

Background: A novel mechanism underlying mitochondrial translocation of Parkin during mitophagy is uncovered.
Results: nNOS plays a significant role in Parkin translocation via interaction with full-length PINK1.
Conclusion: Parkin recruitment through the interaction of nNOS with full-length PINK1 occurs during CCCP-induced mitophagy.
Significance: Optimum levels of NO are sufficient for triggering the translocation of Parkin, even in the absence of PINK1, suggesting the feasibility of NO-based pharmacotherapy.
Keywords: Mitophagy, Nitric Oxide, Parkin, Parkinson Disease, PTEN-induced Putative Kinase 1 (PINK1), nNOS
Abstract
The failure to trigger mitophagy is implicated in the pathogenesis of familial Parkinson disease that is caused by PINK1 or Parkin mutations. According to the prevailing PINK1-Parkin signaling model, mitophagy is promoted by the mitochondrial translocation of Parkin, an essential PINK1-dependent step that occurs via a previously unknown mechanism. Here we determined that critical concentrations of NO was sufficient to induce the mitochondrial translocation of Parkin even in PINK1 deficiency, with apparent increased interaction of full-length PINK1 accumulated during mitophagy, with neuronal nitric oxide synthase (nNOS). Specifically, optimum levels of NO enabled PINK1-null dopaminergic neuronal cells to regain the mitochondrial translocation of Parkin, which appeared to be significantly suppressed by nNOS-null mutation. Moreover, nNOS-null mutation resulted in the same mitochondrial electron transport chain (ETC) enzyme deficits as PINK1-null mutation. The involvement of mitochondrial nNOS activation in mitophagy was further confirmed by the greatly increased interactions of full-length PINK1 with nNOS, accompanied by mitochondrial accumulation of phospho-nNOS (Ser1412) during mitophagy. Of great interest is that the L347P PINK1 mutant failed to bind to nNOS. The loss of nNOS phosphorylation and Parkin accumulation on PINK1-deficient mitochondria could be reversed in a PINK1-dependent manner. Finally, non-toxic levels of NO treatment aided in the recovery of PINK1-null dopaminergic neuronal cells from mitochondrial ETC enzyme deficits. In summary, we demonstrated the full-length PINK1-dependent recruitment of nNOS, its activation in the induction of Parkin translocation, and the feasibility of NO-based pharmacotherapy for defective mitophagy and ETC enzyme deficits in Parkinson disease.
Introduction
Mitophagy, the selective autophagy of mitochondria, is a critical quality control mechanism required to eliminate damaged or excess mitochondria (1–3). Defective mitophagy has been suggested to be one of the major pathological mechanisms underlying mitochondrial dysfunction in autosomal recessive forms of Parkinson disease (PD),2 such as those caused by mutations in PINK1 or Parkin (1, 4, 5). According to the prevailing model of mitophagy, PINK1 and Parkin play key roles in priming damaged mitochondria for further degradation by acting as a sensor and effector pair (6–8). However, the mechanism underlying the recruitment of Parkin by PINK1 on damaged mitochondria is largely unknown.
The PINK1-Parkin signaling model has become a new paradigm for the mechanistic investigation of mitophagy induced by mitochondrial damage in mammals, in which the removal of damaged mitochondria is primarily mediated by PINK1, a mitochondrial serine/threonine kinase, and Parkin, a cytosolic E3 ubiquitin ligase (1). Specifically, the loss of mitochondrial membrane potentials by uncouplers such as CCCP and valinomycin, mitochondrial DNA mutations, or staurosporine (1, 9, 10) stabilizes and facilitates the rapid accumulation of full-length PINK1 on the mitochondrial outer membrane. In wild-type cells, PINK1 is normally imported to the inner mitochondrial membrane and rapidly cleaved by proteases, including presenilin associated rhomboid-like, for further degradation in the cytoplasm (6, 11–13). Therefore, PINK1 serves as a molecular switch to indicate a loss of mitochondrial membrane potential. PINK1 deficiency results in a near complete loss of Parkin translocation and mitophagy (6, 14, 15), suggesting a critical role for PINK1 in Parkin translocation via a previously unidentified mechanism. The E3 ligase activity of Parkin is stimulated by its phosphorylation (16–18) or S-nitrosylation in the cytosol (19) or activated by phosphorylation of ubiquitin by PINK1 (20, 21), which might promote the ubiquitination of various mitochondrial substrates such as voltage-dependent anion channel (VDAC), mitofusins, or Miro (22–24). Therefore, mitochondrial translocation and activation of Parkin play an essential role in priming mitochondria, which occurs via the ubiquitination of specific mitochondrial substrates, to induce autophagy by interaction with Ambra 1 (25). Although the induction of Parkin translocation is a critical step in the activation of the PINK1-Parkin signaling pathway, the specific mechanisms of PINK1-mediated mitochondrial translocation of Parkin remain largely unknown.
PINK1 deficiency leads to mild to severe decreases in mitochondrial complex activities in PD patient-derived fibroblast cultures (26), PINK1-null mice and fruit flies (27–30) and PINK1-deficient dopaminergic neuronal cells (31) and loss of phosphorylation of complex I subunit NdufA10 at Ser250 (32). We have demonstrated previously that PINK1 deficiency also results in a mitochondrial complex IV deficit because of reduced expression of the complex IV assembly factor leucine-rich pentatricopeptide repeat containing (LRPPRC) and dysregulated NO signaling (33) and that low levels of NO treatment help PINK1-deficient dopaminergic neurons restore the function of their defective PINK1-LRPPRC-Hsp60-complex IV signaling axis (33). However, it remains unclear how the functional loss of PINK1 leads to a disturbance in NO signaling and how dysregulated NO signaling affects various mitochondrial functions.
This report is the first to identify the novel function of optimum levels of NO in mitophagy induction, which occurs via the triggering of Parkin translocation. We showed that increased interaction of full-length PINK1 with nNOS and its activation are involved in induction of Parkin translocation and mitophagy and that precise levels of NO allow PINK1-null dopaminergic neuronal cells to recover from defective mitophagy and ETC enzyme deficits. Our findings demonstrate that mitochondrial dysfunction in PINK1-deficient dopaminergic cells can be rescued significantly upon NO treatment, at least in part because of the restoration of Parkin translocation and mitophagy.
EXPERIMENTAL PROCEDURES
Materials
Carbonyl cyanide p-chlorophenylhydrazone (CCCP), sodium nitroprusside (SNP) dehydrate, S-nitroso-N-acetyl-dl-penicillamine (SNAP), Nω-nitro-l-arginine methyl ester, N-[(4S)-4-amino-5-[(2-aminoethyl)amino]pentyl]-N′-nitroguanidine tris(trifluoroacetate) (NAAN), and 5,5′-dithiobis(2-nitrobenzoic acid) were purchased from Sigma-Aldrich, and Phos-tag acrylamide was from Wako Chemicals. Antibodies against TOM 20 (catalog no. sc-11415), GAPDH (catalog no. sc-32233), Actin (catalog no. sc-1616), HA (catalog no. sc-805), and prohibitin (catalog no. sc-28259) were purchased from Santa Cruz Biotechnology. Other antibodies were purchased from the following companies: c-Myc (catalog no. 631206) from Clontech; Parkin (catalog no. 2132), COX IV (catalog no. 4844), cytochrome c (catalog no. 4272), mitofusin-2 (MFN-2, catalog no. 9482), and VDAC (catalog no. 4866) from Cell Signaling Technology; p-nNOS (catalog no. ab5583) and nNOS (catalog no. ab76067) from Abcam; PINK1 (catalog no. BC100-494) from Novus Biologicals; and FLAG (catalog no. F3165) from Sigma-Aldrich.
Plasmids, Cell Cultures, and Transient Transfection
The pcDNA3-PINK1-FLAG, pCMVTnT-PINK1-myc, pCMVTnT-PINK1-L347P-myc, pcDNA3-FLAG-nNOS, and pcDNA3-nNOS-myc plasmids were provided by Dr. Takahashi, Dr. M. R. Cookson, Dr. K. Itoh, and Dr. G. Rameau, respectively. The wild-type dopaminergic neuronal cell line SN4741 and PINK1-null dopaminergic neuronals cells, which were derived from the same transgenic animal lines with the C57BL/6 genetic background (31, 34), were cultured at 33 °C with 5% CO2 in RF medium (DMEM (Invitrogen) supplemented with 10% FBS (HyClone), 1% glucose, penicillin (100 units/ml)-streptomycin (100 g/ml) (Invitrogen, catalog no. 15070-063), and l-glutamine (2 mm) (Invitrogen)). Transient transfection analyses were performed as described previously (33).
nNOS-null Mice and Preparation of Primary MEFs
nNOS-null mice (catalog no. 002986) were purchased from The Jackson Laboratory and have been described previously (35). They were bred to C57BL6/J mice for at least 10 generations. Mice were housed in clear plastic cages in a temperature- and humidity-controlled environment with a 12-h light/dark cycle and maintained on an ad libitum diet of lab chow and water. The nNOS-null mice and wild-type controls used in this study were obtained by crossing an nNOS+/− male and an nNOS+/− female and had the C57BL6/J genetic background. The following primer sets were used for genotype analysis: 5′-TCAGATCTGATCCGAGGAGG-3′ and 5′-TTCCAGAGCGCTGTCATAGC-3′ (117 bp) for nNOS (NOS1) and 5′-GCCCTGAATGAACTGCAGGACG-3′ and 5′-CACGGGTAGCCAACGCTATGTC-3′ (∼500 bp) for the knockout state (neo). To prepare primary MEF embryonic day 12.5 embryos were harvested from pregnant mice, dissected in 0.05% trypsin/EDTA solution, incubated for 15 min at 37 °C, and plated on a 100-mm culture dish. The primary MEFs were then genotyped, maintained, and passaged in DMEM with 10% FBS.
Immunoprecipitation
Immunoprecipitation assay were performed as described previously (33). Briefly, transfected HEK293T cells were incubated in lysis buffer (Thermo Fisher Scientific Inc.) containing protease and phosphatase inhibitors for 30 min on ice. Protein extracts were incubated with anti-FLAG M2-agarose (Sigma-Aldrich) overnight at 4 °C. The precipitated beads were washed three times with PBS, and the bound immunoprecipitated proteins were eluted from the beads by boiling in sample buffer (60 mm Tris-HCl (pH 6.8), 25% glycerol, 2% SDS, 14.4 mm β-mercaptoethanol, and 0.1% bromphenol blue). The eluted proteins were analyzed by 10% SDS-polyacrylamide gel electrophoresis and then subjected to immunoblot analysis. For endogenous protein IP, SN4741 dopaminergic neuronal cells were incubated in the same lysis buffer for 30 min on ice, and the cell lysate was incubated with the indicated protein antibody and protein G-Sepharose overnight at 4 °C. The beads were pelleted by centrifugation, washed with lysis buffer several times, and resuspended in sample buffer for immunoblot analysis.
Immunoblot Analysis
Cells were washed with ice-cold PBS and lysed for 10 min on ice in buffer containing 50 mm Tris (pH 8.0), 150 mm NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5 mm EDTA supplemented with phosphatase inhibitors (0.2 mm Na3VO4, and 4 mg/ml NaF), and protease inhibitors (Complete mixture, Roche). Protein lysates were separated on SDS-polyacrylamide gels (7–14%) and transferred to PVDF membranes. To detect phospho-ubiquitin using SDS-PAGE gels, Phos-tag acrylamide (10 μm final, Wako Chemicals) and Mncl2 (20 μm final) were used. The following primary antibodies were used: anti-phospho-nNOS (1:1000), anti-nNOS (1:1000), anti-Parkin (1:1000), anti-TOM20 (1:5000), anti-prohibitin (1:5000), anti-GAPDH (1:2000), anti-Myc (1:2000), anti-FLAG (1:2000), and anti-Actin (1:5000). After washing, the blots were probed with anti-rabbit (1:2000, Bio-Rad), anti-mouse (1:2000, Bio-Rad), or anti-goat (1:2000, Bio-Rad) IgG-HRP secondary antibodies and visualized by ECL (GE Healthcare, catalog no. RPN2232) as described previously (33). Densitometric quantification of bands was performed using an image analyzer system using Multi Gauge version 2.3 software.
Isolation of Mitochondria
Mitochondria were prepared as described previously (31, 33). Briefly, cells were washed with ice-cold PBS and suspended in 10 mm ice-cold Tris buffer (pH 7.6) with a protease inhibitor mixture. The cells were mechanically disrupted using a 1-ml syringe. After the subsequent addition of ice-cold 1.5 m sucrose, the cells were centrifuged at 700 × g for 10 min at 2 °C. The supernatant was centrifuged at 14,000 × g for 10 min at 2 °C, and the resulting pellet was washed and resuspended in 10 mm ice-cold Tris (pH 7.6) containing a protease inhibitor mixture. Some pellets were lysed for quantification, and the remainder was stored on ice until analysis.
Measurement of Mitochondrial ETC Enzyme Activity
Measurement of the activity of mitochondrial ETC enzymes was performed using a method described previously (31, 36–38) with minor modifications.
Complex I Assay
Activity was measured spectrophotometrically at 600 nm in an incubation volume of 240 μl containing 25 mm potassium phosphate, 3.5 g/liter BSA, 60 μm 2,6-dichloroindophenol, 70 μm decylubiquinone, 1.0 μm antimycine-A, and 3.2 mm NADH (pH 7.8). Mitochondrial samples (1 μg/10 μl) were preincubated in the buffer without NADH for 3 min at 37 °C. 5 μl of 160 mm NADH was then added to the samples, and the absorbance was measured at 30-s intervals for 5 min at 37 °C. After 5 min, 2.5 μl of rotenone (a 1 mm solution in dimethyl sulfoxide that was subsequently diluted to 100 μm in 10 mm Tris (pH 7.6)) was added, and the absorbance was measured at 30-s intervals for 5 min at 37 °C.
Complex II Assay
Activity was measured spectrophotometrically at 600 nm in an incubation volume of 240 μl containing 80 mm potassium phosphate, 1 g/liter BSA, 2 mm EDTA, 0.2 mm ATP, 10 mm succinate, 0.3 mm potassium cyanide, 60 μm 2,6-dichloroindophenol, 50 μm decylubiquinone, 1 μm antimycin A, and 3 μm rotenone (pH 7.8). Mitochondrial samples (1 μg/10 μl) were preincubated without succinate and potassium cyanide for 10 min at 37 °C. 20 μl of 1.5 m succinate and 0.75 μl of 0.1 m KCN were then added, and the absorbance was measured at 30-s intervals for 5 min at 37 °C. Blanks contained 5 mm malonate, which was added prior to the preincubation.
Complex IV and Citrate Synthase (CS) Assay
Complex IV activity was measured spectrophotometrically at 550 nm in an incubation volume of 240 μl containing 30 mm potassium phosphate, 2.5 mm dodecylmaltoside, and 34 μm ferrocytochrome c (pH 7.4). Mitochondrial samples (1 μg/10 μl) were added to buffer, and the absorbance was immediately measured at 30-s intervals for 5 min at 30 °C. Blanks contained 1 mm KCN. CS activity was measured spectrophotometrically at 412 nm in an incubation volume of 240 μl containing 50 mm Tris-HCl (pH 7.5), 0.2 mm 5,5′-dithiobis-(2-nitrobenzoic acid), 0.1 mm acetyl-CoA, and 2.5 mm oxaloacetate. Mitochondrial samples (1 μg/10 μl) were preincubated in buffer without oxaloacetate for 5 min at 30 °C. Subsequently, 2.5 μl of 250 mm oxaloacetate was added, and the absorbance was measured at 30-s intervals for 5 min at 37 °C. For the controls, an equivalent volume of water was added instead of oxaloacetate.
CS Assay for Analyzing Mitochondrial Mass
MitoTracker staining is dependent on the specific membrane potential, whereas a CS activity assay using whole cells is a more precise method for quantitatively measuring mitochondrial mass (41). CS activity assays for the quantitative measurement of mitochondrial mass were performed as described previously (23, 42). After various treatments for 24 h, cells were washed with PBS and lysed in 0.25% (v/v) Triton X-100 in PBS supplemented with protease and phosphatase inhibitors. After the removal of debris by centrifugation, the CS activity over time was determined by measuring the oxidation of 5,5′-dithiobis(2-nitrobenzoic acid) in a spectrophotometer (absorbance at 412 nm) at 30 °C in the presence of acetyl CoA and oxaloacetate. The enzyme activity was expressed as nanomoles per minute per milligram of protein.
Fluorescence Confocal Microscopy
SN4741 and PINK1-null dopaminergic neuronal cells were cultured on poly-l-lysine-coated (Sigma-Aldrich) 2-well slides, transfected with the indicated plasmid, fixed with 4% paraformaldehyde for 10 min at room temperature, and then washed with PBS. Slides were blocked with 1% BSA in PBS with 0.1% Triton X-100 prior to immunostaining. After incubation with primary FLAG antibody (1:500), the cells were labeled with Alexa Fluor 488 goat anti-mouse antibody (1:1000). The culture slides were mounted with Vectashield aqueous mounting medium (Vector Laboratories) and analyzed using a Zeiss LSM510 META laser-scanning confocal microscope (Carl Zeiss). Image processing and analysis were performed using Zeiss LSM510 version 2.3 software.
Statistical Analysis
Data are expressed as the mean ± S.E. on the basis of the results of three to eight independent experiments. Two-sample comparisons were performed using Student's t test, and multiple comparisons were conducted using one-way analysis of variance followed by Bonferroni's multiple comparison test.
RESULTS
NO-induced Mitochondrial Translocation of Parkin
In our previous study (33), dysregulated NO signaling was implicated in PINK1 deficiency-related ETC enzyme deficits, including complex IV deficits, that were significantly restored by NO. Because NO signaling linked to mitochondrial function appeared to be defective in PINK1 deficiency, we first investigated whether NO treatment was able to restore defective mitophagy in PINK1-null dopaminergic neuronal cells. Surprisingly, treatment with NO donors such as SNP (50 nm for 2 h) and SNAP (100 nm for 2 h) triggered a significant Parkin translocation to PINK1-deficient mitochondria, whereas CCCP (10 μm for 2 h) treatment did not induce Parkin translocation (Fig. 1A). In contrast, both SNP (20 nm) and CCCP (10 μm) were sufficient to induce Parkin translocation in a wild-type dopaminergic neuronal cell line, SN4741 (Fig. 1B). Treatment with higher concentrations of SNP (100–200 μm), at which occurrence of nitrosative toxic stress has been observed previously (34, 35), did not induce Parkin translocation (Fig. 1C). Moreover, SNP treatment did not alter Parkin expression levels in wild-type cells (Fig. 1D). Finally, we monitored mitophagy by using quantitative methods such as immunoblotting for mitochondrial proteins and measurement of mitochondrial mass by CS activity (23, 41, 42) in PINK1-null dopaminergic neuronal cells. Mitophagy induction and Parkin translocation by SNP were quantitated by TOM20 (an outer mitochondrial membrane protein), COX IV (an inner mitochondrial membrane protein), and cytochrome c (an intermembrane space protein) immunoblotting (Fig. 1E); CS assays (Fig. 1F); and confocal microscopy using mCherry-Parkin (Fig. 1, G and H). Mitophagy induction was further confirmed by ubiquitination of MFN-2 by SNP (Fig. 1I). However, SNP did not induce phosphorylation of ubiquitin (Fig. 1J). These results further suggest a novel functional role of NO in Parkin translocation even in the absence of PINK1.
FIGURE 1.

NO induced the mitochondrial translocation of Parkin in PINK1 deficiency. A and B, PINK1-null (A) and wild-type (B) dopaminergic neuronal cells, SN4741, were treated with CCCP (10 μm for 2 h) and the NO donors SNP (20–50 nm for 2 h) and SNAP (50–100 nm for 2 h), and mitochondria (Mito) were isolated as indicated and as described under “Experimental Procedures.” Mitochondrial lysates were then prepared and analyzed by immunoblotting using anti-Parkin or anti-TOM 20 antibody. C, wild-type cells were treated with higher concentrations of SNP (100–200 μm for 2 h), and mitochondrial lysates were analyzed by immunoblotting using anti-Parkin or anti-TOM 20 antibody. D, wild-type cells were treated with SNP (50 nm for 2 h) and CCCP (10 μm for 2 h), and whole cell lysates were analyzed by immunoblotting using anti-Parkin antibody. Representative results are shown in A–D. E, both PINK1-null and wild-type cells were treated with either CCCP (10 μm for 24 h) or SNP (50 nm for 24 h), and whole cell lysates were then prepared and analyzed by immunoblotting using an antibody against TOM20 (an outer mitochondrial membrane protein), COX IV (an inner mitochondrial membrane protein), and cytochrome c (an intermembrane space protein). Representative results are shown. The intensities of the TOM 20, COX IV, and cytochrome c bands (normalized against that of GAPDH) were determined from three independent experiments by densitometry. ADU, arbitrary data unit. F, both PINK1-null and wild-type cells were treated with either CCCP or SNP, and a CS activity-based mitochondrial mass assay was performed as described under “Experimental Procedures.” G and H, PINK1-null (G) and wild-type cells (H) were transfected with mCherry-Parkin (red) and pMito-ECFP (cyan) and treated with SNP (50 nm for 2 h) and CCCP (10 μm for 2 h). The mCherry-Parkin translocation was quantified by counting at least 100 cells under a confocal microscope as described under “Experimental Procedures.” CON, control. I, wild-type cells were treated with SNP (50 nm for 2 h) and CCCP, and mitochondrial lysates were analyzed by immunoblotting using anti-MFN-2 antibody. Ubiquitinated MFN-2 is marked by an arrowhead. J, both PINK1-null and wild-type cells were transfected with HA-ubiquitin (Ub) plasmid and treated with CCCP and SNP. Phospho-ubiquitin was resolved by Phos-tag SDS-PAGE gel electrophoresis, followed by immunoblotting using an antibody against HA, as described under “Experimental Procedures.” Representative results are shown. All values are mean ± S.E. from at least three independent experiments. *, p < 0.05; **, p < 0.01; n.s., not significant (difference between control cells and treated cells).
Functional Role of nNOS in Parkin Translocation
To determine whether altered regulation of nNOS underlies dysregulated NO signaling in PINK1 deficiency, the inhibitory effects of a pan-NOS inhibitor, Nω-nitro-l-arginine methyl ester (10–100 μm), and an nNOS-specific inhibitor, NAAN (0.01–10 μm), were assessed by quantitative measurement of Parkin translocation during CCCP-induced mitophagy. As shown in Fig. 2, A and B, both Nω-nitro-l-arginine methyl ester and NAAN treatment diminished Parkin translocation to the basal level observed in the control. This inhibitory effect was confirmed by confocal microscopy (Fig. 2C). Of great interest is that nNOS inhibition in wild-type cells resulted in significant decreases in ETC enzymes and complex I, II, and IV activities of about 40%, 35%, and 45%, respectively (Fig. 2D). Next, the potential role of nNOS in mitophagy was confirmed further by employing nNOS-null MEF cells (Fig. 2E, left panels), which expressed similar levels of Parkin as wild-type MEF cells and accumulated full-length PINK1 by CCCP (Fig. 2E, right panels). As shown in Fig. 2F, the CCCP-induced Parkin translocation to mitochondria was almost completely abolished by nNOS-null mutation. Moreover, CCCP-induced mitophagy was very significantly inhibited by nNOS-null mutation when mitophagy was monitored by a TOM20, MFN-2, and VDAC immunoblot assays (Fig. 2G). As expected, nNOS-null mutation also resulted in the significant reduction in complex I, II, and IV activities, as shown in Fig. 2H. Together, these results suggest that nNOS mediates Parkin translocation during CCCP-induced mitophagy and that nNOS plays an important role in mitochondrial function.
FIGURE 2.

Parkin translocation was mediated by nNOS. A, wild-type cells were pretreated with either a pan-NOS inhibitor, Nω-nitro-l-arginine methyl ester (l-NAME, 10–100 μm) or an nNOS-specific inhibitor, NAAN (1 μm), for 12 h, and Parkin translocation by CCCP (10 μm for 2 h) was analyzed. Representative results are shown. The intensity of the Parkin band (normalized against that of prohibitin) was determined from three independent experiments by densitometry. ADU, arbitrary data unit. B, various concentrations of the nNOS-specific inhibitor NAAN (0.01∼10 μm) were treated, and Parkin translocation by CCCP was analyzed by immunoblotting. C, wild-type cells were transfected with mCherry-Parkin (red) and pMito-ECFP (cyan) and treated with CCCP in the absence and presence of NAAN (1 μm, pretreatment for 12 h). The mCherry-Parkin translocation was quantified by counting at least 100 cells under a confocal microscope as described under “Experimental Procedures.” Representative results are shown. Mito, mitochondria; CON, control. D, wild-type cells were pretreated with an nNOS-specific inhibitor, NAAN (1 μm), for 12 h, and ETC enzyme activities and complex I, II, and IV activities were measured as indicated and as described under “Experimental Procedures.” E, PCR reactions using genomic DNAs from primary MEF cells were performed with NOS1 and Neo primer sets (left panel, top), as indicated and as described under “Experimental Procedures.” Primary nNOS-null MEF cells were also analyzed by immunoblotting using anti-nNOS antibody (left panel, bottom). nNOS-null and wild-type MEF cell lysates were prepared as described under “Experimental Procedures,” and the expression levels of Parkin and PINK1 were analyzed by immunoblotting using anti-Parkin (right panel, top) and anti-PINK1 antibodies (right panel, bottom). F, after CCCP (10 μm for 3 h) treatment, nNOS-null and wild-type MEF cell lysates were prepared, and Parkin translocation was analyzed. G, after CCCP (10 μm) treatment, nNOS-null and wild-type MEF cell lysates were analyzed by immunoblotting using an antibody against TOM20, MFN-2, and VDAC. Representative results are shown. The intensities of the TOM20, MFN-2, and VDAC bands (normalized against that of either GAPDH or actin) were determined from three independent experiments by densitometry. H, ETC enzyme activities and complex I, II, and IV activities were measured in nNOS-null MEF cells as described under “Experimental Procedures.” All values are mean ± S.E. from at least three independent experiments. *, p < 0.05; ***, p < 0.005 (difference between control cells and treated cells). $$, p < 0.01; $$$, p < 0.005 (results of unpaired Student's t test for treatments). n.s., not significant.
Increased Interaction of nNOS with Full-length PINK1 Accumulated during Mitophagy
Because the phosphorylation of nNOS (at Ser1412) is an important posttranslational mechanism of nNOS activation, we examined whether mitophagy induced the phosphorylation of nNOS by PINK1. As shown in Fig. 3A, left panel), endogenous phospho-nNOS (phosphorylated at Ser1412) was accumulated to a relatively high density on CCCP-treated (10 μm for 2 h) mitochondria in wild-type cells but not in PINK1-deficient cells, suggesting a potential role for PINK1 in nNOS phosphorylation. Moreover, the nNOS expression level was not altered by PINK1-null mutation (Fig. 3A, right panel). This effect was further confirmed by the overexpression of PINK1 in PINK1-deficient cells, resulting in significant recovery from the loss of phospho-nNOS as well as Parkin accumulation on PINK1-deficient mitochondria during mitophagy (Fig. 3B, top panel). In contrast, overexpression of the L347P mutant, having reduced kinase activity (43, 44) and loss of interaction with Hsp90/Cdc37 chaperones (45), could not help significantly in the recovery from the loss of phospho-nNOS accumulation (Fig. 3B, bottom panel). A significant fraction of the overexpressed nNOS was localized on mitochondria only after CCCP treatment in wild-type dopaminergic neuronal cells (Fig. 3C). In healthy mitochondria, full-length PINK1 (∼62 kDa) was rapidly converted to processed PINK1 (∼52 kDa), which is rapidly degraded by mitochondrial proteases and proteasome (6, 11, 13). Therefore, we tested whether nNOS interacted directly with the full-length PINK1 (∼62 kDa) that accumulated during CCCP-induced mitophagy. As expected, the interaction of nNOS with the full-length PINK1 (∼62 kDa) was very significantly increased as the full-length PINK1 accumulated during CCCP-induced mitophagy (Fig. 3D). A reverse IP assay confirmed this increased interaction when mitophagy was induced (Fig. 3E). The overexpressed nNOS also showed an increased interaction with overexpressed processed PINK1 (∼52 kDa), which is rapidly degraded under normal physiological conditions (6, 11, 13). As shown in Fig. 3F, left panel, the same molecular interaction was confirmed further by demonstrating the direct interaction of endogenous nNOS with the endogenous full-length PINK1 (∼62 kDa) accumulated by CCCP treatment, whereas no interaction occurred between endogenous nNOS and TOM20 (Fig. 3F, right panel). In contrast, nNOS did not interact with the L347P PINK1 mutant, which has reduced kinase activity and no interaction with Hsp90/Cdc37 chaperones (43–45), compared with the wild-type PINK1 (Fig. 3G), despite the significant accumulation of full-length L347P mutant by CCCP treatment, suggesting that the L347P mutation might be a novel mutation affecting its nNOS binding capability. Therefore, our data indicate that CCCP treatment increased the interaction of nNOS with the full-length PINK1 that had significantly accumulated on damaged mitochondria and might result in a subsequent enhancement of phospho-nNOS accumulation.
FIGURE 3.

Endogenous nNOS interacted with full-length PINK1 accumulated during mitophagy. A, both PINK1-null and wild-type cells were treated with CCCP (10 μm for 24 h), and mitochondrial (Mito) lysates were analyzed by immunoblotting using an antibody against phospho-nNOS at Ser1412 (left panel). Both PINK1-null and wild-type cell lysates were subjected to immunoblotting using anti-nNOS antibody (right panel). B, PINK1-null cells were transfected with either FLAG-tagged wild-type PINK1 or L347P mutant, and cell lysates were prepared. Phospho-nNOS accumulation was analyzed by immunoblotting using anti-phospho-nNOS antibody. Representative results are shown in A and B. C, wild-type cells were transfected with FLAG-tagged nNOS (green) and Su9-RFP (mitochondrial marker, red), followed by CCCP treatment. The colocalization of nNOS with mitochondrial Su9-RFP by CCCP was monitored by confocal microscopy. Representative images are shown. CON, control. D, HEK293T cells were transfected with myc-tagged nNOS and FLAG-tagged PINK1 plasmids as indicated. Immunoprecipitation was performed as indicated and as described under “Experimental Procedures.” IB, immunoblot. E, HEK293T cells were transfected with FLAG-tagged nNOS and myc-tagged PINK1 plasmids, and a reverse IP assay was performed as indicated. F, cell lysates of wild-type cells were prepared in the presence and absence of CCCP and incubated with nNOS or TOM 20 antibody and protein G-Sepharose overnight at 4 °C as indicated and as described under “Experimental Procedures.” A co-IP assay using either the endogenous nNOS and full-length PINK1 (left panel) or the endogenous nNOS and TOM20 (right panel) was performed. TOM20 was used as a negative control protein. G, HEK293T cells were transfected with myc-tagged nNOS and FLAG-tagged L347P mutant plasmids as indicated. An IP assay was performed as indicated. Representative images are shown in D--G. At least three independent experiments were performed.
Recovery from ETC Enzyme Deficits by NO in PINK1-deficient Mitochondria
Finally, we tested whether the induction of Parkin translocation and mitophagy by NO treatment could rescue PINK1-null dopaminergic neuronal cells from mitochondrial ETC enzyme deficits. As expected, both SNP (50 nm for 24 h) and SNAP (100 nm for 24 h) were able to help PINK1-null dopaminergic neuronal cells recover significantly from mitochondrial ETC deficits and resulted in large increases in complex I, II, and IV activities of 60–70%, 60–80%, and 60–70%, respectively (Fig. 4). Therefore, the induction of Parkin translocation by NO may help improve, in part, certain mitochondrial dysfunction caused by defective mitophagy.
FIGURE 4.
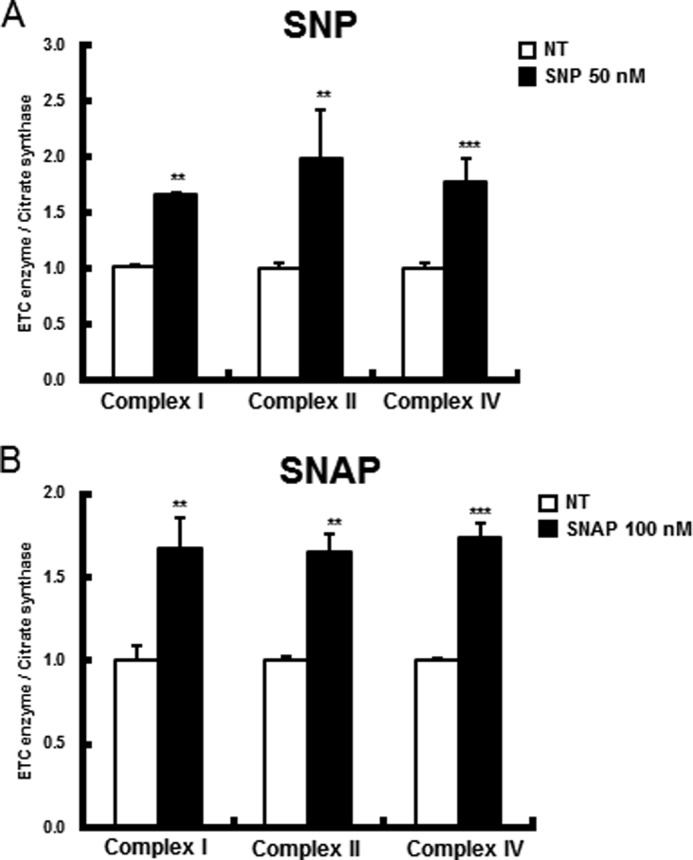
In PINK1 deficiency, NO aided in recovery from mitochondrial ETC enzyme deficits. PINK1-null dopaminergic neuronal cells were treated with SNP (50 nm for 24 h) (A) and SNAP (100 nm for 24 h) (B). ETC enzyme activities and complex I, II, and IV activities were measured as indicated and as described under “Experimental Procedures.” All values are mean ± S.E. from at least three independent experiments. **, p < 0.01; ***, p < 0.005 (differences between control cells and NO-treated cells).
DISCUSSION
We identified a novel mechanism underlying the mitochondrial recruitment of Parkin by PINK1 during CCCP-induced mitophagy. Our data show that PINK1 plays a key role in the recruitment of Parkin through an increased interaction of full-length PINK1, accumulated during mitophagy, with nNOS, whereas the L347P PINK1 mutant failed to bind to nNOS. Moreover, nNOS-null mutation resulted in the same mitochondrial ETC enzyme deficits as PINK1-null mutation. At the same time, phospho-nNOS (Ser1412), a strong NO producer, accumulated significantly on the mitochondria in a PINK1-dependent manner during mitophagy. Similarly, optimum levels of NO were sufficient for triggering the translocation of Parkin and mitophagy, and these occurred even in the absence of PINK1, suggesting a potential role for PINK1 in generating NO signaling for mitophagy. Therefore, appropriate levels of NO treatment enable PINK1-null dopaminergic neuronal cells to circumvent PINK1 deficiency and overcome specific mitochondrial dysfunction caused by PINK1 deficiency, which further implicates the feasibility of pharmacotherapy in certain familial forms of PD.
One major discovery of this study is that optimum levels of NO are critical to regaining the mitochondrial translocation of Parkin and subsequent mitophagic degradation of damaged mitochondria in PINK1-null dopaminergic neuronal cells, indicating that a novel NO signaling mechanism underlying Parkin translocation might be defective in PINK1 deficiency. A specific functional role of NO in the induction of Parkin translocation to mitochondria has not been recognized previously. We provide strong evidence demonstrating that Parkin translocation to mitochondria occurs at precise concentrations (between 20 nm and 10 μm) of NO donor in the absence of PINK1 (Fig. 1). In contrast, treatment with higher concentrations of SNP (100–200 μm) did not induce Parkin translocation (Fig. 1C). The mechanism underlying this biphasic response of Parkin to different NO doses is not clear. However, in accordance with these results, previous studies employing higher concentrations of NO donors (i.e. 50–200 μm) resulted in either a lack of Parkin translocation or nitrosative toxic stress (19, 39, 40). Therefore, NO treatment at non-toxic levels appears to be critical to the induction of Parkin translocation. This notion of defective NO signaling in PINK1 deficiency is consistent with our previous study (33), which demonstrated the partial rescue of some mitochondrial ETC deficits in PINK1-null dopaminergic neuronal cells by optimal concentrations of an NO signaling activator, ginsenoside Re, and an NO donor, SNP, but not by their higher concentrations. Therefore, we speculate that optimum levels of NO treatment might provide PINK1-null dopaminergic neuronal cells with appropriate NO signaling to bypass the NO production step during Parkin translocation.
We also discovered that the increased interaction of nNOS with the accumulated full-length PINK1 and its activation might be prerequisites for Parkin translocation during CCCP-induced mitophagy. First, the mitochondrial translocation of Parkin was significantly suppressed by both pharmacological inhibition and by gene knockout of nNOS in wild-type cells (Fig. 2). The interaction of nNOS with the accumulated full-length PINK1 significantly increased during mitophagy (Fig. 3, D–F) because, during this process, full-length PINK1 is protected from degradation by mitochondrial protease(s), resulting in the strong accumulation of full-length PINK1 on the mitochondrial outer membrane (6, 11, 13). Subsequently, phospho-nNOS (Ser1412) accumulated on mitochondria during mitophagy, whereas this accumulation was almost absent in PINK1-deficient mitochondria and in healthy mitochondria without CCCP-induced mitophagy. Additionally, the loss of phospho-nNOS on PINK1-deficient mitochondria could be reversed significantly by wild-type PINK1 but not by the PINK1-L347P mutant, which has reduced kinase activity and loss of nNOS binding capability during CCCP-induced mitophagy (Fig. 3, B and G), as indicated by the observation that a significant proportion of cytosolic nNOS was translocated to the mitochondria only after CCCP treatment (Fig. 3C). Therefore, PINK1 might temporally phosphorylate multiple proteins, such as nNOS and ubiquitin, to orchestrate Parkin translocation and Parkin activation, respectively. Parkin translocation may be a sufficient step to initiate mitophagy because of the basal E3 ligase activity, whereas Parkin activation by phospho-ubiquitin (20, 21) might be a necessary step to accelerate mitophagy. In fact, SNP did not cause phosphorylation of ubiquitin (Fig. 1J). Therefore, simple induction of Parkin translocation by NO may provide a sufficient Parkin E3 ligase activity to trigger mitophagy in the absence of PINK1. Moreover, the L347P PINK1 mutant lost nNOS binding capability in addition to loss of interaction with Hsp90/Cdc37 chaperones (45) and reduced kinase activity (43, 44). However, the precise mechanisms underlying cytosolic nNOS translocation to the mitochondria and its phosphorylation and NO-induced Parkin translocation remain to be elucidated. Taken together, our data demonstrate that the mitochondrial accumulation of full-length PINK1 by mitophagy could enhance its interaction with nNOS, which might be followed by increased NO production via phospho-nNOS accumulation and the activation of NO signaling for Parkin recruitment on damaged mitochondria. Therefore, phosphorylation of nNOS and activation of NO signaling for Parkin translocation might be also defective under PINK1-deficient conditions.
This study also addressed whether NO treatment could rescue PINK1-null dopaminergic neuronal cells from one of the major mitochondrial dysfunctions (i.e. mitochondrial ETC enzyme deficits) in which defective mitophagy has been implicated as one of the major culprits (46). Our previous studies (31, 33) also demonstrated that PINK1 deficiency resulted in dramatic reductions in ETC activity, including complexes I, II, and IV, and showed a partial rescue of complex IV deficits through up-regulation of specific chaperones, such as LRPPRC, Hsp90 and Hsp60, by the reactivation of NO signaling. In accordance with these results, in this study, optimal levels of NO donors, such as SNP and SNAP, helped PINK1-null dopaminergic neuronal cells to recover significantly from mitochondrial ETC deficits (Fig. 4), and this effect might be, in part, due to the efficient removal of damaged mitochondria (31) and/or the increased turnover of selective ETC enzyme subunits (47). Moreover, both pharmacological inhibition and genetic knockout of nNOS resulted in a significant reduction in ETC enzyme activity (Fig. 2, D and H), further implying an important role of the PINK1-nNOS-NO signaling axis in mitochondrial function. Alternatively, NO is known to induce mitochondrial biogenesis (48, 49) and mitochondrial dynamics (50). Nitrite, the oxidation product of NO, modulates the mitochondrial oxidative phosphorylation rate and mitochondrial fusion (51, 52). Therefore, important contributions of increased mitochondrial biogenesis, altered mitochondrial dynamics, and/or their intimate cross-talk with mitophagy (53) to recovery cannot be ruled out. At present, no effective neuroprotective measures are available to alleviate mitochondrial deficits in PD, despite the implication of mitochondrial dysfunction as one of the major pathological mechanisms underlying neuronal loss (5, 46, 54). Therefore, our findings may enable novel pharmacotherapies for specific mitochondrial dysfunction in PINK1 deficiency, possibly through NO-based restoration of Parkin translocation and mitophagy.
In summary, we demonstrated that PINK1 plays a key role in the recruitment of Parkin through the interaction of nNOS with the accumulated full-length PINK1, localized accumulation of phospho-nNOS (Ser1412), and enhancement of NO production on mitochondria during CCCP-induced mitophagy. Furthermore, NO treatment triggered the translocation of Parkin and mitophagy in PINK1 deficiency and also allowed PINK1-null dopaminergic neuronal cells to significantly recover from mitochondrial ETC enzyme deficits. Therefore, we present the first demonstration of a novel role for NO in induction of Parkin translocation and reveal the feasibility of pharmacotherapy for defective mitophagy in some familial forms of PD.
This research was supported by Grant 2013-008773 from NRF, Republic of Korea.
- PD
- Parkinson disease
- CCCP
- carbonyl cyanide p-chlorophenylhydrazone
- ETC
- electron transport chain
- SNP
- sodium nitroprusside
- SNAP
- S-nitroso-N-acetyl-dl-penicillamine
- NAAN
- N-[(4S)-4-amino-5-[(2-aminoethyl)amino]pentyl]-N′-nitroguanidine tris(trifluoroacetate)
- MEF
- mouse embryonic fibroblast
- IP
- immunoprecipitation
- VDAC
- voltage-dependent anion channel
- LRPPRC
- leucine-rich pentatricopeptide repeat containing.
REFERENCES
- 1. Narendra D., Tanaka A., Suen D. F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sandoval H., Thiagarajan P., Dasgupta S. K., Schumacher A., Prchal J. T., Chen M., Wang J. (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al Rawi S., Louvet-Vallée S., Djeddi A., Sachse M., Culetto E., Hajjar C., Boyd L., Legouis R., Galy V. (2011) Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 334, 1144–1147 [DOI] [PubMed] [Google Scholar]
- 4. Geisler S., Holmström K. M., Treis A., Skujat D., Weber S. S., Fiesel F. C., Kahle P. J., Springer W. (2010) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6, 871–878 [DOI] [PubMed] [Google Scholar]
- 5. Schapira A. H. (2012) Targeting mitochondria for neuroprotection in Parkinson's disease. Antioxid. Redox Signal. 16, 965–973 [DOI] [PubMed] [Google Scholar]
- 6. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Youle R. J., Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ashrafi G., Schwarz T. L. (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suen D. F., Narendra D. P., Tanaka A., Manfredi G., Youle R. J. (2010) Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. U.S.A. 107, 11835–11840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ha J. Y., Kim J. S., Kim S. E., Son J. H. (2014) Simultaneous activation of mitophagy and autophagy by staurosporine protects against dopaminergic neuronal cell death. Neurosci. Lett. 561, 101–106 [DOI] [PubMed] [Google Scholar]
- 11. Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deas E., Plun-Favreau H., Gandhi S., Desmond H., Kjaer S., Loh S. H., Renton A. E., Harvey R. J., Whitworth A. J., Martins L. M., Abramov A. Y., Wood N. W. (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Greene A. W., Grenier K., Aguileta M. A., Muise S., Farazifard R., Haque M. E., McBride H. M., Park D. S., Fon E. A. (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rakovic A., Grünewald A., Seibler P., Ramirez A., Kock N., Orolicki S., Lohmann K., Klein C. (2010) Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum. Mol. Genet. 19, 3124–3137 [DOI] [PubMed] [Google Scholar]
- 15. Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., Magrané J., Moore D. J., Dawson V. L., Grailhe R., Dawson T. M., Li C., Tieu K., Przedborski S. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H. I., Campbell D. G., Gourlay R., Burchell L., Walden H., Macartney T. J., Deak M., Knebel A., Alessi D. R., Muqit M. M. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2, 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shiba-Fukushima K., Imai Y., Yoshida S., Ishihama Y., Kanao T., Sato S., Hattori N. (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2, 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim Y., Park J., Kim S., Song S., Kwon S. K., Lee S. H., Kitada T., Kim J. M., Chung J. (2008) PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 377, 975–980 [DOI] [PubMed] [Google Scholar]
- 19. Ozawa K., Komatsubara A. T., Nishimura Y., Sawada T., Kawafune H., Tsumoto H., Tsuji Y., Zhao J., Kyotani Y., Tanaka T., Takahashi R., Yoshizumi M. (2013) S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 3, 2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kane L. A., Lazarou M., Fogel A. I., Li Y., Yamano K., Sarraf S. A., Banerjee S., Youle R. J. (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E. A., Trempe J. F., Saeki Y., Tanaka K., Matsuda N. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 [DOI] [PubMed] [Google Scholar]
- 22. Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 [DOI] [PubMed] [Google Scholar]
- 23. Gegg M. E., Cooper J. M., Chau K. Y., Rojo M., Schapira A. H., Taanman J. W. (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y. L., Selkoe D., Rice S., Steen J., LaVoie M. J., Schwarz T. L. (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Humbeeck C., Cornelissen T., Hofkens H., Mandemakers W., Gevaert K., De Strooper B., Vandenberghe W. (2011) Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 31, 10249–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoepken H. H., Gispert S., Morales B., Wingerter O., Del Turco D., Mülsch A., Nussbaum R. L., Müller K., Dröse S., Brandt U., Deller T., Wirth B., Kudin A. P., Kunz W. S., Auburger G. (2007) Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 25, 401–411 [DOI] [PubMed] [Google Scholar]
- 27. Gautier C. A., Kitada T., Shen J. (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morais V. A., Verstreken P., Roethig A., Smet J., Snellinx A., Vanbrabant M., Haddad D., Frezza C., Mandemakers W., Vogt-Weisenhorn D., Van Coster R., Wurst W., Scorrano L., De Strooper B. (2009) Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 1, 99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu W., Acín-Peréz R., Geghman K. D., Manfredi G., Lu B., Li C. (2011) Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. U.S.A. 108, 12920–12924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Amo T., Saiki S., Sawayama T., Sato S., Hattori N. (2014) Detailed analysis of mitochondrial respiratory chain defects caused by loss of PINK1. Neurosci. Lett. 580, 37–40 [DOI] [PubMed] [Google Scholar]
- 31. Shim J. H., Yoon S. H., Kim K. H., Han J. Y., Ha J. Y., Hyun D. H., Paek S. H., Kang U. J., Zhuang X., Son J. H. (2011) The antioxidant Trolox helps recovery from the familial Parkinson's disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion 11, 707–715 [DOI] [PubMed] [Google Scholar]
- 32. Morais V. A., Haddad D., Craessaerts K., De Bock P. J., Swerts J., Vilain S., Aerts L., Overbergh L., Grünewald A., Seibler P., Klein C., Gevaert K., Verstreken P., De Strooper B. (2014) PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344, 203–207 [DOI] [PubMed] [Google Scholar]
- 33. Kim K. H., Song K., Yoon S. H., Shehzad O., Kim Y. S., Son J. H. (2012) Rescue of PINK1 protein null-specific mitochondrial complex IV deficits by ginsenoside Re activation of nitric oxide signaling. J. Biol. Chem. 287, 44109–44120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Son J. H., Chun H. S., Joh T. H., Cho S., Conti B., Lee J. W. (1999) Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J. Neurosci. 19, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang P. L., Dawson T. M., Bredt D. S., Snyder S. H., Fishman M. C. (1993) Targeted disruption of the neuronal nitric oxide synthase gene. Cell 75, 1273–1286 [DOI] [PubMed] [Google Scholar]
- 36. Janssen A. J., Trijbels F. J., Sengers R. C., Smeitink J. A., van den Heuvel L. P., Wintjes L. T., Stoltenborg-Hogenkamp B. J., Rodenburg R. J. (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 53, 729–734 [DOI] [PubMed] [Google Scholar]
- 37. Cooperstein S. J., Lazarow A. (1951) A microspectrophotometric method for the determination of cytochrome oxidase. J. Biol. Chem. 189, 665–670 [PubMed] [Google Scholar]
- 38. Sugden P. H., Newsholme E. A. (1975) Activities of citrate synthase, NAD+-linked and NADP+-linked isocitrate dehydrogenases, glutamate dehydrogenase, aspartate aminotransferase and alanine aminotransferase in nervous tissues from vertebrates and invertebrates. Biochem. J. 150, 105–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chung K. K., Thomas B., Li X., Pletnikova O., Troncoso J. C., Marsh L., Dawson V. L., Dawson T. M. (2004) S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304, 1328–1331 [DOI] [PubMed] [Google Scholar]
- 40. Yao D., Gu Z., Nakamura T., Shi Z.Q., Ma Y., Gaston B., Palmer L. A., Rockenstein E. M., Zhang Z., Masliah E., Uehara T., Lipton S. A. (2004) Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. U.S.A. 101, 10810–10814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding W. X., Yin X. M. (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gómez-Sánchez R., Gegg M. E., Bravo-San Pedro J. M., Niso-Santano M., Alvarez-Erviti L., Pizarro-Estrella E., Gutiérrez-Martín Y., Alvarez-Barrientos A., Fuentes J. M., González-Polo R.A., Schapira A. H. (2014) Mitochondrial impairment increases FL-PINK1 levels by calcium-dependent gene expression. Neurobiol. Dis. 62, 426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pridgeon J. W., Olzmann J. A., Chin L. S., Li L. (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5, e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beilina A., Van Der Brug M., Ahmad R., Kesavapany S., Miller D. W., Petsko G. A., Cookson M. R. (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. U.S.A. 102, 5703–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moriwaki Y., Kim Y. J., Ido Y., Misawa H., Kawashima K., Endo S., Takahashi R. (2008) L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci. Res. 61, 43–48 [DOI] [PubMed] [Google Scholar]
- 46. McCoy M. K., Cookson M. R. (2012) Mitochondrial quality control and dynamics in Parkinson's disease. Antioxid. Redox. Signal 16, 869–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vincow E. S., Merrihew G., Thomas R. E., Shulman N. J., Beyer R. P., MacCoss M. J., Pallanck L. J. (2013) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 6400–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nisoli E., Clementi E., Paolucci C., Cozzi V., Tonello C., Sciorati C., Bracale R., Valerio A., Francolini M., Moncada S., Carruba M. O. (2003) Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899 [DOI] [PubMed] [Google Scholar]
- 49. Nisoli E., Falcone S., Tonello C., Cozzi V., Palomba L., Fiorani M., Pisconti A., Brunelli S., Cardile A., Francolini M., Cantoni O., Carruba M. O., Moncada S., Clementi E. (2004) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A. 101, 16507–16512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miller M. W., Knaub L. A., Olivera-Fragoso L. F., Keller A. C., Balasubramaniam V., Watson P. A., Reusch J. E. (2013) Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am. J. Physiol. Heart Circ. Physiol. 304, H1624–H1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hendgen-Cotta U. B., Merx M. W., Shiva S., Schmitz J., Becher S., Klare J. P., Steinhoff H. J., Goedecke A., Schrader J., Gladwin M. T., Kelm M., Rassaf T. (2008) Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 105, 10256–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khoo N. K., Mo L., Zharikov S., Kamga-Pride C., Quesnelle K., Golin-Bisello F., Li L., Wang Y., Shiva S. (2014) Nitrite augments glucose uptake in adipocytes through the protein kinase A-dependent stimulation of mitochondrial fusion. Free Radic. Biol. Med. 70C, 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu J., Wang K. Z., Chu C. T. (2013) After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 9, 1663–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thomas B., Beal M. F. (2010) Mitochondrial therapies for Parkinson's disease. Mov. Disord. 25, S155–S160 [DOI] [PMC free article] [PubMed] [Google Scholar]