

Background: Activated Gαs is internalized and increases microtubule dynamics. The role of Gαs in neuronal growth remains unclear.
Results: Gαs promotes neurite outgrowth in PC12 cells and hippocampal neurons.
Conclusion: Neurite outgrowth is at least partially dependent on Gαs-mediated increases in microtubule dynamics.
Significance: Gαs plays a direct role in neurite formation and branching.
Keywords: G Protein, Growth Factor, Microtubule, Neurite Outgrowth, Neurotrophin
Abstract
Signals that activate the G protein Gαs and promote neuronal differentiation evoke Gαs internalization in rat pheochromocytoma (PC12) cells. These agents also significantly increase Gαs association with microtubules, resulting in an increase in microtubule dynamics because of the activation of tubulin GTPase by Gαs. To determine the function of Gαs/microtubule association in neuronal development, we used real-time trafficking of a GFP-Gαs fusion protein. GFP-Gαs concentrates at the distal end of the neurites in differentiated living PC12 cells as well as in cultured hippocampal neurons. Gαs translocates to specialized membrane compartments at tips of growing neurites. A dominant-negative Gα chimera that interferes with Gαs binding to tubulin and activation of tubulin GTPase attenuates neurite elongation and neurite number both in PC12 cells and primary hippocampal neurons. This effect is greatest on differentiation induced by activated Gαs. Together, these data suggest that activated Gαs translocates from the plasma membrane and, through interaction with tubulin/microtubules in the cytosol, is important for neurite formation, development, and outgrowth. Characterization of neuronal G protein dynamics and their contribution to microtubule dynamics is important for understanding the molecular mechanisms by which G protein-coupled receptor signaling orchestrates neuronal growth and differentiation.
Introduction
Heterotrimeric G proteins play an important role in signal transduction by transferring signals from cell surface receptors to intracellular effector molecules. A series of studies from this laboratory have demonstrated direct binding interactions between Gα or Gβγ subunits and tubulin or microtubules (1–4), suggesting that G proteins modify microtubule assembly and subsequent cytoskeletal rearrangement. Activated Gαs acts as an intracellular messenger to regulate microtubule dynamics and promote neurite outgrowth (5).
Cell shape change and the underlying restructuring of the cytoskeleton contribute to the commitment of cells to grow, undergo apoptosis, or differentiate. Reorganization of the cytoarchitecture regulates signaling pathways, including the mobilization of intracellular calcium, the small GTPase Ras, and activation of tyrosine kinases, such as ERK and JNK (6–9). Consistent with the activation of signal pathways, specific transcription factors are activated by cytoskeletal restructuring (10, 11). NGF, acting through receptor tyrosine kinase, differentiates PC12 cells into a “neuronal” phenotype (12, 13). Membrane receptors, including receptor tyrosine kinases and adhesion receptors, activate intracellular signal cascades that promote cytoskeletal changes in the growing neurite (12, 14). The dynamic structures at the tip of the neurites, known as growth cones, control the directionality of the neurite growth by rapidly remodeling the actin cytoskeleton through cyclical extension of filopodia and lamellipodia (15, 16). The neurite protrusion is determined by the presence of specific areas of the PM2 that cluster neurite-inducing signals (14). Specific membrane microdomains, known as lipid rafts, may act as platforms for concentration of signaling molecules important for cell adhesion, axon guidance, and synaptic transmission in neuronal cells (17). Lipid rafts contain various growth factor receptors, including the PDGF receptor, EGF receptor, and TrkA, as well as other signaling proteins such as G protein-coupled receptors (GPCRs), heterotrimeric G proteins, and Src family kinases and may respond to extracellular signals by inclusion or exclusion of signaling proteins (18). Concomitant with Gαs activation is the internalization of Gα subunits, resulting in an increased association with microtubules in cells bearing newly developing neurites (4).
Gαs is activated by a subset of GPCRs for monoamine and peptide neurotransmitters and neuromodulators. Unlike Gαi and Gαq, Gαs internalizes upon activation (19, 20), and this internalization is mediated via lipid rafts (17). Internalized, activated Gαs associates with microtubules (21).
In this study, we explore possible roles for the Gαs subunit at intracellular sites during growth and differentiation, specifically during neurite formation. The objectives of this study are to understand the dynamic shuttling of Gαs between the PM and intracellular structures, trafficking of Gαs-containing complexes toward the tips of a differentiating neurite, and the relationship of these phenomena to neurite outgrowth. GFP-Gαs localizes at the growth cones of differentiated PC12 cells, primarily in the membrane ruffle, lamellipodia, and filopodia, which are regions that are more enriched with dynamic microtubules. Overexpression of constitutively activated GαsQ227L (GsQL) and NGF enhance the formation of dendritic spines and neuronal growth in PC12 neurites and hippocampal neurons. Importantly, the sites enriched with Gαs enable the formation of protrusive structures that eventually develop into neurites. In addition, we found that, throughout neuronal development, Gαs increases its association with microtubules and is targeted to the leading edges of developing neurites of PC12 and primary rat hippocampal neurons. This interaction between Gαs, microtubules, and membranes may be regulated in coordination with dynamic functions of other signaling molecules during processes such as cell differentiation and neuronal growth.
EXPERIMENTAL PROCEDURES
Cell Cultures
The PC12 cells used in this study were obtained from the ATCC or were a gift from Dr. Puneet Opal (Department of Neurology, Northwestern University, Chicago, IL). PC12 cells were grown in 75-cm2 tissue culture flasks (Falcon, BD Biosciences) at 37 °C in a 5% CO2 humidified atmosphere in Dulbecco's modified Eagle's medium (Cellgro, Mediatech, Inc.) supplemented with 4.5 g/liter glucose, 10% bovine calf serum (Hyclone, Logan, UT), 5% heat-inactivated horse serum (Lonza, Mapleton, IL), and 100 mg/ml penicillin-streptomycin (Gibco BRL, Life Technologies). Glass coverslips (Fisher Scientific Products, Itasca, IL) were thoroughly washed with deionized water and cleaned in concentrated HNO3, HCl, and 100% ethanol for 1 h in an ultrasound water bath. Thereafter, glass coverslips were dried and sterilized under UV light in a biosafety cabinet for 15 min. The coverslips were coated with 1% poly-lysine (30,000–70,000 kDa, dissolved in 0.1 m borate buffer) for 3 h in incubator at 37 °C. For immunofluorescence studies, cells were plated on coverslips and cultured overnight in the above medium. Cells were plated at a density of 2 × 106 cells/cm2. Where indicated, cells were treated with medium supplemented with 50 ng/ml NGF (Invitrogen) for 3 days, and media were changed on day 2. For neurite outgrowth studies, PC12 cells were plated in two 12-well plates (BD Biosciences) at a density of 7 × 104 cells/cm2 in 1 ml of the above plating media. On day 2, half of the plating media were replaced with either differentiation medium (DMEM containing 0.5% fetal bovine serum) or differentiation medium containing 50 ng/ml NGF to induce cell differentiation. On day 3, media were completely replaced with Opti-MEM I (Life Technologies). Opti-MEM I-conditioned cells were transfected with designated plasmids following the instructions of the manufacturer (Life Technologies). On day 4, half of the media were replaced with either pure differentiation media or differentiation media that contained NGF in the concentrations noted above. On day 5, cells were fixed, stained, and imaged. Images were quantified using ImageJ (National Institutes of Health). Transfections were repeated at least three times, and 120–180 cells were examined for each condition.
Neuronal Cultures
Hippocampal neurons were prepared from newborn rat hippocampus (postnatal day 0); cultured in Neurobasal medium (Fisher Life Sciences-Gibco BRL) supplemented with B-27 supplement (Fisher Life Sciences-Gibco BRL), 1 mm glutamine, and 2.5 m cytosine-d-arabinofuranoside; and plated on poly-lysine-coated coverslips or glass bottom microwell dishes (MatTek Corp.) at a density of 1.5 × 106 cells/dish. Neurons were either transiently transfected with plasmids or infected with adenoviruses as described in Ref. 21.
Constructs
The GFP-Gαs fusion protein-encoding plasmid has been described previously (20) and was used in the monomeric GFP form (22). The full-length constitutively active mutant Q227L Gs protein α subunit plasmid was a gift from Dr. Tohru Kozasa (University of Illinois at Chicago). The plasmid encoding NC1 (chimera 3) protein has been described previously (23).
Transfection and Expression of GFP Plasmids for Live Cell Imaging
PC12 cells were plated onto poly-lysine coated T plates (Bioptechs, Inc.) and transfected with 1 μg of GFP-Gαs using LipofectamineTM 2000 reagent (Gibco BRL, Life Technologies) 24 h after plating. The next day, the medium was removed, and cells were treated with 50 ng/ml NGF for 48 h. After 48 h, T plates were imaged at 37 °C using a Deltavision system (Applied Precision, Inc.).
Hippocampal neurons were plated on T plates and transfected either with LipofectamineTM 2000 or NeuroPORTERTM (Gene Therapy Systems Inc.) after 2 days in vitro. For neurite outgrowth studies, PC12 cells were cultured until 80% confluency, trypsinized, washed three times with PBS, and resuspended in buffer to be electroporated using the Neon transfection system (Invitrogen) following the protocol of the manufacturer. Cells were electroporated with the designated plasmids following the instructions of the manufacturer (Life Technologies) and plated onto poly-d-lysine-coated dishes. After 24 h, the medium was replaced, where indicated, with differentiation medium containing 50 ng/ml NGF (Invitrogen) to induce cell differentiation. 10 μg of DNA/1 million cells was used. After 2 h, dishes were mounted in the VivaView system for all further experimental work.
Deconvolution and Time-lapse Fluorescence Microscopy
Images were captured with a DeltaVision system built on an Olympus IX-70 base. Z stacks were deconvolved using SoftWoRx® Suite 2.0 software (Applied Precision, GE Healthcare). Sections were captured every 200 nm. Typically, 15 iterations on the basis of a measured point spread function, calculated from 0.1 and 1 μm of fluorescent beads (Molecular Probes, Life Technologies) were used. The fluorescence intensity was measured and quantified by Volocity image analysis software (Improvision, Lexington, MA). Total intensity was measured in relative fluorescence intensity units. For neurite outgrowth studies, time-lapse imaging was acquired every 2 h with the VivaView FL incubator fluorescence system for 15–20 h. Multiple locations in up to eight samples were imaged simultaneously with fluorescence (GFP) and differential interference contrast (DIC). Sections were captured every 1.1 μm and analyzed with MetaMorph (Olympus).
Immunocytochemistry
PC12 cells were plated on 12-mm coverslips in 12-well tissue culture plates. They were washed twice with phosphate-buffered saline (pH 7.4) enriched with calcium and magnesium (PBS+/+, Life Technologies) before and after permeabilization. Transfected cells and hippocampal neurons were fixed with 3.7% paraformaldehyde (Fisher Sciences, Waltham, MA) dissolved in PBS+/+ for 15 min at room temperature. The staining was performed as described in Ref. 4. Briefly, after washing three times for 10 min each in PBS with 0.1% Triton X-100, cells were incubated for 20 min with blocking buffer (5% nonfat milk and 0.1% Triton X-100 in PBS+/+), followed by three 10-min washes in PBS. To block the nonspecific binding in 4-d cultured neurons, we used PBS with 5% goat or horse serum for 1 h at room temperature and mouse anti-tubulin (DM1A) 1:1000 (Sigma) in PBS with 1% goat or horse serum at 4 °C overnight. His-tagged polyclonal rabbit antibody was from Cell Signaling Technology (Danvers, MA). Mouse monoclonal anti-Gs protein α subunit (clone N192/12) antibody was from NeuroMab (Davis, CA). Rhodamine-conjugated goat anti-mouse antibody (10 mg/ml) was purchased from Pierce. Alexa Fluor 594 donkey anti-mouse IgG (H+L, 1:400) was purchased from Molecular Probes. Stained cells and neurons were mounted onto slides with Vectashield (Vector Laboratories, Burlingame, CA) and viewed under the Deltavision system (Applied Precision, Inc., Seattle, WA) or a Zeiss LSM 510 Meta confocal microscope. For neurite growth studies, PC12 cells were incubated in blocking buffer (2.5% nonfat milk and 1% Triton X-100 in PBS+/+) for 30 min at room temperature. Samples were then incubated with His6 polyclonal rabbit antibody (1:75 dilution) and/or mouse monoclonal anti-Gαs (1:400 dilution) dissolved in blocking buffer for 18 h at 4 °C. The next day, cells were washed three times with blocking buffer for 5 min. Thereafter, cells were incubated with Alexa Fluor 594 goat anti-mouse and/or Alexa Fluor 488 goat anti-rabbit (each 1:300 dilution, Molecular Probes) secondary antibodies for 30 min. The coverslips were washed three times in the blocking buffer with 1% Triton X-100 in PBS+/+ for 5 min, rinsed in distilled water, and mounted on slides (Fisher Sciences) with 10 μl of ProLong antifade reagent (Molecular Probes). PC12 cells were imaged with a Zeiss Axio Observer.Z1 epifluorescence microscope under ×63 magnification with a Zeiss AxioCam camera. The images were processed with Zeiss AxioVision software.
Neurite Outgrowth Assay in PC12 Cells
Images were quantified using ImageJ (National Institutes of Health). Transfections were repeated at least three times, and 120–180 cells were examined for each condition. The PC12 cell neurites were tracked with ImageJ software (National Institutes of Health) or MetaMorph (Olympus). Two morphological parameters were analyzed: the number of neurites that originated from the cell body, including the number of branch points, and their absolute average length.
Image Acquisition and Analysis of Neuronal Morphology
Images of hippocampal neurons transfected with GFP-Gαs and NC1 were captured with a laser-scanning confocal microscope (LSM 510 Meta, Zeiss, Oberköchen, Germany). Z stacks of optical sections were then obtained on the confocal microscope, with the thickness of the optical sections varying from 0.4–1.0 μm. The Z series were deconvolved and reconstructed in three dimensions with Volocity (Improvision, Lexington, MA) software. Four-day cultured hippocampal neurons were infected with Ad/Gαs and Ad/GsQL. Thirty-six hours after infection, images of cultured hippocampal neurons were captured using a digital fluorescence microscope equipped with a 100-Watt mercury arc lamp and with an interline charge-coupled device camera (1300 YHS, Roper Scientific, Trenton, NJ) driven by IP Lab imaging software (Scanalytics, Inc., Suitland, VA). Data were processed and quantified using IP Lab software. Images were reconstructed in three dimensions with Neurolucida (version 7, MBF BioScience, Williston, VT) software. Final image processing and labeling was done with Adobe Photoshop (San Jose, CA). To quantify the length of cellular processes, all neurons with detectable fluorescence were analyzed for the presence of neurites, which, in this study, were defined as thin protrusions that were at least one cell diameter in length. We analyzed individual neurons by laying lines along cellular processes. Dendrites were measured up to the end of the tip, excluding the soma. The number of dendrites was counted from first branches. At least ten neurons from three independent transfection experiments were analyzed for NC1 experiments. Images from four experiments were analyzed for activated GsQL experiments.
Statistical Procedures
The obtained data were analyzed in Microsoft Excel and Prism5 (GraphPad, San Diego, CA) and presented as mean ± S.E. of the mean. Unpaired Student's t tests, corrected when necessary for unequal variances, were used to determine whether means differed from zero or other null values and to compare values from different populations. NGF and Q227L effects were evaluated by unpaired Student's t tests and one-way ANOVA. Two-way ANOVA was used to calculate statistical significance in 5-day NGF-treated PC12 cells.
RESULTS
Localization of Gαs during Neuronal Differentiation
To fully understand the function of G proteins in cellular differentiation, it is a prerequisite to establish their intracellular localization. We set out to define the subcellular localization of the GFP-Gαs fusion protein in PC12 cells. GFP is inserted within the NH2-terminal domain of Gαs. This construct has been used previously to study the internalization of activated Gαs (17). To determine whether the behavior of the endogenous Gαs is similar to the distribution pattern of a fluorescent derivative of that protein, we transiently transfected PC12 cells in culture with GFP-Gαs (Figs. 1, A and B) and determined its subcellular localization by fluorescence microscopy. Distribution of Gαs has been characterized previously and is abundant in the cytoplasm and near the cell surface (4). About 20% of the cells were transfected, and the expression of GFP-Gαs was about 2- to 3-fold greater than that of the endogenous protein (20). Besides the characteristic ring of fluorescence at the cell membrane, punctate fluorescence is evident throughout the cytoplasm (Fig. 1A, arrowhead). To understand the nature of these structures, we selected a specific region in the cell (Fig. 1C). The z series of this region was volume-projected and rotated 180° around the y axis (supplemental Movie 1). Cytoplasmic Gαs appears as distinctive circular discs that are localized to tubular intracellular structures, which have been identified previously as microtubules (21).
FIGURE 1.
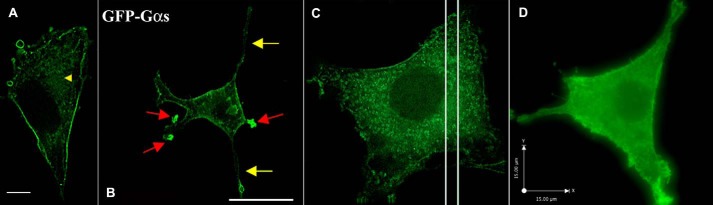
Subcellular localization of Gαs in PC12 cells. A and B, PC12 cells were transfected with GFP-Gαs (A) and then treated with 50 ng/ml NGF for 48 h (B). Cells were fixed for 10 min with paraformaldehyde and processed for deconvolution microscopy. Gαs translocates to the neurites (yellow arrows) and to membrane ruffles of the newly formed extensions (red arrows) after NGF treatment. Sections were captured every 200 nm. Scale bar = 15 μm. These results suggest that, during neuronal differentiation, Gαs redistributes toward areas of highly dynamic cytoskeletal activity, such as the growing tip of neurites. C, a representative example of clustered GFP-Gαs distribution in PC12 cells within a selected specific region. D, a representative example of GFP-Gαs distribution in PC12 cells after 48 h of NGF treatment.
NGF stimulation leads first to sprouting of multiple neurites and then to their outgrowth and finally, to their subsequent differentiation in PC12 cells (Fig. 1B). We observed the archetypal form of outgrowth that has been described previously (24). It involves two steps: the reorganization of the cytoskeleton, with longitudinal distribution of microtubules and concentration of actin filaments in the filopodia and lamellipodia of growth cones (25, 26), and the enlargement of the cell surface sustained by the exocytosis of cytoplasmic vesicles (27–29). The initial features of neurite growth, such as sprouting, are observable after 3 h of NGF treatment. NGF treatment induced a greater portion of GFP-Gαs associated with the PM compared with untreated cells (compare Fig. 1, A and B). GFP-Gαs cells treated with NGF for 48 h display prominent fluorescence in the membrane and along the length of all processes (Fig. 1B, yellow arrows). Although NGF treatment diminished GFP-Gαs in punctate structures in the cytoplasm, strong fluorescence was concentrated at the leading tip of NGF-induced protrusive structures (Figs. 1B, red arrows, and 2, A and B). To examine Gαs-enriched structures in detail, we first acquired z stack images of a NGF-treated PC12 cell (Fig. 1D). Analysis of this image demonstrates enrichment of Gαs in membrane ruffles and lamellipodia of growth cones and several microspikes extending from the cell membrane or neurites. The localization of Gαs in ruffles and other protrusive structures of the growth cone suggests the involvement of Gαs in the NGF-mediated regulation of cytoskeleton reorganization in growing neurites.
FIGURE 2.

Movement of GFP-Gαs in living cells. A–C, PC12 cells were transfected with GFP-Gαs and then treated with 50 ng/ml NGF for 48 h. Images were captured with epifluorescence and deconvolved. Sections were imaged every 200 nm. Analysis of images from a z series reveal that GFP-Gαs concentrates at the leading edge of newly formed cellular protrusions and lamellipodia (A, arrow) and tips of filopodia (B, arrow). A, a series of volume-projected images with maximum intensities from the time-lapse recording of GFP-Gαs movement. Time-lapse imaging was done for 1.5 h every 15 min. Time points (hours:minutes) are selected to illustrate the intermediates of neurite formation. Scale bar = 15 μm. B, GFP-Gαs was transported along the formed cellular processes, concentrating at neurite tips. After NGF treatment, it is present at the leading edges of filopodia (arrow) as well. Scale bar = 15 μm. C and D, ratiometric analysis of images of cells transfected with GFP-Gαs against GFP alone (C, right panel). NGF-treated cells were transfected with either GFP alone or GFP-Gαs. C, fluorescence intensity was measured in six randomly selected regions of the cell body (CB) and growth cone (GC). D, fluorescence intensity of cells transfected with GFP alone or GFP-Gαs. The mean intensities from the cell body and growth cone were calculated from 6 cells/group. Two-way ANOVA was used for statistical analysis. Significance was determined using paired Student's t test. **, p <0.01 between cells that were transfected with GFP alone and cells that were transfected with GFP-Gαs. All data are mean ± S.D.
Real-Time Imaging of Intracellular and Growth Cone-enriched GFP-Gαs in Living PC12 Cells
GFP fusion proteins allow live monitoring of different intracellular components within the cell body and their delivery to varied locations, including the tips of the cellular extensions. Although G protein α and βγ subunits have been classically thought to act only at the PM, several reports suggest important roles for G protein subunits at intracellular locations (30–32). G protein localization is dynamic, and evidence exist that G protein subunits can translocate reversibly from the PM to intracellular structures, such as endosomes and Golgi (33, 34). An earlier study suggested that internalized Gαs recycled to the PM in vesicles upon agonist stimulation (35). To understand the exact locations of internalized Gαs and trafficking/recycling of Gαs dynamics of the GFP-Gαs PC12 cells were examined for up to 3 days after NGF treatment. Time-lapse imaging of differentiated cells reveals a dynamic movement of Gαs-rich vesicle-like structures. These circular structures are abundant throughout the cell body and resemble the lipid raft vesicles in which Gαs has been shown to internalize (17). In addition to the intracellular (supplemental Movie 1) localization, GFP-Gαs accumulated at the tips of the growth cones (Fig. 2, A and B). NGF promoted rapid lamellipodia formation, observed as thin sheets of membrane and cytoplasm at the leading and peripheral zones of the growth cone (Fig. 2A), and filopodia, observed as thin projections ranging from 10–25 μm in length (Fig. 2B, arrow). Gαs concentrated at the tips of filopodia (Fig. 2B, arrow). Time-lapse imaging provided a high-resolution view of the motile growth cone and the dynamics of Gαs occurring at the leading edges of the distal protrusions. High concentrations of Gαs relocalize from the neuritic shaft (Fig. 2A, 0:00) to the very tip of the lamellipodia in 15 min (Fig. 2A, 0:15). Within the next 15–30 min (Fig. 2A, 0:30), Gαs moved out from the ruffles of lamellipodia and returned to the neurite shaft. To examine a real concentration of Gαs at the tips of the neurite, we performed a ratiometric analysis of the images of cells transfected with GFP-Gαs against a volume marker (cells that were transfected with GFP alone (control cells) (Fig. 2C). We calculated separately the fluorescence intensity in six regions, both in the cytoplasm and growth cone. Note that the fluorescence intensity of GFP in the cell body of control cells is 8 times higher than in the growth cone (Fig. 2D). In contrast, GFP-Gαs targets to the filopodia and leading edges of the membrane upon neuronal differentiation. The fluorescence intensity of GFP-Gαs in the cell body decreases 1.5 times relative to the growth cone areas (Fig. 2D). These observations suggest that signals initiating neurite outgrowth induce delivery of Gαs-containing membrane complexes from the cell body toward areas of cellular outgrowth, such as the leading edges of the growth cone and filopodia.
Gαs Marks Future Neurite Initiation Sites in Response to NGF Treatment
We tracked GFP-Gαs during the period of response to NGF in PC12 cells. Analysis of a series of volume-projected time-lapse recordings of differentiated cells expressing GFP-Gαs exhibits a recruitment of GFP-Gαs to specific regions at the membrane of incipient neurites (Fig. 3). NGF treatment increases the size of GFP-Gαs puncta in leading edges of neurites (Fig. 3A, 1 and 2) within 15 min. The GFP-Gαs-enriched structures transformed into protrusions that differentiated into a developing neurite in about 3 h (compare Fig. 3, A, 1 and 2, and L, 1 and 2). Formation of neurites also correlated with rapid increases of GFP-Gαs in the leading edge of lamellipodia. Only newly formed protrusions show a significant increase of GFP-Gαs accumulation, which correlated positively with the presence of microtubules. GFP-Gαs is mobile in mature lamellipodia and can transit in and out on the order of minutes. The accumulation of GFP-Gαs at these selective regions (Fig. 3, A–C, 1 and 2) provides formation of a consolidated area that undergoes further transformation into the neuritic shaft (Fig. 3E, 2). Gαs-enriched outgrowths are formed at the leading edge (Fig. 3H, 2). Subsequently, the projections elongate, and the neurites persist for at least an additional 2 h, suggesting that Gαs participates in the initiation and formation of new projections (compare Fig. 3, E, 1 and 2, and L, 1 and 2).
FIGURE 3.

Gαs distributes to growing tips during neurite formation. A–L, a series of volume-projected images with maximum intensities from the time-lapse recording of GFP-Gαs movement. Time-lapse imaging was done for 3 h every 15 min. Time points (hours:minutes) are selected to illustrate the intermediates of neurite formation. A, 0:00, Gαs-enriched 1 and 2 growth cone extensions are accumulated at the base of a new protrusion. L, 2:45, both the 1 and 2 extensions form independent protrusive structures and neurites. Scale bar = 15 μm.
Both Constitutively Active Gαs and NGF-mediated Signaling Promote Neuronal Growth
It does appear that activation of Gαs increases microtubule dynamics by increasing dynamic behavior of microtubules, leading to neurite growth in PC12 cells (21). The relationship of NGF to this process remains unresolved. To reconcile the effects of NGF signaling and activation of Gαs on neuronal growth, PC12 cells were transfected with constructs expressing either constitutively active GsQLGFP or GsGFP (control) and were then differentiated with NGF (GsGFP + NGF). The changes in cell morphology and translocation of activated Gαs or Gαs were imaged over 16 h (Fig. 4C and supplemental Movies 2–9, DIC and GFP). The DIC images in the left panels in Fig. 4C represent the morphology of cells at the 0 and 16-h time points, whereas the right panels in both columns show the localization of Gαs in those cells. In addition, we evaluated neurite length and the number of neurites per cell. The length of neurites and number of branches were quantified from the DIC images. Neurites that were fasciculated or could not be accurately assigned to specific cells were omitted from analysis. The number of neurites per cell was determined by counting the neurites that extended directly from the cell body and the number of points along the neurites where one neurite gave rise to another. Equivalent numbers of cells were selected for quantification per condition and for each time point. Cells expressing GsQL and NGF-treated cells had more extensive neurites and greater neurite length per cell than control cells (Fig. 4A). This effect was more pronounced and statistically significant in GsQL cells at all time points after the first 2 h of imaging. Note that neurites were 2.51 times longer in cells expressing GsQL than in control cells at 16 h. The neurite-promoting effects of NGF are of a slower onset than those of Gαs. Either NGF-treated cells or those expressing GsQL had more neurites than control cells, but this effect was not statistically significant (Fig. 4B). NGF treatment had a greater effect on the number of neurites at later time point than cells that express activated Gαs (Fig. 4B). The number of neurites in cells treated with NGF was 1.7 times higher than in control cells and 1.2 times higher than in GsQL cells. Furthermore, NGF significantly induced accumulation of GFP-Gαs at the neurite tips (Fig. 4D). These data suggest that Gαs translocates toward regions enriched in more dynamic microtubules.
FIGURE 4.

NGF and constitutively activated GsQL increase neurite length and number in PC12 cells, and this is inhibited by interference with Gαs association with tubulin. PC12 cells were electroporated with GsGFP (control), GsQLGFP (constitutively active GαsQ227L), NC1 (a dominant-negative G protein chimera that inhibits GFP-Gαs association with tubulin) with GsGFP and GsQLGFP with NC1. Cells were induced with NGF after 24 h of protein expression where indicated. Live cell imaging was performed using the VivaView system (see “Experimental Procedures”). C, cells were imaged under DIC and fluorescence (GFP) for 16 h. We identified transfected cells by the intensity of GFP staining. GsQL protein is extensively accumulated in the cell “soma” and in the peripheral regions of the growth cones enriched in actin-based cytoskeleton (21). Images are represented at 0 min and 16 h. Scale bar = 50 μm. A and B, statistical analysis of neuronal growth per cell. Control cells were compared with cells expressing GsQLGFP and GsGFP stimulated with NGF. The average total length (A) and number of neurites (B) were measured. The effect of NGF and GsQL on the number of neurites was insignificant. The data for neurite length were analyzed by column statistics followed by unpaired Student's t test. **, two-tailed p < 0.05. Data are expressed as mean ± S.E. Significance was calculated by Student's t test. **, p < 0.01 (control versus GsQL, time points 10, 14, and 16). One-way ANOVA was used to determine significance among conditions (p = 0.0001). D, statistical analysis of the percentage of neurites expressing GFP at the tips. NGF induced the translocation of Gαs to the neurite tip compared with control cells. Data are mean ± S.E. Significance was calculated by Student's t test. **, two-tailed p <0.05 (control versus GsGFP + NGF). E and F, a dominant-negative G protein chimera that inhibits GFP-Gαs association with tubulin attenuates the number and length in PC12 cells. PC12 cells were induced to differentiate in low-serum medium containing NGF (50 ng/ml) for 4 days. 72 h after plating, cells were transiently transfected with plasmids encoding the control (GFP), a Giα1-Gt chimera that binds to tubulin and blocks the activation of tubulin GTPase by Gαs (NC1), and constitutively active GsQL. We identified NC1-expressing cells by staining with a His tag antibody. E, quantitative analysis of the mean average length per cell. F, average neurite number per cell. Transfections were repeated at least three times with 120–180 cells scored per conditions in each experiment. Error bars represent S.E. G, statistical analysis of mean average neurite number and length per cell. Two-way ANOVA followed with Bonferroni post test was used to determine statistical significance among conditions. H and I, effects of 8-bromo-cyclic AMP on PC12 cells expressing GsQLGFP and NC1. PC12 cells were electroporated with GsQLGFP alone or GsQLGFP with NC1 simultaneously (GsQLGFP + NC1) and treated or untreated with 8-bromo-cyclic AMP (100 μm) for 48 h. H, images were taken at ×40 magnification using a Zeiss AX10 microscope system. Scale bar = 20 μm. I, the average total length and number of neurites were measured. For the analysis, 11 cells were selected from the untreated group (−8-bromo-cyclic AMP) and 14 cells from the group treated with 8-bromo-cyclic AMP (+8-bromo-cyclic AMP). The effect of 8-bromo-cyclic AMP on the number of neurites was insignificant (p = 0.109). The data for neurite length were analyzed by column statistics followed by Student's t test (two-tailed p < 0.05). Data are expressed as mean ± S.E. Significance was calculated by Student's t test (p = 0.0014 with or without 8-bromo-cyclic AMP).
A Dominant-negative Protein That Blocks Tubulin Association with Gαs Attenuates the Growth and Branching of Neurites
Previous studies have suggested that cytoplasmic Gαs is preferentially associated with microtubules in response to NGF treatment (4). In addition, we observed that expression of a dominant-negative Gαi-transducin chimera (NC1) that blocks Gαs binding to tubulin and Gαs activation of tubulin GTPase, attenuating microtubule-based cellular projections in COS-1 cells (23). Here NC1 was used to determine the role of Gαs in neurite outgrowth (Fig. 4, C and E–G). PC12 cells expressing GFP alone (control) or NC1 and/or constitutively active GsQL constructs were compared. The images in Fig. 4C show the distribution of GsQL protein over a 16-h time span. Although Gαs is largely membrane-bound, GsQL accumulates in the cell body and in the peripheral regions of the “growth cones” enriched in cytoskeleton. NGF increased the length and number of neurites in PC12 cells (Fig. 4, E–F). Neurite extensions are two times longer in NGF-treated PC12 cells relative to undifferentiated cells (Fig. 4E). Quantitative analysis showed that overexpression of GsQL resulted in a 57% increase in neurite length and 31.6% increase in branching. Cells expressing GsQL and treated with NGF had the longest and highest number of neurites (Fig. 4, E–G). Neurite length was 2.1 times longer, and these cells displayed 2 times more branches compared with untreated PC12 cells. Expression of NC1 suppressed neurite growth and decreased length compared with control cells. The statistical analysis of the mean average of length and number of neurites per cell is presented in Fig. 4G. Finally, NC1 suppressed neurite length in cells expressing GsQL alone and in cells expressing GsQL and treated with NGF. NC1expression decreased the length of neurites by 16% in untreated cells expressing GsQL. The length of neurites in cells expressing both GsQL and NC1 simultaneously and treated with NGF is 24% shorter than in cells not expressing NC1. In addition, treatment with 8-bromo-cyclic AMP restored the number of neurites/cell and neurite length in cells where the effects of GsQLGFP were attenuated by NC1 (Fig. 4, H and I).
These findings also suggest a “second messenger” role for activated Gαs in increasing microtubule dynamics and functions, such as neurite outgrowth. To explore the possibility that Gαs might regulate aspects of neuronal development, including neurite branching or dendritic spine formation, cultured neurons from rat hippocampus were used as our study model. Neurons were infected with adenovirus expressing wild-type (control) or constitutively activated GsQL with GFP. The effect of these proteins on neuronal morphology was analyzed with fluorescence microscopy. The activated GsQL significantly increased neurite branching, but wild-type Gαs did not (Fig. 5, A and B). Furthermore, activated GsQL expression stimulated dendritic spine formation, especially increasing filopodia-like spines (Fig. 5B, bottom panel). Neurite outgrowth was analyzed by quantifying four independent measures: the number of primary dendrites, the number of ends, nodes, and the mean length of primary dendrites (Fig. 5, C and D). The number of primary neurites per cell was determined by counting the neurites that extended directly from the cell body (Fig. 5A, top panel). The number of ends was quantified by counting the number of all ends of primary dendrites including all branches (Fig. 5A, bottom panel), whereas the number of nodes was the points along the neurites where one neurite gave rise to another (Fig. 5D, top panel). Overexpression of GsQL increased the length and branches of primary dendrites 2-fold, suggesting that activated, internalized Gαs might increase the dynamics of microtubules and promote neurite outgrowth (Fig. 5, A–D).
FIGURE 5.

Activated Gαs increases neuronal growth. A–D, overexpression of constitutively activated Gαs enhances neurite formation in hippocampal neurons. Hippocampi were isolated from newborn mice. Four-day cultured hippocampal neurons were infected with virus containing the indicated Gαs. Twenty-four hours after infection, images were captured, reconstructed, and analyzed. A, neuron reconstruction from microscope images and analysis were done using Neurolucida software, version 7 (MBF BioScience). Images were taken at high magnification (×63). B, activated Gαs (GsQL) increases the spine-like projections in cultured neurons. Top panel, neurons expressing wild-type Gαs (control). Bottom panel, neurons expressing constitutively activate Gαs. Thirty-six hours after infection, spine-like projections are seen in neurons expressing GsQL. Images represent one of three experiments. Scale bar = 20 μm. C and D, quantification of neuronal morphology. For the analysis, four neurons from each group were imaged using a Zeiss AX10 microscope. Neurite outgrowth was analyzed using Neurolucida software. A detailed morphometric analysis was performed, quantifying the number of primary dendrites, nodes, ends, and mean length of primary dendrites. The data were analyzed by column statistics followed by Student's t test (*, two-tailed p < 0.05). Data are mean ± S.E. E and F, 4-day cultured neurons were cotransfected with NC1 and GFP-Gαs constructs. Neurons were fixed with paraformaldehyde, permeabilized, stained with His tag antibody to detect overexpression of NC1, and processed for deconvolution microscopy. Green fluorescent images (bottom panel, center) indicate NC1 expression. Red fluorescent images indicate cells stained with tubulin antibody. F, neurite length was measured in seven neurons from three independent transfection experiments by a person blind to experimental conditions. The data were analyzed by column statistics followed by Student's t test (**, two-tailed p < 0.01). Data are mean ± S.E.
To test the role of Gαs and its interaction with microtubules on neuronal development, we introduced NC1 and GFP-Gαs into 4-day cultured hippocampal neurons. Immunostaining for the His6 tag on NC1 reveals that this protein is cytosolic because of the NH2-terminal His6, which prevents association with Gβγ (Fig. 5E, bottom panel, center). NC1 also accumulates in the central domain of growth cones known to contain microtubules, at least during the early stages of their existence (36). Expression of NC1 attenuated the process of neurite extension in developing hippocampal neurons (Fig. 5F). Neurites in NC1-transfected cells were smaller (23.1 μm, n = 7) than in controls (61.4 μm, n = 7), and, overall, fewer neurites were formed (Fig. 5, E and F).
DISCUSSION
Signals generated by receptor tyrosine kinases and GPCRs are involved in growth and neuronal differentiation, and microtubules play a role in this process. We have previously observed Gαs association with microtubules and suggested that the increase in microtubule dynamics initiated by Gαs was associated with neurite outgrowth in PC12 cells. This study was designed to determine whether internalized Gαs might play a requisite role in neurite outgrowth and elongation. It was necessary to directly compare the overexpression of Gαs and NGF-induced differentiation in the same cellular phenotype. We analyzed the movement of cytosolic and membranous GFP-Gαs particles and the formation of neurites during NGF-induced cell differentiation. Transiently expressed GFP-Gαs has a clear preference to the cell membrane and appears at the periphery of newly formed processes, coinciding with membrane ruffles. The data presented in Fig. 2D show that there is a concentration of Gαs at the tips of the neurites and that it is not due to increased volume or membrane addition at that site.
Particular attention in this paper is given to the analysis of GFP-Gαs trafficking and its role in neurite formation because internalized Gαs promotes neurite outgrowth, and this is partially cAMP independent (21). In the previous study (21), we examined the relationship between activated Gαs and neurite outgrowth. Overexpression of GsQL, but not wild-type Gαs, increased neurite outgrowth in both normal and PKA-deficient PC12 cells. Furthermore, in those PKA-deficient cells, activation of Gαs by cholera toxin increased neurite outgrowth, but increasing cAMP by forskolin or N 6,O2′-dibutyryladenosine 3′:5′-cyclic monophosphate (Bt2-cAMP) had no effect. Activation of Epac was also without effect. The failure of the membrane-permeable cAMP analogue 8-bromo-cyclic AMP or 8-(4-chlorophenylthio) adenosine-3′,5′-cyclic monophosphate (8-CPT-cAMP) to promote neurite outgrowth in PKA-deficient cells suggests that the effects of activated Gαs are independent of PKA and Epac pathways and that Gαs can signal independently of this canonical cAMP/PKA pathway to modulate cytoskeleton-related morphologic changes. cAMP also did not translocate Gαs from the plasma membrane, an event required for association with microtubules and the initiation of neurite outgrowth. It was clear that the differentiating effects of GsQL do not require activation of any of the cAMP sensors protein kinase A, Epac, or cAMP but, rather, depend on direct alterations on microtubules via completely independent signaling pathways. This suggestion is supported by data showing that 8-bromo-cAMP rescues the dominant-negative inhibition of neurite outgrowth and formation of neurites in cells expressing both GsQLGFP and NC1 simultaneously.
These data do not suggest that cAMP lacks a role in neurite outgrowth. The PKA-deficient cells described above do not show the same extent of neurite outgrowth as control cells. Furthermore, a recent study (37) showed cAMP and MEK orchestration of neurite outgrowth. There appears to be a complex relationship between cAMP and NGF in the promotion of neurite outgrowth (38). Like cAMP, the promotion of neurite outgrowth is more rapid than that seen with NGF (Fig. 4, E–G). It is not clear how this cAMP-independent/Gαs-mediated pathway and the cAMP-dependent/ERK-mediated pathway coincide. Certainly this holds an interesting potential for a future study.
Activated Gαs is the preferred conformation for association with tubulin and subsequent activation of tubulin GTPase (39), and internalized Gαs appears to maintain the active conformation (40). This selective accumulation of Gαs at the leading edges of the protrusion suggests increased signaling in those regions. Lipid rafts may act as platforms for clustering neurite-inducing signaling molecules and may respond to extracellular signals by inclusion or exclusion of signaling proteins (18).
It has been shown recently that microtubule invasions of spines play an important role throughout the life of hippocampal neurons by dynamically regulating levels of PSD-95 in spine heads in response to increases in BDNF (41). These observations suggest that targeting of G proteins to specialized subcellular microdomains of newly forming protrusive structures, such as tips of filopodia and lamellipodia, is important for neuronal differentiation. Gαs itself has been suggested as a critical component of synaptic development (42).
Another major event in neuronal development is the reorganization of microtubules toward the future direction of neurite outgrowth (36). It has been reported that microtubule assembly and disassembly dynamics underlie multiple processes, including neurite outgrowth and branching and growth cone steering and forward movements (43). Gαs has been demonstrated to internalize in lipid raft vesicles subsequent to GPCR activation, and internalized Gαs has been shown to associate with microtubules (21) and to regulate microtubule dynamics (39). Gαs is able to promote the dynamic instability of microtubules by binding to the growing end of a microtubule and activating tubulin GTPase, therefore destabilizing microtubules, increasing the number of dynamic microtubules, and stimulating neurite outgrowth (21, 36, 39).
The dynamic phenomena analyzed here are central for understanding how specific signals propagate from receptors to the motile structures at the tips of advancing neurites. A picture is emerging in which scaffolding might be involved in the propagation of intracellular signals in a way so that sorting of Gαs to intracellular structures may be a requisite signaling component (44). Neurite outgrowth is a complex multistep event. Nonetheless, inhibition of the normal interaction between Gαs and tubulin significantly attenuates both the number and length of cellular extensions (in epithelial cells) and neurites in PC12 cells and hippocampal neurons. Conversely, overexpression of activated Gαs increases spine-like projections and neurite length and number, implicating Gαs in neuronal morphogenesis. Although the nature of this signal still remains to be determined, these data suggest that the interaction between Gαs and tubulin is relevant for the initiation sites, formation, elongation, and branching of developing neurites in PC12 cells and neurons. Together, these results provide evidence for involvement of the G protein in cytoskeleton-related mechanisms underlying the continuous reorganization of microtubules at the growth cone extensions and the interplay between microtubule dynamic stability and synapse formation and activity.
Here we provide a novel mechanism for activated, internalized Gαs to increase neurite outgrowth, suggesting a mechanism for GPCR activation to increase synaptogenesis. This process involves increased microtubule dynamics, growth directionality, and cross-talk between microtubules and the actin cytoskeleton. Although interplay between the receptor tyrosine kinases responsible for NGF effects with GPCRs has been proposed (45), it is not clear how this intercalates with the results presented here because NGF-induced neurite outgrowth proceeds independently of Gαs, and attenuation of Gαs-induced increases in microtubule dynamics does partially attenuate NGF actions. The mechanism for this remains to be elucidated.
Supplementary Material
This work was supported by Veterans Affairs merit award BX11049.

This article contains supplemental Movies 1–9.
- PM
- plasma membrane
- GPCR
- G protein-coupled receptor
- DIC
- differential interference contrast
- ANOVA
- analysis of variance.
REFERENCES
- 1. Wang N., Yan K., Rasenick M. M. (1990) Tubulin binds specifically to the signal-transducing proteins, Gs α and Gi α 1. J. Biol. Chem. 265, 1239–1242 [PubMed] [Google Scholar]
- 2. Roychowdhury S., Panda D., Wilson L., Rasenick M. M. (1999) G protein α subunits activate tubulin GTPase and modulate microtubule polymerization dynamics. J. Biol. Chem. 274, 13485–13490 [DOI] [PubMed] [Google Scholar]
- 3. Roychowdhury S., Rasenick M. M. (1997) G protein β1γ2 subunits promote microtubule assembly. J. Biol. Chem. 272, 31576–31581 [DOI] [PubMed] [Google Scholar]
- 4. Sarma T., Voyno-Yasenetskaya T., Hope T. J., Rasenick M. M. (2003) Heterotrimeric G-proteins associate with microtubules during differentiation in PC12 pheochromocytoma cells. FASEB J. 17, 848–859 [DOI] [PubMed] [Google Scholar]
- 5. Joiner M. L., Lisé M. F., Yuen E. Y., Kam A. Y., Zhang M., Hall D. D., Malik Z. A., Qian H., Chen Y., Ulrich J. D., Burette A. C., Weinberg R. J., Law P. Y., El-Husseini A., Yan Z., Hell J. W. (2010) Assembly of a β2-adrenergic receptor-GluR1 signalling complex for localized cAMP signalling. EMBO J. 29, 482–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jalali S., Li Y. S., Sotoudeh M., Yuan S., Li S., Chien S., Shyy J. Y. (1998) Shear stress activates p60src-Ras-MAPK signaling pathways in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 18, 227–234 [DOI] [PubMed] [Google Scholar]
- 7. Denker S. P., McCaffery J. M., Palade G. E., Insel P. A., Farquhar M. G. (1996) Differential distribution of α subunits and β γ subunits of heterotrimeric G proteins on Golgi membranes of the exocrine pancreas. J. Cell Biol. 133, 1027–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malek A. M., Izumo S. (1996) Mechanism of endothelial cell shape change and cytoskeletal remodeling in response to fluid shear stress. J. Cell Sci. 109, 713–726 [DOI] [PubMed] [Google Scholar]
- 9. Ishida T., Takahashi M., Corson M. A., Berk B. C. (1997) Fluid shear stress-mediated signal transduction: how do endothelial cells transduce mechanical force into biological responses? Ann. N.Y. Acad. Sci. 811, 12–23; discussion 23–14 [DOI] [PubMed] [Google Scholar]
- 10. Kheradmand F., Werner E., Tremble P., Symons M., Werb Z. (1998) Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science 280, 898–902 [DOI] [PubMed] [Google Scholar]
- 11. Rosette C., Karin M. (1995) Cytoskeletal control of gene expression: depolymerization of microtubules activates NF-κ B. J. Cell Biol. 128, 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang E. J., Reichardt L. F. (2001) Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang E. J., Reichardt L. F. (2003) Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 [DOI] [PubMed] [Google Scholar]
- 14. da Silva J. S., Dotti C. G. (2002) Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 3, 694–704 [DOI] [PubMed] [Google Scholar]
- 15. Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 [DOI] [PubMed] [Google Scholar]
- 16. Pollard T. D., Blanchoin L., Mullins R. D. (2000) Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29, 545–576 [DOI] [PubMed] [Google Scholar]
- 17. Allen J. A., Yu J. Z., Donati R. J., Rasenick M. M. (2005) β-Adrenergic receptor stimulation promotes G β s internalization through lipid rafts: a study in living cells. Mol. Pharmacol. 67, 1493–1504 [DOI] [PubMed] [Google Scholar]
- 18. Simons K., Toomre D. (2000) Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 [DOI] [PubMed] [Google Scholar]
- 19. Wedegaertner P. B., Bourne H. R., von Zastrow M. (1996) Activation-induced subcellular redistribution of Gs α. Mol. Biol. Cell 7, 1225–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu J. Z., Rasenick M. M. (2002) Real-time visualization of a fluorescent Gαs: dissociation of the activated G protein from plasma membrane. Mol. Pharmacol. 61, 352–359 [DOI] [PubMed] [Google Scholar]
- 21. Yu J. Z., Dave R. H., Allen J. A., Sarma T., Rasenick M. M. (2009) Cytosolic Gαs acts as an intracellular messenger to increase microtubule dynamics and promote neurite outgrowth. J. Biol. Chem. 284, 10462–10472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Czysz A. H., Schappi J. M., Rasenick M. M. (2014) Lateral diffusion of Gα in the plasma membrane is decreased after chronic but not acute antidepressant treatment: role of lipid raft and non-raft membrane microdomains. Neuropsychopharmacology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen N. F., Yu J. Z., Skiba N. P., Hamm H. E., Rasenick M. M. (2003) A specific domain of Giα required for the transactivation of Giα by tubulin is implicated in the organization of cellular microtubules. J. Biol. Chem. 278, 15285–15290 [DOI] [PubMed] [Google Scholar]
- 24. Aletta J. M., Greene L. A. (1988) Growth cone configuration and advance: a time-lapse study using video-enhanced differential interference contrast microscopy. J. Neurosci. 8, 1425–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pfenninger K. H. (2009) Plasma membrane expansion: a neuron's Herculean task. Nat Rev. Neurosci. 10, 251–261 [DOI] [PubMed] [Google Scholar]
- 26. Witte H., Bradke F. (2008) The role of the cytoskeleton during neuronal polarization. Curr. Opin. Neurobiol. 18, 479–487 [DOI] [PubMed] [Google Scholar]
- 27. Pfenninger K. H., Laurino L., Peretti D., Wang X., Rosso S., Morfini G., Cáceres A., Quiroga S. (2003) Regulation of membrane expansion at the nerve growth cone. J. Cell Sci. 116, 1209–1217 [DOI] [PubMed] [Google Scholar]
- 28. Chaineau M., Danglot L., Galli T. (2009) Multiple roles of the vesicular-SNARE TI-VAMP in post-Golgi and endosomal trafficking. FEBS Lett. 583, 3817–3826 [DOI] [PubMed] [Google Scholar]
- 29. Chieregatti E., Meldolesi J. (2005) Regulated exocytosis: new organelles for non-secretory purposes. Nat. Rev. Mol. Cell Biol. 6, 181–187 [DOI] [PubMed] [Google Scholar]
- 30. Hewavitharana T., Wedegaertner P. B. (2012) Non-canonical signaling and localizations of heterotrimeric G proteins. Cell Signal. 24, 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vilardaga J. P., Gardella T. J., Wehbi V. L., Feinstein T. N. (2012) Non-canonical signaling of the PTH receptor. Trends Pharmacol. Sci. 33, 423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Popova J. S., Rasenick M. M. (2003) G β γ mediates the interplay between tubulin dimers and microtubules in the modulation of Gq signaling. J. Biol. Chem. 278, 34299–34308 [DOI] [PubMed] [Google Scholar]
- 33. Marrari Y., Crouthamel M., Irannejad R., Wedegaertner P. B. (2007) Assembly and trafficking of heterotrimeric G proteins. Biochemistry 46, 7665–7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saini D. K., Chisari M., Gautam N. (2009) Shuttling and translocation of heterotrimeric G proteins and Ras. Trends Pharmacol. Sci. 30, 278–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hynes T. R., Mervine S. M., Yost E. A., Sabo J. L., Berlot C. H. (2004) Live cell imaging of Gs and the β2-adrenergic receptor demonstrates that both αs and β1γ7 internalize upon stimulation and exhibit similar trafficking patterns that differ from that of the β2-adrenergic receptor. J. Biol. Chem. 279, 44101–44112 [DOI] [PubMed] [Google Scholar]
- 36. Dent E. W., Callaway J. L., Szebenyi G., Baas P. W., Kalil K. (1999) Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J. Neurosci. 19, 8894–8908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emery A. C., Eiden M. V., Mustafa T., Eiden L. E. (2013) Rapgef2 connects GPCR-mediated cAMP signals to ERK activation in neuronal and endocrine cells. Sci. Signal. 6, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heidemann S. R., Joshi H. C., Schechter A., Fletcher J. R., Bothwell M. (1985) Synergistic effects of cyclic AMP and nerve growth factor on neurite outgrowth and microtubule stability of PC12 cells. J. Cell Biol. 100, 916–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davé R. H., Saengsawang W., Lopus M., Davé S., Wilson L., Rasenick M. M. (2011) A molecular and structural mechanism for G protein-mediated microtubule destabilization. J. Biol. Chem. 286, 4319–4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G., Sunahara R. K., El-Samad H., Huang B., von Zastrow M. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu X., Ballo L., Pietila L., Viesselmann C., Ballweg J., Lumbard D., Stevenson M., Merriam E., Dent E. W. (2011) BDNF-induced increase of PSD-95 in dendritic spines requires dynamic microtubule invasions. J. Neurosci. 31, 15597–15603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wolfgang W. J., Clay C., Parker J., Delgado R., Labarca P., Kidokoro Y., Forte M. (2004) Signaling through Gs α is required for the growth and function of neuromuscular synapses in Drosophila. Dev. Biol. 268, 295–311 [DOI] [PubMed] [Google Scholar]
- 43. Gordon-Weeks P. R. (2004) Microtubules and growth cone function. J. Neurobiol. 58, 70–83 [DOI] [PubMed] [Google Scholar]
- 44. Efendiev R., Bavencoffe A., Hu H., Zhu M. X., Dessauer C. W. (2013) Scaffolding by A-kinase anchoring protein enhances functional coupling between adenylyl cyclase and TRPV1 channel. J. Biol. Chem. 288, 3929–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pyne N. J., Pyne S. (2011) Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: out of the shadow? Trends Pharmacol. Sci. 32, 443–450 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.