

Abstract
Setdb1, an H3-K9 specific histone methyltransferase, is associated with transcriptional silencing of euchromatic genes through chromatin modification. Functions of Setdb1 during development have been extensively studied in embryonic and mesenchymal stem cells as well as neurogenic progenitor cells. But the role of Sedtdb1 in myogenic differentiation remains unknown. In this study, we report that Setdb1 is required for myogenic potential of C2C12 myoblast cells through maintaining the expressions of MyoD and muscle-specific genes. We find that reduced Setdb1 expression in C2C12 myoblast cells severely delayed differentiation of C2C12 myoblast cells, whereas exogenous Setdb1 expression had little effect on. Gene expression profiling analysis using oligonucleotide micro-array and RNA-Seq technologies demonstrated that depletion of Setdb1 results in downregulation of MyoD as well as the components of muscle fiber in proliferating C2C12 cells. In addition, exogenous expression of MyoD reversed transcriptional repression of MyoD promoter-driven lucif-erase reporter by Setdb1 shRNA and rescued myogenic differentiation of C2C12 myoblast cells depleted of endogenous Setdb1. Taken together, these results provide new insights into how levels of key myogenic regulators are maintained prior to induction of differentiation.
Keywords: C2C12 myoblast cells, MyoD, myogenic differentiation, Setdb1
INTRODUCTION
Differentiation of myogenic progenitor cells (myoblasts) into terminally differentiated myotube is tightly controlled by the coordinated actions of the myogenic regulatory factors (MRFs) MyoD, Myf5, myogenin, and Mrf4 that are parts of basic helix-loop-helix (bHLH) transcription factor family (Sabourin and Rudnicki, 2000; Sartorelli and Caretti, 2005). Gene targeting experiments and biochemical analysis established roles for these MRFs in skeletal muscle formation during embryonic development (Hasty et al., 1993; Rudnicki et al., 1993; Zhang et al., 1995). Among these MRFs, MyoD and Myf5 are primary MRFs that play key roles in the initiation and maintenance of skeletal muscle differentiation (Rudnicki et al., 1993; Tapscott, 2005) whereas myogenin and Mrf4, acting at downstream of MyoD and Myf5, are involved in muscle differentiation and the formation of myofibers. Upon the initiation of differentiation, MyoD interacts with E proteins and MEF2 transcription factors to activate muscle-specific genes and this initial change in gene expression is followed by the withdrawal from cell cycle, silencing of genes required for cell proliferation, and finally the formation of terminally differentiated myotubes (Lassar et al., 1991; Molkentin et al., 1995).
Functions of MyoD are known to be modulated by a number of cell type specific or ubiquitous transcription factors. Especially, the roles of chromatin modifying enzymes that mediate post-translational modifications of histone and/or non-histone substrates have been increasingly appreciated as key regulators of myogenic differentiation (Blum and Dynlacht, 2013; Sartorelli and Juan, 2011). Histone deacetylases I (HDAC I) interacts with MyoD and occupies the promoter or enhancer of muscle-specific genes to prevent premature differentiation (Mal and Harter, 2003; Mal et al., 2001). In differentiating skeletal muscle cells, this negative regulation is countered by the actions of histone acetyltransferases p300/CBP and PCAF which acety-late histones and MyoD through interaction with MyoD and thereby initiate myogenic differentiation program (Dilworth et al., 2004; Puri et al., 1997a; 1997b). Similar to the regulation by acetylation/deacetylation, methylation/demethylation of his-tones has also been shown to play an important role in regulating myogenic differentiation. SET7 that mediates monomethylation of H3-K4 and SETD3 that can methylate both H3-K4 and H3-K36 have been shown to interact with MyoD to target muscle specific genes, which is correlated with the differentiation of myoblast cells (Eom et al., 2011; Tao et al., 2011). On the other hand, histone methyltransferases, which are associated with repressed chromatin, appear to function as negative regulators of myogenic differentiation. Suv39h1, in cooperation with MyoD, represses muscle genes expression prior to differentiation, whereas G9a histone methyltransferase negatively regulates muscle differentiation via methylation of nonhistone substrates such as MEF2D and MyoD (Choi et al., 2014; Ling et al., 2012; Mal, 2006). The Polycomb protein Ezh2, which tri-methylates histone H3-K27 interacts with YY1 in undif-ferentiated myoblast cells to target specific muscle regulatory genes, leading to inhibition of differentiation (Caretti et al., 2004). In contrast, UTX mediates demethylation of H3-K27 at muscle-specific genes and LSD1 counteracts the function of G9a on MEF2D, both of which lead to the promotion of myogenesis (Choi et al., 2014; Seenundun et al., 2010). Coincidently, the functions of histone modifying enzymes in myogenic differentiation are correlated with their activities in transcription. Histone modifying enzymes that are thought to mark repressed chromatin act in general as negative regulators of differentiation, whereas epigenetic regulators associated with active transcription seem to promote myogenic differentiation of C2C12 myoblast cells.
Setdb1, also known as ESET or KMT1E, is a bifurcated SET domain-containing histone methyltransfease that catalyzes trimethylation of H3-K9 at euchromatic genes, leading to the transcriptional repression (Schultz et al., 2002). Loss of Setdb1 results in peri-implantation lethality in mice and impairs survival of embryonic stem cells in mice (Dodge et al., 2004). In embryonic stem (ES) cells, Setdb1 is associated with repression of developmental regulators and thus depletion of Setdb1-destabilizes ES cell state and impairs its role in lineage specification into extraembryonic trophoblasts (Bilodeau et al., 2009; Yuan et al., 2009). Functions of Setdb1 as a developmental regulator are not limited to ES cells. Setdb1 is involved in silencing of non-neuronal genes in neural progenitor cells and ablation of Setdb1 impairs early neurogenesis and thereby results in severe defects in brain development (Tan et al., 2012). Of particular interest, Setdb1 plays pivotal roles in lineage specification of mesenchymal stem cells (Takada et al., 2009). Activation of Setdb1 by non-canonical Wnt signaling inhibits PPARγ, leading to the shift into osteoblastic cell lineage from mesenchymal stem cells (Takada et al., 2007). In addition, conditional deletion of Setdb1 in mesenchymal cells has revealed that Setdb1 is required for hypertrophic differentiation of chondrocytes (Yang et al., 2013). However, the role of Setdb1 in myogenic progenitor cells has not been tested.
In this study, we have identified Setdb1 as an important regulator in myogenic differentiation. Depletion of endogenous Setdb1 impaired the ability of C2C12 myoblast cells to differentiate into terminally differentiated myotubes, whereas exogenous Setdb1 expression had little effect on myogenic potential. Biochemical and expression profiling analyses demonstrated that loss of myogenic potential by Setdb1 shRNA in C2C12 myoblast cells was correlated with transcriptional repressions of both MyoD and muscle-specific genes prior to differentiation, of which the expressions increase upon the induction of differentiation. Finally, exogenous expression of MyoD reversed the inhibition of myogenic differentiation caused by Setdb1 depletion in C2C2 myoblast cells, suggesting that Setdb1 is required for myogenic differentiation by maintaining levels of MyoD and muscle-specific genes in proliferating C2C12 myoblast cells.
MATERIALS AND METHODS
Plasmids
Full-length Flag-Setdb1 was generated from a plasmid containing full-length Setdb1 cDNA (kindly provided by Dr. F. J. Rauscher III, Wistar Institute, USA) using polymerase chain reaction (PCR) and cloned into pcDNA3 mammalian expression vector following digestion with EcoRI and NotI restriction enzymes. Full-length mouse Suv39h1 was purchased from Korea human gene bank (mMU006450). Coding sequences for Suv39h1 were amplified with a forward primer containing a BamH1 restriction site and a Flag epitope tag and a reverse primer with an XhoI restriction site. Amplified products were digested with BamHI and XhoI and cloned into pcDNA3 vector. Full-length myc-tagged MyoD was kindly provided by Dr. K. Y. Lee, Chonnam National University, Korea). Complete sequences of all PCR-amplified constructs were confirmed. For retroviral infection, full-length Flag-Setdb1, Flag-Suv39h1, and MyoD were subcloned into pLZRS-BMN or pLZRS-IRES-GFP retroviral vector using restriction enzymes, EcoRI and NotI for Setdb1 and BamH1 and XhoI for both Suv39h1 and MyoD. The plasmid (pMyoD-Luciferase containing F3/-2.5 fragment and MyoD promoter) used in the reporter assay has been previously described (Song and Lee, 2011).
Cell cultures, differentiation assays, and retroviral gene transfer
Mouse C2C12 myoblast cells and C3H 10T1/2 mesenchymal cells were maintained in DMEM (WelGENE) supplemented with 10% fetal bovine serum (WelGENE) and antibiotics in humidified atmosphere with 5% CO2 at 37°C. Myogenic differentiation was initiated as previously described (Song and Lee, 2011) and differentiation was assessed by the appearance of myotubes as well as expression of myosin heavy chain (MHC). Production of retrovirus and infection into C2C12 myoblast cells were carried out as previously described (Song and Lee, 2011).
Cell growth assay and cell cycle analysis
Cell proliferation was analyzed by the naphthol blue-black (NBB) method. Briefly, 2 × 103 cells were seeded in 24-well plates and cultured in a CO2 incubator at 37°C. Cells were fixed with 10% formalin (Sigma) at indicated time points and stained with 0.1% NBB staining solution containing 9% acetic acid and 0.1 M sodium acetate. After 30 min of staining, cells were extracted with 200 μl of 50 mM NaOH and absorbance of each sample was measured at 595 nm. Experiments were performed at least three times with triplicate for each time point. For cell cycle analysis, 5 × 105 proliferating myoblast cells were harvested, fixed with 75% ethanol solution, and then labeled with 20 mg/ml of propidium iodide and 20 mg/ml of RNase A for 30 min. Samples were then analyzed on FACS-Caliber flowcytometer (BD Biosciences) and relative proportion of cells at each stage of cell cycle was determined and displayed by using FlowJo V 7 software.
Western blot analysis and immunofluorescence
Western blot analysis was performed as previously described (Song and Lee, 2011). Antibodies used in this study were monoclonal anti-Flag M2 (Sigma-Aldrich), monoclonal anti-Myc (kindly provided by Dr. Abate-shen, Columbia University, USA), monoclonal anti-Actin (Sigma), anti-MyoD (Santacruz), anti-myogenin (Santacruz) and two rabbit polyclonal anti-Setdb1 (Proteintech group and Santacruz). For immunofluorescence, C2C12 cells were grown on 18-mm coverslip to 10% confluency with DMEM supplemented with 10% fetal bovine serum and myogenic differentiation was induced by serum withdrawal. After 72 h, cells were fixed in 4% paraformaldehyde-phosphate- buffered saline (PBS) for 10 min at room temperature and incubated in ice-cold methanol for 20 min at −20°C. After permeabilization with 1% Triton X-100 in PBS for 10 min, coverslips were blocked with 10% goat serum in PBS (Invitrogen) for 30 min, followed by incubation with monoclonal anti-MHC (Developmental hybridoma), and subsequently with Alexa 488-conjugated secondary antibody (Molecular Probes). Immunofluorescence was visualized with an Olympus IX71 inverted microscope equipped with a U-RFL-T burner and TH4-200 halogen lamp. All images were taken with a 20 X objective and processed using Adobe Photoshop premium suite CS3 software. Numbers of MHC-positive nuclei in myotubes were counted from at least 10 different randomly chosen areas until a total cell number of 1,000.
RNA preparation and quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was isolated from C2C12 cells following the manufacturer’s instructions. RNA was then further purified with RNeasy mini kit (Qiagen) following treatment with DNase I (Promega) to eliminate genomic DNA. For quantitative reverse transcription PCR (qRT-PCR) analysis, cDNA was synthesized by using 3 μg of RNA with superscript II cDNA synthesis kit (Invitrogen) following the manufacturer’s protocol. Quantitative real-time RT-PCR was carried out on an ABI 7300 Real-Time PCR system (Applied Biosystems) with SYBR Green I and iTaq polymerase (Intron). Relative expression was first quantified using standard curve method and data were normalized to GAPDH mRNA. Primers used in this study were as follows. Setdb1; 5′-CTTCTGGCTCTGACGGTG-3′ and 5′-GGAAGCCATGTTGGTTGATT-3′, Myogenin; 5′-CACTGGAGTTCGGTCCCAA-3′ and 5′-TGTGGGCGTCTGTAGGGTC-3′, MyoD; 5′-TCAGGTGCTTTGAGAGATCGAC-3′ and 5′-CGAAAGGACAGTTGGGAAGAGT-3′, GAPDH; 5′-TGCGACTTCAACAGCAACTC-3′ and 5′-GCCTCTCTTGCTCAGTGTCC-3′.
Gene expression profiling analysis
Gene expression profiling analyses using both oligonucleotide microarray and RNA-Seq were performed at Macrogen (Macrogen Inc., Korea). For microarray experiments, total RNAs prepared from six groups of three independently cultured C2C12 myoblast cells were labeled with biotin using an Illumina TotalPrep RNA Amplification Kit (Ambion). Following fragmentation, labeled RNAs were hybridized to a MouseWG-6 v2.0 Expression BeadChip that contains more than 45,200 transcripts (Illumina, USA). Array data were processed and analyzed using the Illumina BeadStudio v3.1.3, ArrayAssistand and R statistical language v.2.4.1 software. For RNA-Seq analysis, mRNA libraries were constructed using the Illumina TruSeq RNA sample preparation kit following the manufacturer’s instruction. Samples were run on an Illumina HiSeq2000 instrument and raw data were processed and analyzed using TopHat and Cufflinks software. Gene ontology analysis was performed by using ANNOVAR software and MGI Gene Ontology Term Finder (http://www.informatics.jax.org/gotools/MGI_Term_Finder.html).
Chromatin immunoprecipitation (ChIP) analysis
Chromatin immunoprecipitation (ChIP) analysis was performed as previously described (Song and Lee, 2011). Briefly, cells were cross-linked with 1% formaldehyde and then neutralized with 0.125 M glycine (Bio-Rad). Chromatins were prepared by sonication and incubated with anti-Flag-M2 agarose (Sigma-Aldrich). Immunoprecipitated DNA was analyzed by PCR followed by agarose gel electrophoresis. Primer sets to amplify MyoD promoter and Actin are previously described (Song and Lee, 2011) and primers to amplify Nnat gene are as follows. Nnat; 5′-TGCTGCTGCAGGTGAGTATGTA-3′ and 5′-TTGCGGCAATTGGGATAGGA - 3′.
Reporter assay
C2C12 cells were grown to 50% confluency in 6-well plates in DMEM with 10% fetal bovine serum and transfected with 0.2 μg of pMyoD-Luciferase containing F3/-2.5 fragment and MyoD promoter, 0.1 μg of pSV-β-Gal and 0.3 μg of Setdb1 shRNA expression vector and/or 0.4 μg of myc-MyoD expression vector. For Figs. 5C and 5D, 0.3 μg of Flag-Setdb1 expression vector was used for transfection. The total amount of plasmid DNA was adjusted to 1.0 μg by adding empty vector (pcDNA3). Cells were harvested at 24 h after transfection and analyzed for luciferase activity following the manufacturer’s protocol (Promega).
Fig. 5.
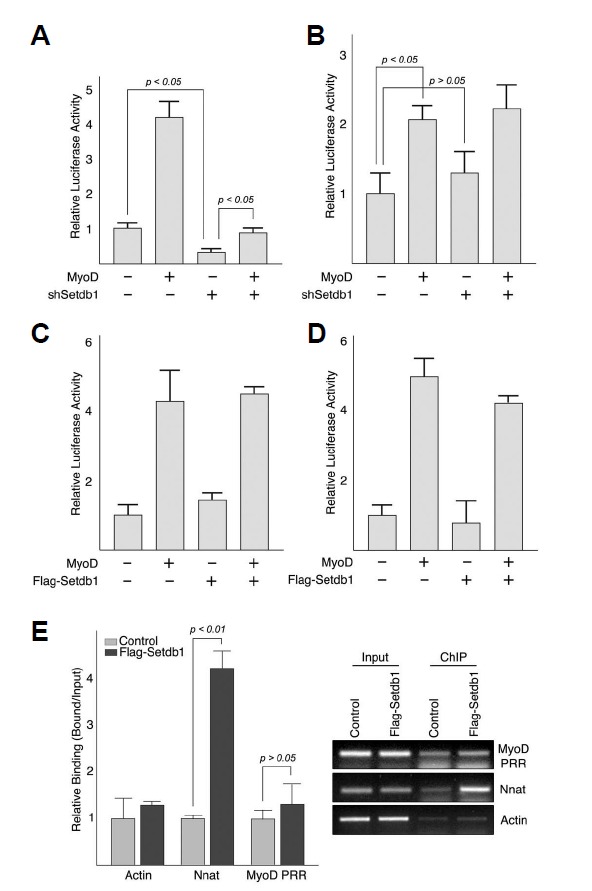
Setdb1 maintains endogenous MyoD expression without interfering transactivation by exogenous MyoD. (A, B) Setdb1 shRNA inhibits MyoD-luciferase reporter in C2C12 cells but not in C3H 10T1/2 cells. C2C12 myoblast cells (A) or C3H 10T1/2 mesenchymal cells (B) were transiently transfected for 24 h with plasmid expressing myc-MyoD and/or Setdb1 shRNA together with a MyoD-luciferase reporter. (C–D) Exogenous expression of Flag-Setdb1 had little effect on MyoD-luciferase reporter in both C2C12 and C3H 10T1/2 cells. C2C12 cells (C) or C3H 10T1/2 cells (D) were transfected with plasmid expressing myc-MyoD and/or Flag-Setdb1. For all reporter analysis, empty vector (pcDNA3) was added to adjust the total amount of transfected DNA to 1.0 μg. Data are presented as relative luciferase activity to the control (empty vector); Shown are representative data of three independent experiments performed in triplicate, and error bars indicate standard deviation. (E) Setdb1 did not bind to MyoD or myogenin promoter. Chromatin Immunoprecipitation (ChIP) was performed in C2C12 cells infected with retroviruses expressing Flag-Setdb1. The empty vector (pLZRS- IRES-GFP) was used as a control. Immunoprecipitated DNA was analyzed by PCR with specific primer sets described in Materials and methods followed by real-time qRT-PCR and agarose gel electrophoresis. Shown are representative data of three independent experiments. Nnat is a known Setdb1 target and was used as a positive control for Setdb1 binding, whereas Actin was used as a negative control.
Establishment of stable cell lines with Setdb1 shRNA
C2C12 cells were grown to 30% confluency in 60-mm plates in DMEM supplemented with 10% fetal bovine serum and transfected with 5 μg of pLKO.1 (control) or pLKO.1 containing shRNA against mouse Setdb1 (purchased from Open Biosystems). Transfected cells were treated with puromycin (5 μg/ml) for 2 weeks and puromycin-resistant clones were tested for endogenous Setdb1 expression by Western blot analysis. Selected clones were maintained for further study in the presence of 2.5 μg/ml of puromycin. Sequence information of Setdb1 shRNA used in this study can be found at http://www.broadinstitute.org/rnai/public/clone/search with the following IDs; TRCN-0000092973 ∼ TRCN000092977.
RESULTS
Depletion of Setdb1 impairs myogenic potential in C2C12 myoblast cell
To investigate the roles of histone modifying enzymes that target lysine 9 of histone H3 in myogenic differentiation, we depleted C2C12 myoblast cells of five H3-K9 specific histone methyltransferases, namely Suv39h1, Suv39h2, GLP, G9a, and Setdb1, by using relevant shRNAs and then tested for myogenic differentiation. Among those tested, we found that transfection of Setdb1 shRNA severely impaired the ability of myoblast cells to differentiate in a serum-deprived growth condition (Fig. 1 and data not shown). Of particular interest, decreased expression of endogenous Setdb1 inhibited differentiation rather than promoted it, whereas depletion of Suv39h1 or G9a that also targets H3-K9 has been shown to increase myogenic potential in C2C12 myoblast cells (Ling et al., 2012; Mal, 2006). To confirm that Setdb1 is indeed required for myogenic differentiation, we first examined the changes in Setdb1 expression during differentiation of C2C12 myoblast cells (Fig. 1A). Quantitative RT-PCR and Western blot analysis showed that levels of both Setdb1 mRNA and protein decrease during differentiation, which is inversely correlated with that of myosin heavy chain (MHC), a marker of terminal differentiation (Fig. 1A). Next, we established stable myoblast cell lines in which endogenous Setdb1 expression is depleted by shRNAs that target different regions of mouse Setdb1 gene and then tested for their myogenic differentiation potential (Fig. 1B and Supplementary Fig. S1). In vitro differentiation assay revealed that C2C12 cells with decreased level of Setdb1 protein displayed severely delayed differentiation even after 72 h in differentiation medium (Fig. 1B). In addition, western blot analysis and immunofluorescence using anti-MHC antibody showed that knockdown of Setdb1 resulted in significantly lower level of myosin heavy chain expression (MHC) as well as myogenic index during an extended differentiation period (96 h), suggesting that Setdb1 is required for the differentiation of C2C12 myoblast cells (Figs. 1C–1E).
Fig. 1.
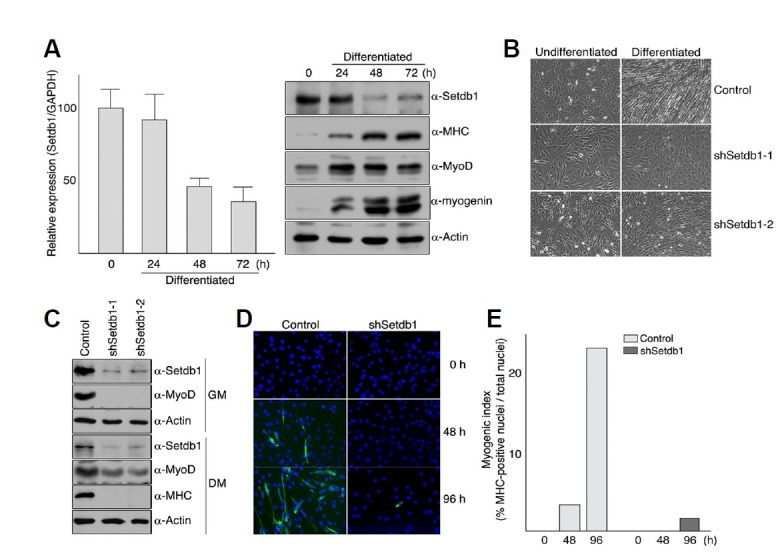
Inhibition of myogenic differentiation by Setdb1 depletion. (A) levels of Setdb1 decrease during C2C12 myoblast differentiation. C2C12 myoblast cells were grown to confluency in DMEM supplemented with 10% fetal bovine serum and differentiation was induced by serum withdrawal. Cells were harvested at the indicated time points and total RNAs or total protein extracts were prepared as described in “Materials and Methods”. RNA was analyzed by quantitative real-time RT-PCR using primers specific for Setdb1 and GAPDH. Relative expression of Setdb1 was determined using the standard curve method and then normalized to GAPDH. Error bars indicate standard deviation (left). Proteins were resolved on 7.5% (for Setdb1 and MHC) or 12% (for MyoD, myogenin, and Actin) SDS-PAGE and detected with antibodies against indicated proteins (right). (B-E) C2C12 myoblast cells with Setdb1 shRNA displayed severely delayed differentiation under serum-deprived conditions. C2C12 myoblast cells stably expressing control vector (pLKO.1) or Setdb1 shRNA were maintained in DMEM containing 10% fetal bovine serum and differentiation was initiated as described in Materials and methods. After 72 h, differentiation was assessed by the appearance of myotubes using photomicrograph (B), expression of MHC as well as endogenous MyoD using Western blot analysis (C), and number of MHC-positive nuclei per 103 cells using immunofluorescence (D, E).
We next investigated whether or not the defects in myogenic differentiation observed in Setdb1-depleted C2C12 cells can be rescued by exogenous expression of Setdb1 that is not targeted by Setdb1 shRNA (Fig. 2). Retroviral infection of human Setdb1 followed by in vitro differentiation appeared to restore myogenic potential in C2C12 cells depleted of Setdb1, which is evident by the formation of myotubes as well as the expressions of both myosin heavy chain (MHC) and MyoD (Fig. 2A). These data suggest that inhibition of differentiation in our established C2C12 cells was due to decreased Setdb1 level and not off-site effects caused by shRNAs. Interestingly, steady state mRNAs levels of both MyoD and myogenin, which are two key regulators of skeletal muscle differentiation, were significantly lower in cells depleted of Setdb1 compared to cells with control myoblast cells in undifferentiated state and these lower levels of expressions are not fully restored in a condition that promotes differentiation (Figs. 2B and 2C). This result implies that the role of Setdb1 in C2C12 cells might be associated with maintaining steady state levels of key myogenic regulatory factors (MRFs) such as MyoD and myogenin.
Fig. 2.
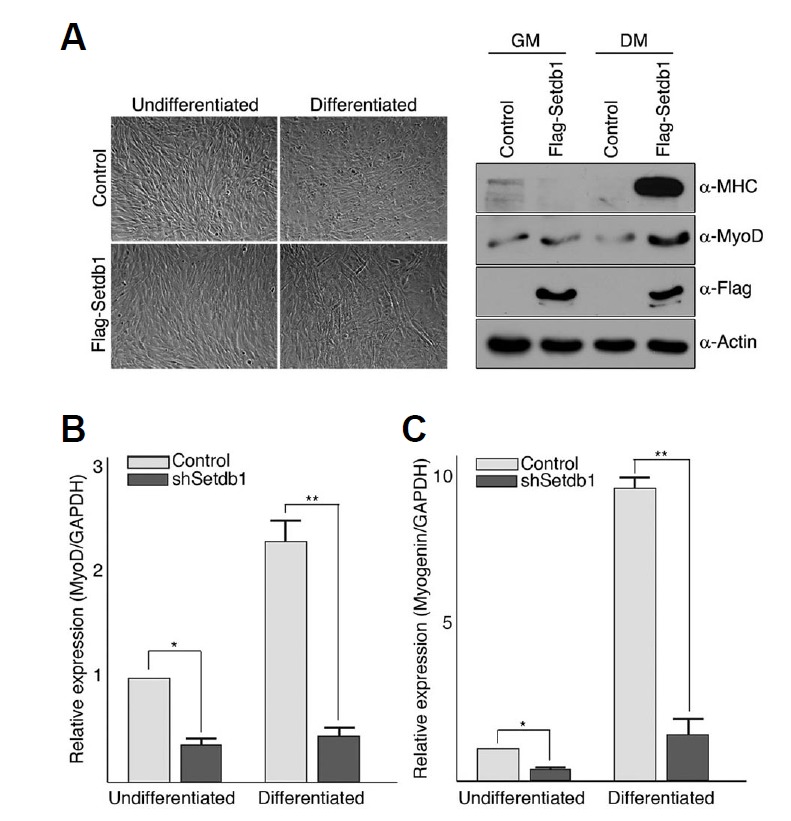
Setdb1 depletion leads to downregulation of myogenic regulatory factors in proliferating C2C12 cells. (A) Exogenous Setdb1 not targeted by Setdb1 shRNA rescues myogenic differentiation in Setdb1-depleted myoblast cells. C2C12 cells depleted of endogenous Setdb1 were infected with retrovirus expressing Flag-Setdb1 which is not targeted by Setdb1 shRNA used for cell line establishment. Empty vector was used as a control. Differentiation was assessed by the appearance of myotubes after 72 h in serum-deprived growth medium (left) and expressions of MHC and MyoD (right). (B, C) Total RNAs were prepared from both control and Setdb1 shRNA-expressing C2C12 cells prior to or 72 h after initiation of differentiation. RNA was analyzed by real-time PCR using primers specific for MyoD (B) and myogenin (C). Results were quantified using the standard curve method and normalized to GAPDH. Data were represented as relative expression. Shown are representative data of at least three independent experiments performed in triplicate, and error bars indicate standard deviation. *p-value < 0.05 and **p-value < 0.01
Setdb1 is required for myoblast cell proliferation
It is often the case that factors that regulate cell cycle progression inhibit myogenic differentiation by preventing exit from the cell cycle (Rao and Kohtz, 1995; Wang et al., 1995). So, we tested whether depletion of Setdb1 affects on cell proliferation by measuring cell growth and relative proportions of cells at different stages of cell cycle using flowcytometry (Figs. 3A and 3B). C2C12 myoblast cells harboring Setdb1 shRNA showed slower growth at low confluence (2 × 103 cells/24-well plate) and higher proportion of cells at G2/M stage of cell cycle compared to control C2C12 myoblast cells (Figs. 3A and 3B). Although it is unclear whether delayed cell growth is directly linked to the inhibition of differentiation, these data demonstrate that Setdb1 is associated with both cell proliferation and differentiation of C2C12 myoblast cells.
Fig. 3.
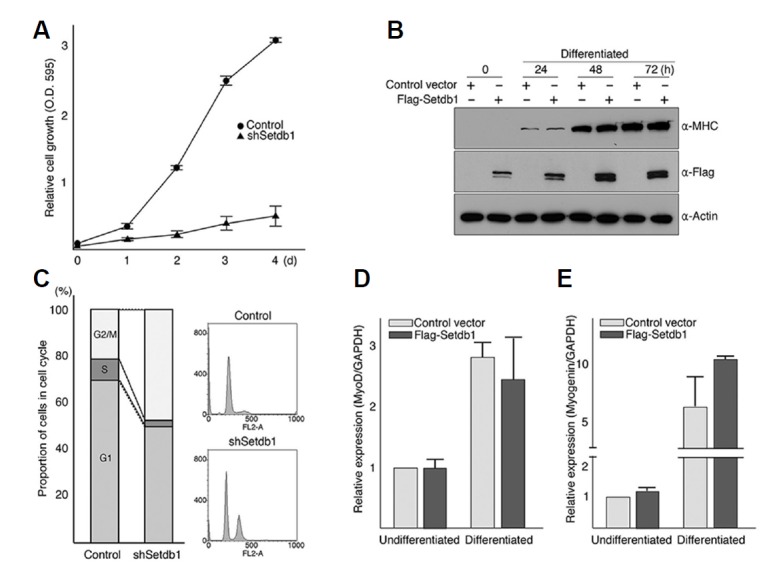
Knockdown of Setdb1 in C2C12 cells delay cell cycle progression. (A) The 2 × 103 cells stably expressing Setdb1 shRNAs or control vector (pLKO.1) were grown in 24-well plates, harvested at indicated time points, and stained with NBB staining solution following fixation with 10% formalin. Stained cells were extracted with 50 mM NaOH and absorbance for each sample was measured at 595 nm. Experiments were performed at least three times with triplicate for each time point. Error bars indicate standard deviation. (B) To measure relative proportion of cells at each stage of the cell cycle, 5 × 105 proliferating myoblast cells with indicated shRNA were labeled with propidium iodide in the presence of RNase A and analyzed using an FACS-Caliber flowcytometer. Data shown are representative of at least three independent experiments performed in triplicate. FlowJo V 7software was used for quantitation and graphic presentation. (C–E) Overexpression of Setdb1 had little or no effect on myogenic differentiation. (C) Undifferentiated C2C12 myoblast cells were infected with retroviruses expressing Flag-Setdb1. Empty vector (pLZRS-IRES-GFP) was used as a control. After 2 consecutive days of infection, cells were grown for 72 h under differentiation condition. Differentiation was assessed by Western blot analysis using antibody against MHC and exogenous expression of Flag-Setdb1 was shown to confirm retroviral infection. (DE) Levels of endogenous MyoD (D) and myogenin (E) were quantified by real-time RT-PCR as described in “Materials and Methods”. Shown are representative data of at least three independent experiments in triplicate, error bars indicate standard deviation.
Overexpression of Setdb1 does not appear to promote differentiation of C2C12 myoblast cells
Next, we tested the effect of Setdb1 overexpression on skeletal muscle cell differentiation (Figs. 3C–3E). Whereas overexpression of Suv39h1 in C2C12 myoblast cells inhibits differentiation as previously reported (Supplementary Fig. S2) (Mal, 2006), retroviral infection of Flag-tagged Setdb1 appeared to have little or no effect on myogenic differentiation up to 72 hours in differentiation medium, which is evident by the comparable expression of myosin heavy chain (MHC) (Fig. 3C) as well as the formation of terminally differentiated myotubes (data not shown). Consistently, levels of mRNAs for MyoD and myogenin did not change significantly by the expression of Flag-Setdb1 prior to and after induction of differentiation (Figs. 3D and 3E), demonstrating that overexpression of functional Setdb1, at least in our experiments, did not perturb the differentiation of C2C12 myoblast cells.
Knockdown of Setdb1 leads to deregulation of genes associated with myogenic differentiation
Considering that Setdb1 is involved in silencing of euchromatic genes and depletion of Setdb1 in C2C12 myoblast cells leads to the transcriptional repression of MyoD and myogenin as well as inhibition of cellular differentiation, it is unlikely that Setdb1 is directly linked to transcriptional repression of MyoD and myogenin. To better understand the underlying mechanisms linking inhibition of differentiation and depletion of Setdb1 in C2C12 myoblast cells, we investigated genes deregulated by
Setdb1 depletion by applying high-density oligonucleotide microarray (Illumina MouseWG-6 v2 expression BeadChip) and direct sequencing of total RNAs (Fig. 4). Gene expression profiles were obtained from three stable cell lines derived from C2C12 myoblast cells, one harboring control vector (pLKO.1) that maintain myogenic potential and two independently established cells with decreased Setdb1 respectively. By using criteria of a two-fold threshold and a false discovery rate (FDR) < 0.05, we first identified 757 genes (347 genes up and 410 genes down) that are differentially expressed between proliferating and differentiating C2C12 myoblast cells (72 h after induction of differentiation) (Fig. 4A), most of which were also presented in previously reported data (Cao et al., 2006; Delgado et al., 2003; Moran et al., 2002; Tomczak et al., 2004). Interestingly, 766 genes (352 genes up and 414 genes down) were found to be differentially expressed in myoblast cells with decreased Setdb1 under same experimental conditions even though they could not differentiate into myotube (Fig. 4A). Comparison of these expression profiles revealed that more than 85% of genes (296/347) induced in differentiated myotubes, which includes myogenin and muscle-specific genes, were not found in the list of genes upregulated in Setdb1 shRNA expressing cells. In addition, 50% of genes (207/414) that are downregulated by serum withdrawal in Setdb1-depleted myoblast cells overlapped with genes decreased during myogenic differentiation, demonstrating that knockdown of Setdb1 in C2C12 cells leads to the deregulation of genes involved in muscle differentiation.
Fig. 4.
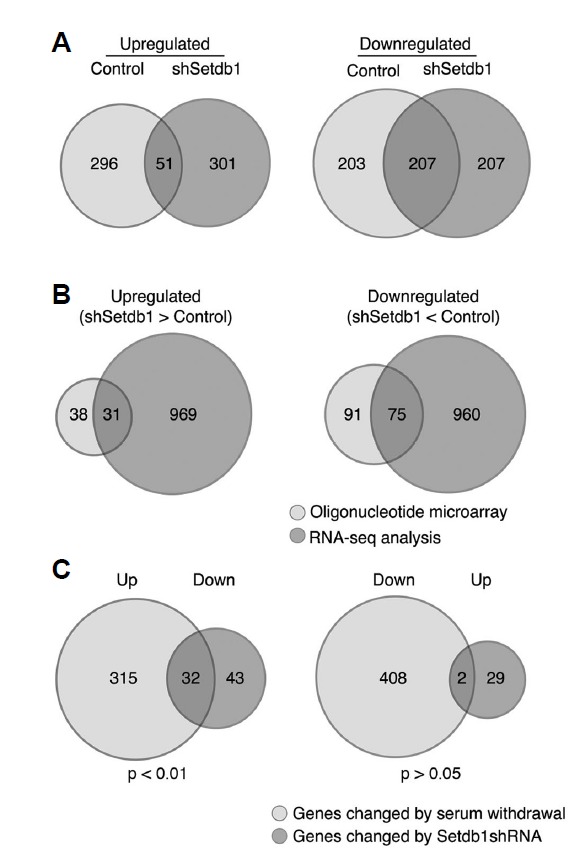
Depletion of Setdb1 leads to downregulation of muscle-specific genes in proliferating myoblast cells. (A) Venn diagrams show overlap of genes altered by serum withdrawal, which initiates myogenic differentiation of control C2C12 cells. Depletion of Setdb1 leads to deregulation of a comparable number of genes without induction of myogenic differentiation. Only a subset of upregulated genes (51/352) in C2C12 cells with Setdb1 depletion increase during normal differentiation (left panel), while 50% of downregulated genes (207/414) overlaps with those reduced by serum withdrawal (right panel). (B) Venn diagrams show upregulated (left) and downregulated (right) genes by Setdb1 depletion that are identified by combination of oligonucleotide microarray (light gray) and RNA-seq analsyis (dark gray). (C) Setdb1 is required to maintain levels of differentiation-dependent genes. Venn diagrams show that 32 of 75 genes downregulated by Setdb1 knockdown are induced during normal myogenic differentiation (left panel), whereas only two upregulated genes overlap with genes reduced during differentiation (right panel). P-values were calculated by hypergeometric test.
Since our previous results show that depletion of Setdb1 is associated with reduced expression of myogenic regulatory factors in proliferating myoblast cells (Fig. 2), we next investigated the effect of Setdb1 depletion on global gene expression in proliferating myoblast cells by applying oligonucleotide arrays and RNA-seq technologies (Fig. 4B). Statistical analysis of expression profiles identified 31 over-represented genes and 75 under-represented genes in C2C12 myoblast cells depleted of Setdb1 (Fig. 4B and Supplementary Table S1). Notably, at least 42 of 75 down-regulated genes were found to be related to muscle differentiation, which includes myogenic regulatory factors (MyoD and myogenin), structural components of muscle fibers (Myl1, Myom2, Tnnt1, and Tnni2), proteins involved in myoblast fusion (Ncam1, Sulf2, and Pgam2), and regulators of mesenchymal cell differentiation (Ebf3, G0s2, and Igfbp5). However, only two genes, Lys6a and PPARγ respectively, of those 31 up-regulated genes in Setdb1 shRNA-expressing myoblast cells have been linked to muscle differentiation (Mitchell et al., 2005; Singh et al., 2007). Of particular interest, we noticed that many genes that are deregulated in Setdb1-depleted proliferating myoblast cells were differentially expressed during normal skeletal muscle differentiation (Fig. 4C and Table 1). Among the 75 genes downregulated by Setdb1 depletion, 32 genes were upregulated during skeletal muscle differentiation (Fig. 4C) and 9 of those 32 genes including myogenin are known as direct targets of MyoD (Blais et al., 2005; Cao et al., 2006). In contrast, only two genes (Aqp1 and PPARγ) of 31 over-represented genes were differentially expressed during myogenic differentiation. These data suggest that function of Setdb1 is, at least in part, to maintain the steady state expression level of MyoD, and failure of which could lead to inhibition of differentiation.
Table 1.
List of differentiation-dependent genes deregulated in Setdb1-depleted C2C12 myoblast cells
| Gene | Description | Fold changes | MyoD binding | |
|---|---|---|---|---|
| Upregulated in Setdb1-depleted myoblast cells | Differentiation | Setdb1 depletion | ||
| Aqp1 | Aquaporin 1 | −13.32 | 2.71 | |
| Pparg | Perpxisome proliferatior activated receptor gamma | −2.28 | 3.23 | |
| Downregulated in Setdb1-depleted myoblast cells | ||||
| Acta1 | Actin, alpha 1, skeletal muscle | 3.43 | −12.23 | + |
| Actc1 | Actin, alpha, cardiac | 6.96 | −72.79 | |
| Actn3 | Actinin alpha 3 | 4.68 | −5.63 | |
| Atp2a1 | ATPase, Ca++ transporting, cardiac muscle, fast twitch 1 | 7.85 | −30.11 | |
| Chrng | Cholinergic receptor, nicotinic, gamma polypeptide | 8.07 | −19.22 | + |
| Drp2 | Dystrophin related protein 2 | 2.18 | −4.29 | |
| Gamt | Guanidinoacetate methyltransferase | 2.52 | −2.13 | |
| Igf2 | Insulin-like growth factor 2 | 2.35 | −50.22 | |
| Igfbp5 | Insulin-like growth factor binding protein 5 | 2.00 | −32.89 | |
| Kbtbd5 | Kelch repeat and BTB (POZ) domain containing 5 | 3.93 | −3.19 | |
| Mb | Myoglobin | 9.97 | −10.38 | |
| Myl1 | Myosin, light polypeptide 1 | 3.55 | −66.34 | |
| Mylpf | Myosin light chain, phosphorylatable, fast skeletal muscle | 3.33 | −34.60 | |
| Myod1 | Myogenic differentiation 1 | 1.62 | −6.05 | + |
| Myog | Myogenin | 2.83 | −13.91 | + |
| Myom1 | Myomesin 1 | 6.18 | −8.92 | + |
| Pdlim3 | PDZ and LIM domain 3 | 2.61 | −3.23 | + |
| Pgam2 | Phosphoglycerate mutase 2 | 8.50 | −3.89 | + |
| Rtn2 | Reticulon 2 (Z-band associated protein), transcript variant B | 2.16 | −5.26 | |
| Sgca | Sarcoglycan, alpha (dystrophin-associated glycoprotein) | 3.27 | −3.88 | |
| Srl | Sarcalumenin | 6.89 | −3.05 | |
| Tgfb3 | Transforming growth factor, beta 3 | 2.33 | −2.89 | |
| Tnnc1 | Troponin C, cardiac/slow skeletal | 3.94 | −23.81 | |
| Tnnc2 | Troponin C2, fast | 7.30 | −76.32 | + |
| Tnnt1 | Troponin T1, skeletal, slow | 3.70 | −101.42 | |
| Tnnt3 | Troponin T3, skeletal, fast | 12.12 | −8.96 | + |
| Tpm2 | Tropomyosin 2, beta | 10.27 | −15.82 | |
Setdb1 maintains MyoD expression in C2C12 myoblast cells
Level of MyoD is believed, if not entirely, to be maintained by positive autoregulation (Thayer et al., 1989). Based on our previous data demonstrating that depletion of Setdb1 leads to the downregulation of endogenous MyoD in proliferating myoblast cells (Figs. 2B and 4), we next tested whether Setdb1 regulates MyoD promoter-dependent transcription using MyoD-luciferase reporter plasmid (Figs. 5A–5D). Transfection of Setdb1 shRNA resulted in a strong repression of MyoD-luciferase reporter and inhibited transactivation by exogenous MyoD in C2C12 myoblast cells, which expresses endogenous MyoD (Fig. 5A). Surprisingly, transfection of Setdb1 shRNA appeared to have little effect on MyoD-luciferase reporter in C3H 10T1/2 cells, which do not express endogenous MyoD (Fig. 5B), implying that the effect of Setdb1 depletion on endogenous MyoD expression might be linked to positive autoregulation of MyoD. On the other hand, cotransfection of Flag-Setdb1 and MyoD had no significant effect on the expression of MyoD-luciferase reporter in either cell types (Figs. 5C and 4D), which is consistent with our previous results (Fig. 3B).
Actions of other H3-K9 specific methyltransferase known to regulate myogenic differentiation are mediated by the interactions with myogenic regulatory factors (MRFs) (Choi et al., 2014; Mal, 2006; Ling et al., 2012). However, it appeared not to be the case for Setdb1 as we failed to detect any significant physical interaction between Setdb1 and MyoD in vitro (data not shown). Moreover, Setdb1 did not bind to the MyoD promoter (Fig. 5E). Together, these data suggest that although Setdb1 is required for the MyoD promoter-dependent transcription, its action on MyoD promoter is likely indirect.
Exogenous MyoD can restore the myogenic differentiation potential in Setdb1-depleted C2C12 myoblast cells
If Setdb1 indirectly regulates endogenous expression of MyoD, it is possible that exogenous expression of MyoD independent of the MyoD promoter could restore the myogenic potential in Setdb1-depleted C2C12 myoblast cells. To test this possibility, we first examined the effect of exogenous MyoD on the MyoD-luciferase reporter in Setdb1-knockdowned C2C12 myoblast cells (Fig. 6A). Consistent with our previous result (Fig. 4A), MyoD-luciferase activity in Setdb1-depleted myoblast cells was consistently lower than that in control C2C12 cells. Interestingly, exogenous expression of MyoD can still transactivate expression of MyoD-luciferase reporter. Finally, we found that retroviral infection of exogenous MyoD resulted in fully differentiated myotubes from Setdb1-depleted C2C12 myoblast cells. This result strongly suggests that Setdb1 plays an important role in maintenance of endogenous MyoD, the perturbation of which could lead to deregulated differentiation (Fig. 6B and Supplementary Fig. S3).
Fig. 6.
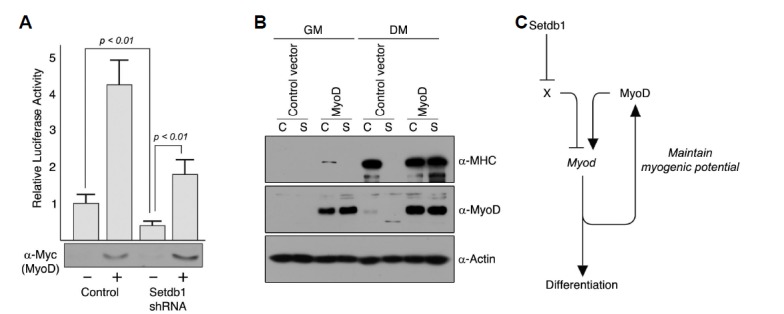
Exogenous MyoD can restore myogenic potential in Setdb1-depleted C2C12 myoblast cells. (A) Exogenous MyoD can transactivate MyoD-luciferase reporter in C2C12 cells depleted of Setdb1. C2C12 cells stably expressing Setdb1 shRNA or control vector were transfected with plasmid expressing myc-MyoD together with a MyoD-luciferase reporter. The empty vector (pcDNA3) was added to adjust the total amount of transfected DNA to 1.0 μg. Data are presented as relative luciferase activity to the control (empty vector) and expression of exogenous Myc-MyoD was verified by Western blot; Data shown are representative of three independent experiments performed in triplicate, and error bars indicate standard deviation. (B) Proliferating C2C12 myoblast cells stably expressing Setdb1 shRNA or control C2C12 cells were infected with retroviruses expressing MyoD. Empty vector (pLZRS-IRES-GFP) was used as a control. Cells were harvested prior to and 72 hours after induction of differentiation and total proteins were extracted. Differentiation was assessed by western blot analysis using antibody against MHC, and exogenous expression of MyoD was shown to confirm retroviral infection (C) Setdb1 is required for myogenic potential via maintenance of endogenous MyoD expression. In our proposed model, we speculate that Setdb1 inhibits repressor (X) of MyoD that could compete with MyoD in normal proliferating C2C12 myoblast cells. However, depletion of Setdb1 leads to inhibition of differentiation as this potential repressor is relieved from suppression.
DISCUSSION
Our current study demonstrated that Setdb1 is necessary for expression of muscle-specific genes and differentiation of myoblast cells into myotubes. Depletion of Setdb1 inhibited myogenic differentiation under serum-deprived growth conditions and gene expression profiling analysis revealed that knockdown of Setdb1 led to decreased expression of genes involved in skeletal muscle differentiation including key myogenic regulatory factor MyoD prior to differentiation. In addition, transcriptional repression of MyoD-luciferase by Setdb1 shRNA was dependent on the expression of endogenous MyoD and exogenous MyoD counteracted the inhibitory effect of Setdb1 depletion. Our data provide new insights into the underlying mechanism of how Setdb1 is involved in regulation of skeletal muscle differentiation.
MyoD is considered as a master regulator of skeletal muscle differentiation because of its ability to induce muscle-specific genes upon the initiation of differentiation and thereby initiate myogenic differentiation in non-muscle lineages (Tapscott, 2005). During embryonic development, myogenic progenitor cells that express MyoD arise from the dorsal part of the somite mesoderm but differentiation is delayed until they reach their destination and receive environmental cues that initiate the myogenic differentiation program (Rudnicki et al., 1993). This is also the case for cell culture model as C2C12 myoblast cells expressing endogenous MyoD remain undifferentiated under normal growth condition. It is quite intriguing and at the same time puzzling that endogenous expression of MyoD is necessary in general but not sufficient for initiation of myogenic differentiation, considering that MyoD expression is positively autoregulated by MyoD itself (Thayer et al., 1989). Although not fully elucidated, there have been many studies on the molecular mechanisms by which MyoD expression or activity is negatively regulated in MyoD-positive cells. Msx1 has been shown to inhibit skeletal muscle differentiation by its activity as a transcription repressor of endogenous MyoD through interactions with linker histone H1b as well as nuclear YB1 transcription factor (Lee et al., 2004; Song and Lee, 2011). Transcriptional activation by MyoD can also be negatively controlled by protein-protein interactions, which includes I-mf that prevents nuclear localization of MyoD (Chen et al., 1996), Id proteins that sequester functional E-box proteins as well as MyoD (Jen et al., 1992), and Twist that inhibits transactivation of MyoD (Spicer et al., 1996). Histone modifying enzymes that determine local chromatin structure have also been recognized as regulators of myogenic differentiation without interfering MyoD binding to the promoter (Mal et al., 2001; 2006; Puri et al., 1997b).
The role of Setdb1 as a developmental regulator has been suggested in studies on ES cell as well as other progenitor cells. Setdb1 contributes to the maintenance of ES cell state presumably by repressing genes involved in developmental processes (Bilodeau et al., 2009). In mesenchymal stem cells, Setdb1 plays a pivotal role in determining cell fates as overexpression of Setdb1 induces osteoblastogenesis and its depletion promotes adipogenesis (Takada et al., 2007). Of particular interest, our findings that depletion of Setdb1 inhibited myogenic differentiation is quite similar to a previous report showing impaired neurogenic differentiation resulted from Setdb1 deletion in mice (Tan et al., 2012). Both processes are controlled by ‘master regulators’, MyoD in myogenic differentiation and NeuroD in neuronal differentiation respectively and inhibition of differentiation by Setdb1 inactivation is accompanied by downregulation of many lineage-specific genes. However, the underlying mechanisms of Setdb1 in differentiation of progenitor cells are still largely unknown.
Given the nature of Setdb1 as a transcriptional repressor and its inhibitory effect on myogenic differentiation upon depletion, we expected to observe derepression of many developmental regulators upon the initiation of myogenic differentiation, which could perturb the normal differentiation program. Instead, our gene expression profiling analysis revealed that Setdb1 knockdown resulted in decreased expression of MyoD and muscle specific genes even prior to initiation of differentiation, suggesting that role of Setdb1 in C2C12 myoblast cells is likely linked to maintenance of myogenic potential. Consistent with our view that Setdb1 maintains level of endogenous MyoD in proliferating myoblast cells, the effect of Setdb1 shRNA on transcription of MyoD-luciferase reporter and transactivation of MyoD appeared to depend on the expression of endogenous MyoD (Figs. 5A and 5B) and exogenous MyoD were able to restore the inhibition of differentiation caused by Setdb1 depletion in C2C12 cells (Fig. 6B).
Moreover, we have not found any evidence of physical interaction between Setdb1 and MyoD both in vitro nor detected Setdb1 binding to MyoD and myogenin promoter (Fig. 5E and data not shown). These data together support the idea that Setdb1 might function at upstream of a double negative regulatory network to regulate endogenous MyoD expression (Fig. 6C). We speculate that Setdb1 negatively regulates the expression or function of transcriptional repressor(s) that compete with positive autoregulation by MyoD. In normal proliferating C2C12 myoblast cells, Setdb1 maintains myogenic potential by suppressing the function of a transcriptional repressor (described as X in our model). However, this repressor is relieved from inhibition in cells lacking Setdb1 and therefore actively represses endogenous MyoD expression, eventually leading to inhibited differentiation. On the other hand, Setdb1 might not be critical In differentiating C2C12 cells, as balanced actions of other histone modifying enzymes known to modulate functions of MyoD shift to promote expression of muscle-specific genes including MyoD itself. This model could also explain why over-expression of Setdb1 did not affect myogenic differentiation in our study.
Finally, although our proposed model is based on the assumption that there might be transcriptional repressor at downstream of Setdb1 that potentially inhibits expression of endogenous MyoD, our expression profiling analysis did not show any significant derepression in genes that are previously known as negative regulator of MyoD transcription. However, we have found that expression of PPARγ was inversely correlated with MyoD and depletion of Setdb1 led to derepression of PPARγ. Since PPARγ has been implicated in fate determination and differentiation of mesenchymal stem cells and perturbation of PPARγ has been shown to inhibit myogenic differentiation (Singh et al., 2007), it would be interesting to test whether PPARγ is at downstream of Setdb1 in the regulation of endogenous MyoD transcription.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2010-0023820).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Bilodeau S., Kagey M.H., Frampton G.M., Rahl P.B., Young R.A. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009;23:2484–2489. doi: 10.1101/gad.1837309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais A., Tsikitis M., Acosta-Alvear D., Sharan R., Kluger Y., Dynlacht B.D. An initial blueprint for myogenic differentiation. Genes Dev. 2005;19:553–569. doi: 10.1101/gad.1281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum R., Dynlacht B.D. The role of MyoD1 and his-tone modifications in the activation of muscle enhancers. Epigenetics. 2013;8:778–784. doi: 10.4161/epi.25441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Kumar R.M., Penn B.H., Berkes C.A., Kooperberg C., Boyer L.A., Young R.A., Tapscott S.J. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 2006;25:502–511. doi: 10.1038/sj.emboj.7600958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caretti G., Di Padova M., Micales B., Lyons G.E., Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004;18:2627–2638. doi: 10.1101/gad.1241904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.M., Kraut N., Groudine M., Weintraub H. I-mf, a novel myogenic repressor, interacts with members of the MyoD family. Cell. 1996;86:731–741. doi: 10.1016/s0092-8674(00)80148-8. [DOI] [PubMed] [Google Scholar]
- Choi J., Jang H., Kim H., Lee J.H., Kim S.T., Cho E.J., Youn H.D. Modulation of lysine methylation in myocyte enhancer factor 2 during skeletal muscle cell differentiation. Nucleic Acids Res. 2014;42:224–234. doi: 10.1093/nar/gkt873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado I., Huang X., Jones S., Zhang L., Hatcher R., Gao B., Zhang P. Dynamic gene expression during the onset of myoblast differentiation in vitro. Genomics. 2003;82:109–121. doi: 10.1016/s0888-7543(03)00104-6. [DOI] [PubMed] [Google Scholar]
- Dilworth F.J., Seaver K.J., Fishburn A.L., Htet S.L., Tapscott S.J. In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci. USA. 2004;101:11593–11598. doi: 10.1073/pnas.0404192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge J.E., Kang Y.K., Beppu H., Lei H., Li E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol. Cell. Biol. 2004;24:2478–2486. doi: 10.1128/MCB.24.6.2478-2486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom G.H., Kim K.B., Kim J.H., Kim J.Y., Kim J.R., Kee H.J., Kim D.W., Choe N., Park H.J., Son H.J., et al. Histone methyltransferase SETD3 regulates muscle differentiation. J. Biol. Chem. 2011;286:34733–34742. doi: 10.1074/jbc.M110.203307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P., Bradley A., Morris J.H., Edmondson D.G., Venuti J.M., Olson E.N., Klein W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- Jen Y., Weintraub H., Benezra R. Overexpression of Id protein inhibits the muscle differentiation program: in vivo association of Id with E2A proteins. Genes Dev. 1992;6:1466–1479. doi: 10.1101/gad.6.8.1466. [DOI] [PubMed] [Google Scholar]
- Lassar A.B., Davis R.L., Wright W.E., Kadesch T., Murre C., Voronova A., Baltimore D., Weintraub H. Functional activity of myogenic HLH proteins requires heterooligomerization with E12/E47-like proteins in vivo. Cell. 1991;66:305–315. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- Lee H., Habas R., Abate-Shen C. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science. 2004;304:1675–1678. doi: 10.1126/science.1098096. [DOI] [PubMed] [Google Scholar]
- Ling B.M., Bharathy N., Chung T.K., Kok W.K., Li S., Tan Y.H., Rao V.K., Gopinadhan S., Sartorelli V., Walsh M.J., et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA. 2012;109:841–846. doi: 10.1073/pnas.1111628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal A.K. Histone methyltransferase Suv39h1 represses MyoD-stimulated myogenic differentiation. EMBO J. 2006;25:3323–3334. doi: 10.1038/sj.emboj.7601229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal A., Harter M.L. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc. Natl. Acad. Sci. USA. 2003;100:1735–1739. doi: 10.1073/pnas.0437843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mal A., Sturniolo M., Schiltz R.L., Ghosh M.K., Harter M.L. A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 2001;20:1739–1753. doi: 10.1093/emboj/20.7.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P.O., Mills T., O’Connor R.S., Kline E.R., Graubert T., Dzierzak E., Pavlath G.K. Sca-1 negatively regulates proliferation and differentiation of muscle cells. Dev. Biol. 2005;283:240–252. doi: 10.1016/j.ydbio.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Molkentin J.D., Black B.L., Martin J.F., Olson E.N. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell. 1995;83:1125–1136. doi: 10.1016/0092-8674(95)90139-6. [DOI] [PubMed] [Google Scholar]
- Moran J.L., Li Y., Hill A.A., Mounts W.M., Miller C.P. Gene expression changes during mouse skeletal myoblast differentiation revealed by transcriptional profiling. Physiol. Genomics. 2002;10:103–111. doi: 10.1152/physiolgenomics.00011.2002. [DOI] [PubMed] [Google Scholar]
- Puri P.L., Avantaggiati M.L., Balsano C., Sang N., Graessmann A., Giordano A., Levrero M. p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 1997a;16:369–383. doi: 10.1093/emboj/16.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P.L., Sartorelli V., Yang X.J., Hamamori Y., Ogryzko V.V., Howard B.H., Kedes L., Wang J.Y., Graessmann A., Nakatani Y., Levrero M. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol. Cell. 1997b;1:35–45. doi: 10.1016/s1097-2765(00)80005-2. [DOI] [PubMed] [Google Scholar]
- Rao S.S., Kohtz D.S. Positive and negative regulation of D-type cyclin expression in skeletal myoblasts by basic fibroblast growth factor and transforming growth factor beta. A role for cyclin D1 in control of myoblast differentiation. J. Biol. Chem. 1995;270:4093–4100. doi: 10.1074/jbc.270.8.4093. [DOI] [PubMed] [Google Scholar]
- Rudnicki M.A., Schnegelsberg P.N., Stead R.H., Braun T., Arnold H.H., Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Sabourin L.A., Rudnicki M.A. The molecular regulation of myogenesis. Clin. Genet. 2000;57:16–25. doi: 10.1034/j.1399-0004.2000.570103.x. [DOI] [PubMed] [Google Scholar]
- Sartorelli V., Caretti G. Mechanisms underlying the transcriptional regulation of skeletal myogenesis. Curr. Opin. Genet. Dev. 2005;15:528–535. doi: 10.1016/j.gde.2005.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V., Juan A.H. Sculpting chromatin beyond the double helix: epigenetic control of skeletal myogenesis. Curr. Top. Dev. Biol. 2011;96:57–83. doi: 10.1016/B978-0-12-385940-2.00003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Ayyanathan K., Negorev D., Maul G.G., Rauscher F.J., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seenundun S., Rampalli S., Liu Q.C., Aziz A., Palii C., Hong S., Blais A., Brand M., Ge K., Dilworth F.J. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010;29:1401–1411. doi: 10.1038/emboj.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J., Verma N.K., Kansagra S.M., Kate B.N., Dey C.S. Altered PPARgamma expression inhibits myogenic differentiation in C2C12 skeletal muscle cells. Mol. Cell. Biochem. 2007;294:163–171. doi: 10.1007/s11010-006-9256-x. [DOI] [PubMed] [Google Scholar]
- Song Y.J., Lee H. YB1/p32, a nuclear Y-box binding protein 1, is a novel regulator of myoblast differentiation that interacts with Msx1 homeoprotein. Exp. Cell Res. 2011;316:517–529. doi: 10.1016/j.yexcr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Spicer D.B., Rhee J., Cheung W.L., Lassar A.B. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- Takada I., Mihara M., Suzawa M., Ohtake F., Kobayashi S., Igarashi M., Youn M.Y., Takeyama K., Nakamura T., Mezaki Y., et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat. Cell Biol. 2007;9:1273–1285. doi: 10.1038/ncb1647. [DOI] [PubMed] [Google Scholar]
- Takada I., Kouzmenko A.P., Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat. Rev. Rheumatol. 2009;5:442–447. doi: 10.1038/nrrheum.2009.137. [DOI] [PubMed] [Google Scholar]
- Tan S.L., Nishi M., Ohtsuka T., Matsui T., Takemoto K., Kamio-Miura A., Aburatani H., Shinkai Y., Kageyama R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development. 2012;139:3806–3816. doi: 10.1242/dev.082198. [DOI] [PubMed] [Google Scholar]
- Tao Y., Neppl R.L., Huang Z.P., Chen J., Tang R.H., Cao R., Zhang Y., Jin S.W., Wang D.Z. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J. Cell Biol. 2011;194:551–565. doi: 10.1083/jcb.201010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott S.J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Tapscott S.J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Thayer M.J., Tapscott S.J., Davis R.L., Wright W.E., Lassar A.B., Weintraub H. Positive autoregulation of the myogenic determination gene MyoD1. Cell. 1989;58:241–248. doi: 10.1016/0092-8674(89)90838-6. [DOI] [PubMed] [Google Scholar]
- Tomczak K.K., Marinescu V.D., Ramoni M.F., Sanoudou D., Montanaro F., Han M., Kunkel L.M., Kohane I.S., Beggs A.H. Expression profiling and identification of novel genes involved in myogenic differentiation. FASEB J. 2004;18:403–405. doi: 10.1096/fj.03-0568fje. [DOI] [PubMed] [Google Scholar]
- Wang J., Helin K., Jin P., Nadal-Ginard B. Inhibition of in vitro myogenic differentiation by cellular transcription factor E2F1. Cell Growth Differ. 1995;6:1299–1306. [PubMed] [Google Scholar]
- Yang L., Lawson K.A., Teteak C.J., Zou J., Hacquebord J., Patterson D., Ghatan A.C., Mei Q., Zielinska-Kwiatkowska A., Bain S.D., et al. ESET histone methyltransferase is essential to hypertrophic differentiation of growth plate chondrocytes and formation of epiphyseal plates. Dev. Biol. 2013;380:99–110. doi: 10.1016/j.ydbio.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P., Han J., Guo G., Orlov Y.L., Huss M., Loh Y.H., Yaw L.P., Robson P., Lim B., Ng H.H. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 2009;23:2507–2520. doi: 10.1101/gad.1831909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Behringer R.R., Olson E.N. Inactivation of the myogenic bHLH gene MRF4 results in up-regulation of myogenin and rib anomalies. Genes Dev. 1995;9:1388–1399. doi: 10.1101/gad.9.11.1388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.