments were performed essentially as in (A–C), except that the inactive caspase-8 mutant was transfected instead of wild-type caspase-8.
The EphA7 extracellular region is required for interacting with caspase-8
We further investigated whether a specific domain in the EphA7 cytoplasmic region is crucial for interacting with caspase-8. Thus, we constructed two different deletion mutants, such as EphA7-D1 and -D2 (Fig. 6A). The EphA7-D1 mutant lacked an entire cytoplasmic region except for the first 10 aa of the juxtamembrane region. In contrast, the EphA7–D2 mutant had an intact juxtamembrane region but no tyrosine kinase domain or COOH-terminal region. Expression of these deletion mutants was highly detected in 293 cells (Fig. 6B). In addition, a cell surface binding assay using ephrinA5-Fc revealed that the cell surface localization of the deletion mutants was indistinguishable from that of wild-type EphA7 (Figs. 6C and 6D). Strikingly, these two EphA7 deletion mutants were equally effective in triggering apoptotic cell death, similar to wild-type EphA7 when they were co-expressed with caspase-8 (Fig. 6E, bars 3 and 4). More importantly, it was observed that the level of caspase-8 co-precipitated with the EphA7 deletion mutants was significantly elevated to a similar degree as observed in wild-type EphA7 (Fig. 6F). Taken together, these findings suggest that the extracellular region of EphA receptors is critical for interacting with caspase-8. We propose that Eph receptors physically interact with a transmembrane protein, which is not yet identified, but acts as a biochemical linker between the Eph receptor and caspase-8.
Fig. 6.
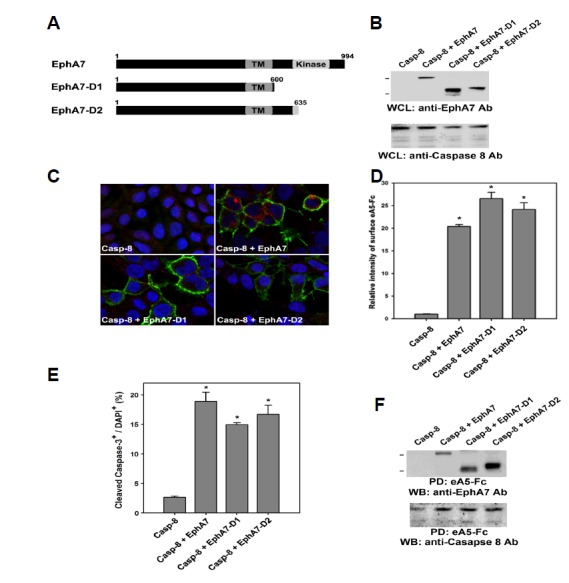
The EphA7 cytoplasmic region is not essential for apoptotic activation of caspase-8. (A) Schematic diagrams showing EphA7 deletion mutants used for expression in HEK293 cells. (B) Each of these deletion mutants was co-transfected with caspase-8 into HEK293 cells. Whole cell lysates were directly subjected to Western blot analysis using anti-EphA7 or anti-caspase-8 antibody 20 h post-transfection. (C) HEK293 cells were co-transfected with the indicated constructs. The cells were subjected to a cell surface binding assay using ephrinA5-Fc 16 h post-transfection, as described in Fig. 6. (D) The data in (C) were quantified. Data represent means ± standard errors. *P < 0.001; paired Student’s t-test. (E) HEK293 cells were co-transfected with the indicated constructs. The cells were subjected to immunocytochemical staining using anti-cleaved caspase3 antibody 16 h post-transfection, as described in Fig. 3. Data represent means ± standard errors. *P < 0.001; paired Student’s t-test. (F) Transfected cells were precipitated with biotinylated ephrinA5-Fc, as described in Figs. 1 and 2. Cell lysates were subjected to Western blot analysis using anti-EphA7 or anti-caspase-8 antibody.
DISCUSSION
Our previous studies showed that EphA7 recruits TNFR1 in response to ephrin-A ligands and that this receptor complex is involved in triggering the apoptotic signaling cascade (Lee et al., 2013). In this report, we provide strong evidence that EphA7 physically interacts with caspase-8 to induce apoptotic cell death in 293 cells. However, the EphA7 deletion mutant, lacking its cytoplasmic region, continued to interact with caspase-8 for apoptotic signaling. This observation suggests that the formation of complex between EphA7 and caspase-8 does not involve a direct interaction and that other transmembrane proteins may be a biochemical linker between EphA7 and caspase-8. Although TNFR1 was a potential biochemical linker candidate between EphA7 and caspase-8, TNFR1 was not endogenously expressed in 293 cells, suggesting that another cell death receptor may be involved in forming the complex between EphA7 and caspase-8 in 293 cells. Alternatively, a novel transmembrane protein rather than cell death receptors may play a role forming a ternary complex with EphA7 and caspase-8.
Notably, EphA8 did not interact with caspase-8 to induce apoptotic signaling pathway in 293 cells unlike other EphA receptors. We also found that EphA8 did not co-precipitate with caspase-8 in 293 cells (HL’s unpublished observation). As EphA8 is homologous to other EphA receptors, an intensive aa sequence comparison between EphA8 and EphA7 may allow us to identify a critical region in the EphA7extracellular domain that is involved in a physical interaction with caspase-8. In contrast, EphA8 has a unique juxtamembrane region (aa 584–603 of the mouse EphA8 protein), and this region is not conserved in other Eph receptors, such as EphA7 and EphA4. Therefore, this unique juxtamembrane region of EphA8 may inhibit formation of the apoptotic complex with caspase-8. It would be interesting to construct various chimeric EphA7/EphA8 receptors to test their ability to interact with caspase-8.
It remains unclear whether formation of an apoptotic complex between the EphA receptor and caspase-8 has physiological relevance in vivo, particularly during embryonic brain development. Interestingly we found that caspase-8 is co-expressed with the EphA4 receptor in olfactory neuroepithelium during embryonic brain development (HL’s unpublished observation). We observed that a subset of caspase8-positive olfactory neuroepithelial cells undergo apoptotic cell death. It would be interesting to investigate whether a null mutation of either EphA4 or capase-8 results in decreasing apoptotic cell death with the concomitant increase in the number of olfactory sensory neurons. In addition, identifying a cell surface protein biochemically linking EphA with caspase-8 would be essential to elucidate the regulatory mechanism of apoptotic cell death vs. cell proliferation during the development of olfactory epithelial tissue.
Acknowledgments
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare grant (A111706) and also by the National Research Foundation of Korea (NRF) grant funded by Korea government (MSIP) (No. 2011-0030074).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Ashkenazi A., Herbst R.S. To kill a tumor cell: the potential of proapoptotic receptor agonists. J. Clin. Invest. 2008;118:1979–1990. doi: 10.1172/JCI34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatright K.M., Salvesen G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Chang D.W., Xing Z., Capacio V.L., Peter M.E., Yang X. Interdimer processing mechanism of procaspase-8 activation. EMBO J. 2003;22:4132–4142. doi: 10.1093/emboj/cdg414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaepe V., Suarez-Gonzalez N., Dufour A., Passante L., Gorski J.A., Jones K.R., Ledent C., Vanderhaeghen P. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature. 2005;435:1244–1250. doi: 10.1038/nature03651. [DOI] [PubMed] [Google Scholar]
- Gu C., Shim S., Shin J., Kim J., Park J., Han K., Park S. The EphA8 receptor induces sustained MAP kinase activation to promote neurite outgrowth in neuronal cells. Oncogene. 2005;24:4243–4256. doi: 10.1038/sj.onc.1208584. [DOI] [PubMed] [Google Scholar]
- Holmberg J., Clarke D.L., Frisen J. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature. 2000;408:203–206. doi: 10.1038/35041577. [DOI] [PubMed] [Google Scholar]
- Hsu H., Huang J., Shu H.B., Baichwal V., Goeddel D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996a;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Hsu H., Shu H.B., Pan M.G., Goeddel D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996b;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Johnson C.E., Kornbluth S. Caspase cleavage is not for everyone. Cell. 2008;134:720–721. doi: 10.1016/j.cell.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Kang T.B., Ben-Moshe T., Varfolomeev E.E., Pewzner-Jung Y., Yogev N., Jurewicz A., Waisman A., Brenner O., Haffner R., Gustafsson E., et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- Kim Y., Park E., Noh H., Park S. Expression of EphA8-Fc in transgenic mouse embryos induces apoptosis of neural epithelial cells during brain development. Dev. Neurobiol. 2013;73:702–712. doi: 10.1002/dneu.22092. [DOI] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt S., Behrmann I., Germer M., Pawlita M., Krammer P.H., Peter M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan C.Y., Roth K.A., Flavell R.A., Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- Lee H., Park E., Kim Y., Park S. EphrinA5-EphA7 complex induces apoptotic cell death via TNFR1. Mol. Cells. 2013;35:450–455. doi: 10.1007/s10059-013-0072-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- Park S. Brain-region specific apoptosis triggered by Eph/ephrin signaling. Exp. Neurobiol. 2013;22:143–148. doi: 10.5607/en.2013.22.3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E., Kim Y., Noh H., Lee H., Yoo S., Park S. EphA/ephrin-A signaling is critically involved in region-specific apoptosis during early brain development. Cell Death Differ. 2013;20:169–180. doi: 10.1038/cdd.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M.E., Krammer P.H. The CD95(APO-1/Fas) DISC and beyond. Cells Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- Pop C., Salvesen G.S. Human caspases: activation, specificity, and regulation. J. Biol. Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Brachert W., Tchikov V., Neumeyer J., Jakob M., WinotoMorbach S., Held-Feindt J., Heinrich M., Merkel O., Ehrenschwender M., Adam D., et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Shin J., Gu C., Park E., Park S. Identification of phosphotyrosine binding domain-containing proteins as novel downstream targets of the EphA8 signaling function. Mol. Cell. Biol. 2007;27:8113–8126. doi: 10.1128/MCB.00794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.B., Liu Y.Q., Cui Y.F. Pathways to caspase activation. Cell Biol. Int. 2005;29:489–496. doi: 10.1016/j.cellbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Zhang J., Cado D., Chen A., Kabra N.H., Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392:296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.