

Background: Epicardial cells are a potential source of progenitor cells for revascularization of the injured heart.
Results: Decreased p63RhoGEF and GEF-H1 and increased Epac, p190RhoGAP, and Rnds activities suppress RhoA signaling in epicardial progenitors.
Conclusion: The embryonic epicardial progenitor state is maintained by signaling pathways that control RhoA activity.
Significance: Manipulation of these signaling molecules might promote cardiac revascularization.
Keywords: Cardiovascular; Guanine Nucleotide Exchange Factor (GEF); Ras Homolog Gene Family, Member A (RhoA); Signal Transduction; Transformation; Epac; Rnds; Epicardial Cells; Epithelial to Mesenchymal Transformation; p190RhoGAP
Abstract
This study identifies signaling pathways that play key roles in the formation and maintenance of epicardial cells, a source of progenitors for coronary smooth muscle cells (SMCs). After epithelial to mesenchymal transition (EMT), mesenchymal cells invade the myocardium to form coronary SMCs. RhoA/Rho kinase activity is required for EMT and for differentiation into coronary SMCs, whereas cAMP activity is known to inhibit EMT in epithelial cells by an unknown mechanism. We use outgrowth of epicardial cells from E9.5 isolated mouse proepicardium (PE) explants, wild type and Epac1 null E12.5 mouse heart explants, adult rat epicardial cells, and immortalized mouse embryonic epicardial cells as model systems to identify signaling pathways that regulate RhoA activity to maintain the epicardial progenitor state. We demonstrate that RhoA activity is suppressed in the epicardial progenitor state, that the cAMP-dependent Rap1 GTP exchange factor (GEF), Epac, known to down-regulate RhoA activity through activation of Rap1 GTPase activity increased, that Rap1 activity increased, and that expression of the RhoA antagonistic Rnd proteins known to activate p190RhoGAP increased and associated with p190RhoGAP. Finally, EMT is associated with increased p63RhoGEF and RhoGEF-H1 protein expression, increased GEF-H1 activity, with a trend in increased p63RhoGEF activity. EMT is suppressed by partial silencing of p63RhoGEF and GEF-H1. In conclusion, we have identified new signaling molecules that act together to control RhoA activity and play critical roles in the maintenance of coronary smooth muscle progenitor cells in the embryonic epicardium. We suggest that their eventual manipulation could promote revascularization after myocardial injury.
Introduction
Cardiovascular disease is the most prevalent cause of morbidity and mortality worldwide. Importantly, early revascularization of the myocardium is essential for recovery of cardiac function after ischemic injury (1, 2), whereas the embryonic epicardium plays an important role in the development of the coronary vasculature (3, 4). The adult epicardium is a potential source of smooth muscle cells (SMCs)3 and pericytes for revascularization after infarction (5). Exciting developments support the possibility that the epicardium can serve as a source of vascular progenitors for cell based therapies (1, 6). This is well illustrated in a study showing that thymosin β4 activates adult epicardial derived cells and facilitates a robust and stable neovascularization of the injured adult heart (7). Furthermore, a seminal paper on zebrafish heart regeneration after resection of the ventricular apex showed that mature epicardium can re-express a developmental sequence, undergo epithelial to mesenchymal transition (EMT), and promote coronary angiogenesis in the regenerating myocardium (2). However, this has been more difficult to observe in adult mammalian hearts. Most investigators have studied mechanisms that promote epicardial EMT in embryonic hearts as a way to gain insight into factors missing in adult hearts. In the present study we take a different perspective and hypothesize that identification of signaling pathways that play key roles in the formation and maintenance of the embryonic epicardium, a source of vascular progenitor cells, will provide new insights into how EMT is controlled in mature epicardial cells.
Growth factors, including bFGF and TGFβ, released from the myocardium are known to regulate EMT (8–10). After EMT, epicardial mesothelial cells populate the sub-epicardial space with mesenchymal cells that invade the myocardium to form the coronary SMCs and cardiac fibroblasts (11–13). RhoA/Rho kinase (ROCK) activity has been shown to be required for EMT and for differentiation into coronary SMCs (14, 15), whereas cAMP activity is known to inhibit EMT in epithelial cells, but the mechanism is unknown (16–18). We have found in adult vascular, airway, and fundus smooth muscle (SM) that cAMP activates the Rap1-GTPase exchange factor (GEF), Epac1, to suppress RhoA activity (19). This cAMP effect is independent of protein kinase A (PKA) activation. Thus, we reasoned that Epac1 activity could contribute to the suppression of RhoA activity to down-regulate EMT, a focus of the present study. Furthermore, the GEFs that exchange GTP for GDP to activate RhoA and drive EMT are unknown in epicardial cells. Conversely, GTP activating proteins (GAPs) hydrolyze GTP to antagonize RhoA activity. Activation of p190RhoGAP has been reported in adult rat epicardial mesothelial cells stimulated with the α4β1 integrin ligand, VCAM-1, to inhibit TGFβ-induced EMT (20), but a role for p190RhoGAP in maintaining the epicardial progenitor phenotype has not been explored.
Additionally, members of the RhoA family of small GTPases called Rnds, proteins having a constitutively GTP-bound state and known to act as RhoA antagonists (21) and to bind and activate p190RhoGAP (22), may also converge to attenuate RhoA signaling in epicardial cells. Rnd3 is ubiquitously expressed, whereas Rnd1 is abundant in brain and liver, and Rnd2 is primarily found in brain and testis (21). Rnd3 has been shown to play an important functional role in uterine SM where its expression is high during midterm pregnancy to depress contractility and falls at the time of delivery, whereas RhoA and ROCK levels increase to enhance contractions and expulsion of the fetus (23). Expression of Rnds or their role in epicardial cells is unknown.
In this study we use outgrowth of epicardial cells from E9.5 isolated mouse proepicardium (PE) explants and from wild type and Epac null E12.5 mouse hearts, adult rat epicardial cells (EMCs) (24), and immortalized mouse embryonic epicardial cells (EECs) (25) as model systems to study signaling pathways that maintain the epicardial progenitor state. We confirm the critical role of RhoA activity in driving EMT in embryonic epicardial cells. We present evidence in epicardial cells for the presence of increased Epac/Rap1, Rnds, and p190RhoGAP activities that antagonize RhoA to suppress EMT. We also show up-regulation of the RhoA-specific GEFs p63RhoGEF and GEF-H1, expression, and activity upon growth factor-induced EMT, which is suppressed by partial silencing of these GEFs. Identification of responsible signaling molecules could provide new molecular targets for eventual manipulation of these SM progenitor cells and should be examined in the future for their roles during revascularization after myocardial injury or hypoxia.
EXPERIMENTAL PROCEDURES
All animal experiments were performed according to protocols reviewed and approved by the University of Virginia Institutional Animal Care and Use Committee and were in compliance with United States Public Health Service and Department of Agriculture guidelines for laboratory animal welfare.
PE Isolation and Explant
E9.5 mouse embryos were microdissected in ice-cold PBS treated with 2× antibiotic/antimycotic solution (Invitrogen). The chest wall membranes were opened, and the PEs were cut at their base to avoid inclusion of the liver primordium using tungsten needles. Isolated PEs were then placed in a drop of prewarmed complete medium containing M199 supplemented with 1× antibiotic/antimycotic, 30 mm glucose, 5 mm glutamine, 1.25 mm putrescine, 1× insulin transferrin selenium, and 10% fetal bovine serum on glass-bottom culture dishes (MatTek Corp.) and incubated at 37 °C in 95% air, 5% CO2.
Embryo Whole Mount Scanning Electron Microscopic Imaging
E9.5 and E10.5 whole embryonic mouse samples were fixed in 2.5% glutaraldehyde, 50 mm sodium cacodylate buffer with 6% sucrose for 24 h followed by a graded EtOH wash (from 50% to 100%). Samples were then dried using AutoSamdri-815 Critical Point Drying Machine and Gold sputter coated in a Bal-Tec Sputter Coater SCD005 apparatus. Images were then taken using a Zeiss Sigma VP HD field emission scanning electron microscope (University of Virginia Advanced Microscopy Facility).
Cell Migration (Collagen Gel Assay)
The ventricular chambers of hearts from E12.5 mouse embryos were dissected and placed epicardial side down on three-dimensional gels containing 1% type I collagen (BD Biosciences) and incubated in M199 media supplemented with 1% antibiotic/antimycotic, 30 mm glucose, 5 mm glutamine, 1.25 mm putrescine, 1% insulin transferrin selenium ITS, and 10% fetal bovine serum for 48 h at 37 °C with/without 30 μm 8-pCPT-2′-O-Me-cAMP (subsequently referred to as 007). Migration of epicardial cells from the heart explants into collagen gels, indicative of EMT, was measured after fixation in 4% paraformaldehyde, rinsing with PBS, then incubation in 5% and 15% sucrose in PBS. The collagen gel around the heart was excised and embedded in Tissue-TEK O.C.T. compound and frozen in liquid N2. 10-mm-thick serial sections were cut at −25 °C at right angles to the surface of the gel. The sections at selected positions relative to the heart were used for counting, with the edge of the heart taken as zero. The selected sections were permeabilized with 0.3% Triton X-100 for 15 min, washed with PBS, and then incubated in PBS with TOPRO (1:40,000) for 15 min followed by a 2 × 10-min wash with PBS. The sections then were imaged with an Olympus confocal microscope (FV300). The total number of cells at the surface and within the collagen gel as well as the number of cells within the gel were counted in each section and expressed as the ratio of the % of cells migrating into the gel to the total number of cells. Migrating cells were taken to represent cells having undergone EMT.
Cell Culture and Treatment
The following sources of cells were used depending on the type of assay required: (i) isolated mouse PE E9.5, (ii) cultured PE outgrowth (72 h), (iii) epicardial outgrowth from E12.5 embryonic heart explants, (iv) EECs from the ImmortoMouse carrying a ts large T-antigen, which undergo growth factor-stimulated EMT provided by and well characterized by Dr. Barnett (25), and (v) adult rat EMCs (from Dr. Bader), which maintain vasculogenic potential and provide a large number of cells for biochemical assays (24). EECs were maintained in DMEM supplemented with 10% heat inactive fetal bovine serum, 1% penicillin/streptomycin, and 10 units/ml of interferon-γ at 33 °C in a humidified 5% CO2 incubator. To induce EMT, the cells were treated with 250 pm TGF-β1 (Humanzyme Inc.) at 37 °C for 24–72 h. EECs were treated with Rho kinase inhibitor Y-27632 (10 μm), Epac activator 007 (50 μm), phosphodiesterase (PDE) inhibitor IBMX (10 μm), or PDE4 inhibitor Rolipram (10 μm) for 1 h before and after TGF-β1 treatment. C3 toxin (3–5 μg/ml) was added 15 h before and after TGF-β1 treatment. To evaluate the localization of Rnd3 and p190, EECs were cultured at 33 °C on coverslips overnight. EMCs were grown in DMEM containing 4.5 mg/ml glucose, 4 mmol/liter l-glutamine with 10% FBS, and 10 μg/ml penicillin/streptomycin. Incubation conditions were 37 °C and 5% CO2. As tubulin and GAPDH were used for normalization before and after treatments with TGF-β1, tubulin and GAPDH protein content/equal number of cells was measured. The number of cells decreased after treatment, and tubulin and GAPDH scaled to this decreased appropriately.
Generation of Epac1 Null Mice
The Epac1 targeting vector was constructed by inserting two loxP sites into Exon 1 and Intron 2 that flanked a portion of Exon 1 and Exon 2. A frt-Neo-frt cassette was inserted into Intron 2 for a positive selection marker. The linearized targeting DNA was electroporated into E14Tg2a embryonic stem cells, then selected and expanded at UNC-Chapel Hill Animal Models Core. The recombinant ES cell clones were microinjected into C57BL/6 blastocysts and transferred to pseudo-pregnant foster mice. Male chimeras were mated with female C57BL/6 mice, and progenies with germ line transmission (frt-neo/+) were confirmed by PCR. Male chimeric mice were then crossed with female Flpe +/+ mice for excision of Neo cassette, and excision was confirmed by PCR. Heterozygous Epac1 (−/+) mice were obtained by breeding with ZP3-Cre carrier mice (a gift form Ann E. Sutherland, University of Virginia). Epac1 null (−/−) and wild type (+/+) mice were generated from crossing heterozygous mice.
Epac Silencing
Predesigned siRNAs (Ambion Silencer, Life Technologies, Carlsbad, CA) targeting Epac1 and Epac2 have been used. A mix of two different sequences for each Epac isoform (RapGEF3 and RapGEF4) or scrambled siRNA were delivered by electroporation (Amaxa Nucleofector). After transfection EECs were cultured for 48 h under control conditions at 33 °C then transferred to 37 °C and stimulated by TGFβ1 for another 48 h to induce EMT. Efficiency of Epac knockdown was confirmed by Western blotting.
Real Time RT-PCR
Individual PE explants were harvested at the time of surgery (0 h) or after times in culture (12, 24, or 36 h). EECs where used at 33 °C as well as after transfer to 37 °C and after the subsequent addition of 250 pm TGF-β1. Total RNA extraction was completed with TRIzol (Invitrogen) as per the manufacturer's instructions. RT-PCR was carried out as described previously (26).
Membrane Fractionation and Co-immunoprecipitation Assays
Membrane and cytosolic proteins were separated as described previously (27). Co-immunoprecipitation assays on lysed cells are described previously (28).
Protein Phosphorylation
Cells were harvested in 10% cold trichloroacetic acid/acetone to preserve the phosphorylation state as described previously (29). For the detection of p190RhoGAP phosphorylation, cell lysates were immunoprecipitated with a mouse monoclonal anti-p190RhoGAP antibody and then blotted with a monoclonal antibody against phosphotyrosine (Cell Signaling Technology).
GTPase Activity Assays
RhoA-GTP was determined by precipitation of active GTP-bound RhoA (RhoA-GTP) with a glutathione S-transferase (GST) fusion protein of the Rho binding domain of the Rho effector Rhotekin (30). Active Rap-GTP pulldown was done with the GST-fused Ras binding domain of RalGDS (31).
RhoGEF Activity Assay
Recombinant GST-fused RhoA G17A mutant protein associated with glutathione Superflow Resin (Life Technologies) was produced and used to capture active RhoGEFs as described previously (32).
Lentiviral Infection of Epicardial Cells for GEFH1 and p63RhoGEF Silencing
For construction of shRNA vectors, a plasmid-based system was used (33). Six different target sequences specific for rat GEF-H1 protein (NCBI Reference Sequence: NM_001012079.1) were selected: 1) GTACCAAGGTCAAGCAGAA; 2) GGAGTCCCTTATTGATGAA; 3) TGAGTGACTTTGAGATGGA; 4) TGAGAACCTTGAAGATTAT; 5) AAAGGGATGTTTCTGATCA; 6) AGACGAAACTGCAGCAGAA. Sequences were first tested for efficacy in HEK293 T cells co-transfected with GEF-H1 and, respectively, with each of the GEF-H1 shRNAs or pLVHTM Empty. As pLVHTM GEF-H1 #1 was the most potent candidate for suppression of GEF-H1, it was used in our study of epicardial cells. To knock down p63RhoGEF, a previously published siRNA sequence (GCCAAGCTGGATGAAGATGAG) was used (28). shRNA constructs include GFP for measurement of transfections efficiency. Vectors pLVHTM, pMD2G, and psPAX2 were kind gifts from Dr. Ian Macara at the University of Virginia. Infected cells were left overnight at control conditions (33 °C, 5% CO2) for rest, and knockdown was induced by doxycycline (5 mg/ml). At 48 h cells were transferred to 37 °C and stimulated by TGFβ1 to induce EMT.
Immunofluorescence Staining
Cultured cells and isolated PE explants were fixed in 4% paraformaldehyde in PBS for 15 min and 48 h, respectively. Cells were permeabilized with 0.03% Triton X-100 for 5 min and then blocked with 3% bovine serum albumin and 1 mm sodium azide and then incubated with primary antibodies diluted in blocking solution overnight at 4 °C. Samples were quickly rinsed with PBS and then incubated for 1 h with secondary antibodies in blocking buffer. All images for a given experiment were acquired under identical laser power and gain settings using an Olympus Fluoview 300 confocal microscope (Olympus).
Protein Ligation Assay (PLA)
A PLA assay was performed in epicardial cells fixed and assayed according to the manufacturer's instructions (Olink).
Western Blotting
Proteins were transferred to polyvinylidene difluoride membranes and blocked with Odyssey Blocking Buffer and probed with primary antibodies in Blocking Buffer. Detection and quantification of the infrared signal were performed using the Odyssey system software (Li-Cor).
Antibodies
The following antibodies were used: mouse monoclonal anti-MYPT1 (1:1000 Western blot (WB); BD Biosciences); anti-RhoA (1:500 WB; Santa Cruz Biotechnology Inc.); anti-Rnd3 (1:100 WB; Millipore); anti-Rnd3 (1:400 for immunolabeling; Santa Cruz Biotechnology Inc.); anti-p190RhoGAP (1:500 WB and 1:1500 for immunolabeling; BD Transduction Laboratories); anti-SMA (SM α-actin, 1:200 for immunolabeling and 1:1000 for WB; Sigma); GAPDH (1:5000 WB; Millipore); rabbit polyclonal anti-MYPT1 Thr-853 (1:500 WB; Millipore); anti-MYPT1 Thr-696 (1:500 WB; Millipore); anti-E-cadherin (1:200 immunolabeling/1:2000 WB: Zymed Laboratories Inc.); anti-SM22 (1:200 immunolabeling/1:5000 WB, Millipore); anti-ZO-1 (1:200 immunolabeling/1:1000 WB: Zymed Laboratories Inc.); anti-LARG (1:100 WB, Santa Cruz Biotechnology Inc.); anti-vinculin (1:500 immunolabeling; Sigma); p63RhoGEF (GEFT) (1:200 WB; Proteintech Group Inc.); anti-vimentin (1:200 WB immunolabeling/1:1000 WB; Sigma); anti-Rap1 (1:500; Santa Cruz Biotechnology Inc.); anti-GEF H1 (1:200 WB; Cell Signaling); goat polyclonal anti-Rnd1 (1:2000 WB, Santa Cruz Biotechnology Inc.; anti-Epac1 (1:500 WB; Abcam); anti-Epac 2 (1:200 WB; Santa Cruz Biotechnology Inc.). Primary antibodies used for immunolabeling were visualized by an Alexa Fluor 488-, 594-, or 633-conjugated secondary antibody (1:1000; Molecular Probes). For WBs, primary antibodies were detected using either a goat anti-mouse Alexa 680 (1:10,000, Invitrogen) or a goat anti-rabbit IRDye800 (1:10,000, Rockland Immunochemicals)-conjugated secondary antibody.
Statistical Analysis
Data are presented as the means ± S.E. Statistical significance with respect to control was determined using the two-tailed Student's t test (Microsoft Excel). The level of significance was set at p < 0.05.
RESULTS
EMT in Epicardial Cell Outgrowth from E9.5 Proepicardia
The PE in E9.5 mouse embryos develops as a mesothelial outgrowth arising from the septum transversum in the region of the atrioventricular junction in the developing looped heart (Fig. 1A, panel a). Cells from the PE “grape-like” structure with villus projections (Fig. 1A, panel b) composed of simple cuboidal mesothelium migrate to the primitive heart where they attach to the developing myocardium (Fig. 1A, panel c). Epicardial outgrowth from explants of E9.5 embryos in the presence of serum formed a tightly packed sheet of epithelial cells with peripheral cells exhibiting lamellipodia and migratory behavior (Fig. 1B). After 36 h in culture the outgrowing epicardial cells expressed SMA and smooth muscle myosin heavy chain mRNA indicative of the onset of differentiation into SMCs (Fig. 1C). Immunolabeling of the PE outgrowth at 48 h showed that cells toward the center (Fig. 1D, panels d, e, and f), but not those at the periphery (Fig. 1D, panels a, b, and c), developed vinculin-containing focal adhesions and subcortical actin (Fig. 1D, panel a) reorganized into stress fibers (Fig. 1D, panels e and f), consistent with epicardial mesenchymal cells expressing SM marker proteins.
FIGURE 1.
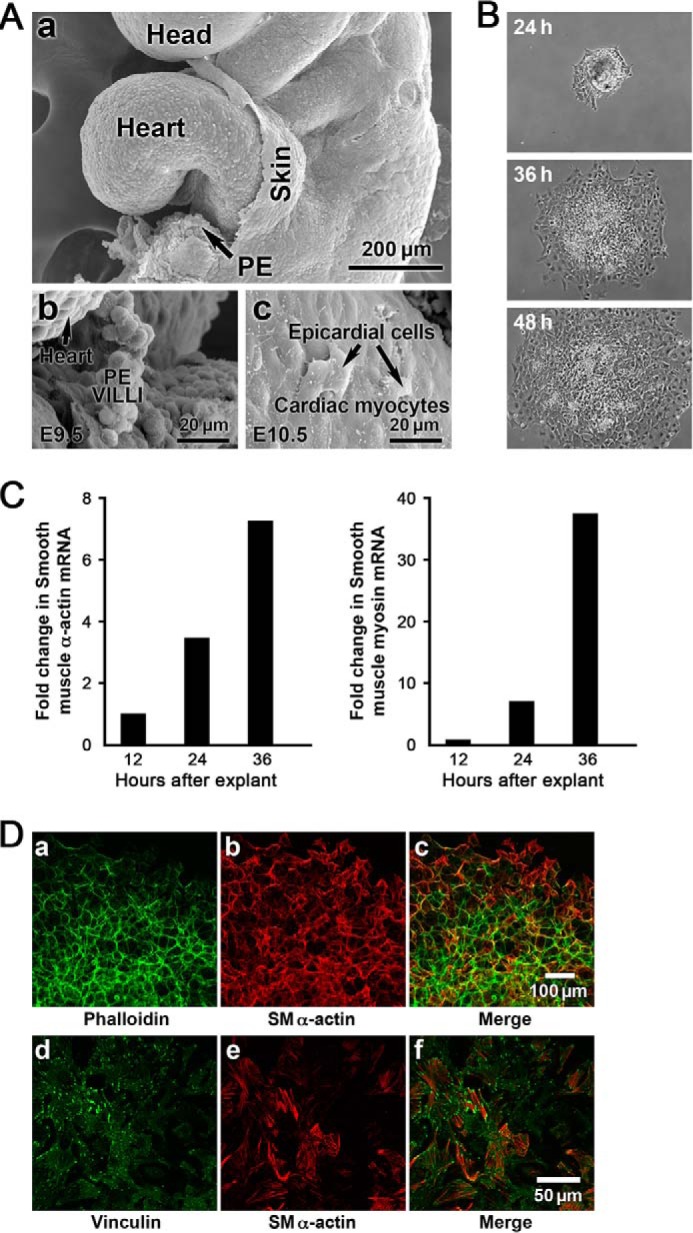
A, structure and characterization of the developing E9.5 mouse PE. The PE develops as a mesothelial outgrowth and arises from the septum transversum in the region of the atrioventricular junction in the developing looped heart. The scanning electron micrographs show the PE as a “grape-like” structure with villus projections (A, panels a and b) composed of cuboidal mesothelium. PE vesicles migrate to the primitive heart, and the epicardial mesothelial cells are shown attached and spread on the E10.5 cardiac myocytes of the heart (panel c)). These mesothelial cells are reported to undergo EMT and migrate into the heart differentiating into SMCs to form the walls of the coronary vessels (not shown). B, PE explants from mouse (E9.5) undergo EMT. At 24 h the PE was attached, and there was an outward migration of epicardial cells in all directions over time. C, SMA and SM myosin heavy chain mRNA expression increased over time. D, immunolabeling of the 48-h explants showed that cells toward the center (panels d, e, and f), but not those at the periphery (panels a, b, and c), developed vinculin-containing focal adhesions (panel d) and that subcortical actin (panel a) was now reorganized into stress fibers (panels e and f) consistent with epicardial mesenchymal cells expressing SM marker proteins.
TGF-β1 Induces EMT in Cultured EECs
Treatment of EECs with 250 pm TGF-β1 at 37 °C also significantly increased their expression of the SM markers SMA as well as SM22 by 24 h, the first time point measured, and this was maintained over 72 h (Fig. 2A). The increased expression was significantly inhibited by the Rho kinase (ROCK) inhibitor Y-27632, the Epac activator 007, and by C3 toxin that ADP-ribosylates and inhibits activation of RhoA. When maintained at 33 °C with interferon-γ (10 unit/ml), EECs displayed an epithelial-like phenotype; small cobblestone-shaped cells that clustered with adjacent cells displaying intense expression of E-cadherin, β-catenin, and ZO-1, especially at the sites of cell-cell contact as shown in (Fig. 2C, panels a, c, and e). Upon treatment with 250 pm TGF-β1 at 37 °C, these cells changed their phenotype to a SMC-like morphology with the appearance of actin stress fibers (panels h and j), cell-cell separation (panels b, d, f, h, and j), with loss of membrane associated E-cadherin (panel b) and increased expression of SMA (panel j) as shown by immunofluorescence staining. More E-cadherin and ZO-1 proteins were observed in the cytosolic compartments of these TGF-β1-treated cells compared with the undifferentiated cells. Western blots showed increased SMA and SM22 protein expression with TGF-β1 treatment (Fig. 2A). Altogether, the morphological changes and mRNA and protein expression data demonstrate that these epicardial cells have undergone EMT in vitro in response to TGF-β1.
FIGURE 2.

TGF-β1 treatment of EEC cells induced RhoA-mediated EMT. A, transcription levels of SMA and SM22 increased over a 24–72-h period in the presence of 250 pm TGF-β1. ROCK inhibitor Y-27632 Epac activator 007 and C3 treatment to inhibit RhoA activity partially suppressed or abolished this effect but were without effect on control cells (data not shown). GAPDH was used as a loading control. Data are presented as the means ± S.E., n = 3–24. Western blots show that SMA and SM22 protein expression also increased with TGF-β1 treatment (lower panel). B, TGF-β1 treatment of EECs for 72 h at 37 °C significantly increased RhoA activity as measured with the Rhotekin activity assay and with the phosphorylation of MYPT1 at the ROCK sites Thr-696 and Thr-853. The vertical lines in the Western blots indicate that an intervening band was deleted. C, distribution of E-cadherin, β-catenin, ZO1, and expression of actin (phalloidin) and SMA detected by immunolabeling of EECs under control conditions and with TGF-β1-induced EMT. Note that TGF-β1 treatment (panels b, d, and f) initiated separation of cell-cell contacts based on E-cadherin, β-catenin, and ZO1. The appearance of actin stress fibers (panels h and j) appeared only with TGF-β1 treatment.
Active Rho-GTP Is Required for EMT in EECs
RhoA activity has been shown to be required for EMT and for differentiation into coronary SMCs (14, 15). Here we demonstrated that RhoA-GTP activity is also required for the transformation of immortalized epicardial cells used in this study. Further support for the critical role of RhoA activity in EMT is shown in Figs. 2A and 5 (panels M–P), where pretreatment with inhibitory C3 toxin that ADP-ribosylates RhoA results in suppression of EMT as evidenced by decreased SM22 and SMA mRNA and protein expression. As shown in Fig. 2B, the Rho-GTP level was low in control cells maintained in the progenitor state. Rho activity is markedly increased when cells were stimulated with TGF-β1 at 37 °C. The activation of RhoA is consistent with the TGF-β1-induced RhoA-dependent increased expression of SMC marker genes, SMA and SM22, the appearance of actin stress fibers, and the ability of the Rho kinase inhibitor, Y-27632, to prevent the increase in SMA and SM22 mRNA (Fig. 2A) in EECs as well as the increased actin stress fibers in the PE epicardial cell outgrowth in cells undergoing EMT (Fig. 1D). The increased RhoA activity in the EECs was accompanied by a significant increase in phosphorylation of myosin light chain phosphatase regulatory subunit, MYPT1 at both Thr-696 and Thr-853, known ROCK sites (Fig. 2B), consistent with increased RhoA/ROCK activity.
FIGURE 5.

Activation of Epac with 007, partial silencing of Epac with shRNA, inhibition of cAMP phosphodiesterase with IBMX, or inhibition of RhoA by C3 toxin altered TGF-β1-induced EMT in EECs. The epicardial cells were treated with 250 pm TGF-β1 with/without 50 μm 007, 10 μm IBMX, or 3–5 μg/ml C3 toxin for 72 h followed by immunolabeling with antibodies for detection of SM22 and ZO1. TGF-β1 but not 007 treatment alone significantly increased the expression of SM22 protein (C versus B) and disrupted the cell-cell contacts as indicated with ZO-1 staining (G versus F), whereas these TGF-β1-induced changes were suppressed by treatment with either 007 (D and H), IBMX (L), or C3 toxin (P). Partial silencing of Epac1 by 45% ± 17 S.E., n = 8 (panel U), resulted in an increase in SM22 in both control and TGF-β1 treated EECs detected by immunohistochemistry (panels Q–T) consistent with Epac suppression of EMT (three independent experiments).
Increased Expression and Activity of the Rho GEFs p63RhoGEF and GEF-H1 with TGF-β1-induced EMT and EMT Suppression upon Partial GEF Silencing
Although Rho/ROCK activity is critical for EMT in epicardial cells, nothing is known about GTP exchange factors that activate the exchange of GTP for GDP on RhoA in these cells. Therefore, we carried out screens for changes in mRNA and protein expression of a group of RhoGEFs that we have found to be prevalent in SM (34). We found a significant increase in p63RhoGEF and GEF-H1 but not in leukemia-associated RhoGEF (LARG), PDZ-RhoGEF (PRG), VAV2, or p115RhoGEF at both the mRNA (data not shown) and protein levels (Fig. 3A). Although the decreased PRG content with TGF-β1 treatment was statistically significant, we do not expect it to contribute significantly to RhoA activation, as the bands are very lightly stained, unlike brain samples (data not shown), suggesting very low protein expression. This is consistent with a qRT-PCR screen of EECs for 25 RhoGEFs where mRNA expression of PRG did not significantly change with treatment and was 14-fold lower than GEF-H1 mRNA and at the margin of detectability (data not shown). The increase in p63RhoGEF and GEF-H1 protein expression suggests that they are likely to be functionally relevant in TGF-β1-induced EMT in epicardial cells. As p63RhoGEF is known to specifically directly couple to the heterotrimeric G-protein, Gαq/11 (28, 35, 36), whereas the coupling of GEF-H1 is less certain, we assayed protein expression levels and found that Gαq/11 or Gα12 proteins were expressed in both control and TGF-β1-treated cells, with no significant increase in protein expression detected in either Gαq/11 or Gα12, whereas Gα13 decreased by ∼25% (data not shown). LARG and PRG activity was not measured, as the detection level for protein expression was low and did not increase with stimulation. Both of these GEFs couple largely through Gα12/13. Therefore, it is unlikely that LARG and PRG play a major role in TGF-β1-induced activation of RhoA in epicardial cells. RhoGEF activity was next assayed using a G17ARhoA mutant to capture active GEFs. GEF-H1 activity were significantly greater after TGF-β1 treatment; although p63RhoGEF activity increased in all experiments (n = 4), it did not reach significance, suggesting that GEF-H1 and possibly p63RhoGEF drive the increased RhoA activity associated with EMT (Fig. 3B). No change in activity or expression level of VAV2 was detected. Gene silencing of either p63RhoGEF or GEF-H1 using lentiviral shRNA vectors (Fig. 3, C and D) significantly suppressed TGF-β1-induced expression of SMA. Control and empty vectors were without effect. Furthermore, silencing of p63RhoGEF did not significantly affect the TGF-β1-induced increase in GEF-H1 and vice versa (Fig. 3C). Note that additional controls are shown in Fig. 2A where TGF-β1 is shown to markedly increase SMA and SM22 protein expression compared with untreated controls. Altogether, p63RhoGEF and GEF-H1 significantly contribute to the activation of RhoA mediated EMT and the differentiation into SM cells.
FIGURE 3.

TGF-β1-induced EMT increased the expression and activity of p63RhoGEF and GEF-H1, but not VAV2 and EMT, and partial silencing of p63RhoGEF and GEF-H1 expression suppressed EMT as indicated by a decrease in SMA expression. A, protein expression, detected by Western blotting, of p63RhoGEF, GEF-H1, but not LARG, VAV2, or p115RhoGEF, significantly increased with TGF-β1 treatment, n = 12–16. Cells were stimulated with TGF-β1 (250 pm) for 72 h. The protein level of PDZRhoGEF (PRG) was decreased. n.s., not significant. B, GEF-H1, but not VAV2 activity, measured with a RhoG17A mutant to capture active RhoGEFs increased with TGF-β1-induced EMT; n = 3. A trend in increase of p63RhoGEF activity did not reach significance. RhoGEF protein levels were normalized to tubulin. Data are presented as the means ± S.E. C and D, lentiviral shRNAs to p63RhoGEF and GEF-H1 and empty vectors (controls) were introduced into mouse embryonic epicardial cells, and the extent of GEF suppression and SMA protein expression was measured by Western blotting 48 h after stimulation with TGF-β1, n = 3. Expression of SMA was significantly decreased with suppression of p63RhoGEF or GEF-H1.
Activation of Epac Suppresses and Silencing of Epac Enhances EMT in Cultured EECs and Cell Migration of Epicardial Cells
We previously demonstrated that activation of the cAMP-dependent, PKA-independent Rap1 GTP exchange factor, Epac, relaxed SM tissues through increased Rap1 activity that resulted in decreased RhoA activity (19). The Epac-specific nucleotide 8-pCPT-2′-O-Me-cAMP, referred to as 007, has been shown to specifically activate Rap1 (37, 38). In view of the importance of increased RhoA activity in EMT, we tested whether 007 blocks epicardial cell EMT. 007 significantly suppressed and treatment with the ROCK inhibitor Y-27632 completely inhibited TGF-β1-induced increases in SMA and SM22 mRNA (Fig. 2A) consistent with our findings in SM tissues (19). We next examined whether molecules in the Epac signaling pathway were expressed in cultured epicardial cells and proepicardia and whether Rap1 activity was present in the progenitor state and responsive to TGF-β1-induced EMT.
Epac1, Rap1, and p190RhoGAP proteins were present in EMCs as well as in isolated mouse PE (Fig. 4A). Active GTP-bound Rap1 was present in EMCs and increased further upon treatment with 007 to activate Epac (Fig. 4B). Rap1 was localized at cell-cell contacts and at the free surface membrane regions as well as at punctate perinuclear sites in 007-treated rat epicardial cells, EMCs (Fig. 4C). Furthermore, 007 inhibited EMT in epicardial cells from E10.5 heart explants grown in the presence of bFGF as evidenced by the compact outgrowth (Fig. 4D, panel c) with compact cells outlined by cortical actin (Fig. 4D, panel d). In the absence of 007, b-FGF treatment resulted in separated migratory cells with processes and stress fibers (Fig. 4D, panels a and b). To circumvent possible nonspecific effects of 007 treatment, epicardial cell migration from six WT and nine Epac null hearts was also examined (Fig. 4G). In the absence of Epac1, cell migration was enhanced with an increase in lamellipodia and a significant increase in cell separation indicated by a significant increase in distance between nuclei (WT 167 cells, Epac KO 73 cells) consistent with accelerated EMT. It is known that FGF promotes epicardial EMT and the migration of these cells into collagen gels (9). Therefore, we additionally measured the effects of 007 on cell migration into collagen gels. We found that the percentage of bFGF-treated epicardial cells from E10.5 heart explants, migrating into collagen gels, was significantly decreased in the presence of 007 (Fig. 4E). Furthermore, TGF-β1 markedly increased immunolabeled SM22 in EECs and disrupted cell-cell contacts as measured with ZO-1 immunolabeling in EECs (Fig. 5, panels C and G), and these effects were suppressed in the presence of 007 (Fig. 5, panels D and H). SM22 expression was enhanced with partial siRNA silencing of Epac in EECs both in basal conditions and after TGF-β1 treatment (Fig. 5, panels Q–T), providing further evidence that Epac signaling suppresses EMT. Pretreatment with the phosphodiesterase inhibitor, IBMX, to increase cAMP markedly suppressed the TGF-β1-induced increase in SM22 mRNA and protein expression (data not shown) and maintained cell-cell contacts imaged with ZO-1 immunolabeling (Fig. 5, panel L). We also found that mRNA expression of the phosphodiesterase PDE4D was increased 3-fold upon treatment of EECs with TGF-β1 (data not shown), consistent with cAMP/Epac signaling contributing to the progenitor phenotype and the loss of this activity with EMT. Finally, active Rap1 was high in the progenitor state and significantly decreased when EECs underwent EMT, as measured with the RalGDS pulldown assay (Fig. 4F). Altogether, these experiments support the hypothesis that cAMP/Epac signaling results in increased Rap1 activity suppressing epicardial EMT and contributing to preserving the SMC progenitor state through decreasing RhoA activity (19, 39).
FIGURE 4.

The cAMP target, Epac, suppresses EMT and cell migration in epicardial cell outgrowth from heart explants. A, Western blot showing that Epac1, p190RhoGAP, and Rap1 proteins were expressed in EMCs and the PE. B, the Epac activator, 007 (50 μm), increased active Rap1·GTP in EMCs. C, immunolabeling showed Rap1 localized to junctional membranes (arrows) and in the cytosol in 007 treated EMCs (panel a) stained with TRITC-labeled phalloidin (panel b) and co-localized with cortical actin (panel c). D, activation of Epac1 with 007 treatment suppressed EMT in epicardial cells grown out from E10.5 heart explants for 48 h in the presence of 50 ng of bFGF showing compact rounder cells outlined by cortical actin (panels c and d). bFGF treatment alone resulted in migratory cells with many processes (panel a) and actin stress fibers (panel b). E, migration of mesenchymal cells grown out from E10.5 heart explants into collagen gels in the presence of 50 ng of bFGF with/without 50 μm 007 for 6 days was retarded in the presence of 007. Counting of cells at different distances from the edge of the heart (distance = 0) is detailed under “Experimental Procedures.” F, Rap1 activity shown as a fraction of total Rap1 at 72 h decreased by 4-fold with TGF-β1-induced EMT in EECs consistent with a fall in Epac activity. Data are the means ± S.E., n = 3. G, epicardial cells grown out from WT and Epac1 null E10.5 heart explants for 48 h in the presence of 50 ng of bFGF, showing that the Epac1 null cells exhibit an increased migration indicated by the greater separation of the nuclei and the increased presence of lamellipodia; data for cell separation were from 6 WT and 9 Epac1 null hearts, number of cells counted were WT = 167 and Epac1 null = 73. The western blot shows absence of Epac1 protein in the Epac1 null cells.
Rnds and p190RhoGAP Activity Contribute to Suppressing RhoA Activity and EMT
As the Rnd proteins, which form a unique subfamily of Rho small GTPases, are known to antagonize RhoA activity through associating with and activating p190RhoGAP (22), we determined whether Rnd proteins play a role in maintaining the low RhoGTPase state of progenitor epicardial cells. Because the regulation of Rnd proteins is dependent on their expression level and post translational modification (21), we first measured the expression of Rnd1 and Rnd3 mRNA and protein levels in EECs. As shown in Fig. 6, A and B, both Rnd1 and Rnd3 mRNA and protein were detected in EECs. At the transcriptional level, Rnd1 but not Rnd3 mRNA was significantly decreased by TGF-β1-induced EMT; p < 0.05. However, at the protein level, after 72 h of TGF-β1 treatment, the expression of both Rnd1 and Rnd3 proteins were significantly decreased; p < 0.05. The decrease in these RhoA antagonists would be expected to result in an increase in RhoA activity in the transformed epicardial cells. Note that p190RhoGAP protein expression level was unchanged with EMT (Fig. 6B).
FIGURE 6.

Rnds 1 and 3 mRNA and protein were expressed in epicardial cells but were suppressed and their membrane/cytosol distribution changed in cells having undergone EMT. A, relative Rnd 1 and 3 mRNA expression in control and TGF-β1-treated EECs; n = 3. n.s., not significant. B, relative Rnd 1 and 3 and p190RhoGAP protein expression in control and TGF-β1-treated EECs n = 3. C, Rnds 1 and 3 were found in both the cytosolic (c) and the Triton-soluble fractions (m) in control EECs but decreased in the membrane fraction with TGF-β1 treatment for 72 h; n = 3. Data are the means ± S.E.
As an increase in RhoA activity activates EMT in epicardial cells and active prenylated RhoA translocates to the membrane (27, 40), we therefore examined whether the distribution of the antagonistic Rnd proteins was altered with TGF-β1 treatment. Rnd 1 and 3 were found in both the control Triton-soluble and the cytosolic fractions (Fig. 6C) and decreased in the Triton-soluble fraction when stimulated with TGF-β1 and expressed as a membrane/cytosolic ratio to correct for the decrease in Rnds upon stimulation (Fig. 6, A and B; n = 3, p < 0.05). Although p190RhoGAP was found predominantly in the cytosolic fraction and did not change in the Triton-soluble fraction with TGF-β1 treatment, this likely reflects the inability to maintain its localization with this fractionation technique, as unlike Rnds, it is neither farnesylated nor prenylated. Altogether, as active prenylated RhoA is membrane-associated, this decrease in antagonistic Rnds in the membrane fraction upon EMT supports the increased RhoA activity associated with EMT.
Based on the ability of Rnds to bind to and activate p190RhoGAP and antagonize RhoA in fibroblasts and epithelial cells (22), we next studied whether p190RhoGAP interacts with Rnd proteins in epicardial cells to suppress RhoA activity and EMT. By immunoprecipitation methods with anti-p190RhoGAP and anti-Rnd3 antibodies, we showed that endogenous p190RhoGAP coprecipitated Rnd3 and vice versa in EMCs (Fig. 7A). To further confirm the interaction, we performed a PLA assay that detects whether the p190RhoGAP and Rnd3 antibodies are in close proximity (<40 nm) in situ. Using rabbit polyclonal Rnd3 and mouse monoclonal p190RhoGAP antibodies, we found that endogenous Rnd3 and p190RhoGAP are in close proximity and that the “complex” was present in the peripheral cytoplasm but not in the nucleus or perinuclear space in epicardial cells (Fig. 7B). Control experiments for the PLA assay showed no signal for colocalization of p190RhoGAP and ROCK (Fig. 7C). Additional controls included standard immunohistochemistry for p190RhoGAP and Rnd3 (Fig. 7, D and E), that localized throughout the cytoplasm and perinuclear region. This suggests that both complex and free proteins exist in the cell and that there is some spatial segregation. In addition to its interaction with and activation by Rnd3, p190RhoGAP is regulated by a second mechanism, phosphorylation at Tyr-1105, that up-regulates GAP activity resulting in hydrolysis of GTP-RhoA (41). Note that we found p190RhoGAP expressed in PE explants, EMCs, and EECs (Figs. 4A, 6B, and 7D). Significant Tyr phosphorylated p190RhoGAP was detected in unstimulated EMCs (Fig. 7G), consistent with the presence of p190RhoGAP activity in the progenitor state. Furthermore, tyrosine-phosphorylated p190RhoGAP significantly decreased in EECs treated with TGF-β1 for 36 h compared with control cells (Fig. 7G), in keeping with the high RhoA activity associated with EMT and its low activity in the epicardial progenitor state.
FIGURE 7.

Rnd3 associates with p190RhoGAP in epicardial cells in the epithelial state; overall signaling scheme. A, anti-p190RhoGAP antibody, but not a nonspecific mouse IgG antibody, coimmunoprecipitated (IP) endogenous p190RhoGAP and Rnd 3. Anti-Rnd3 antibody, but not a nonspecific IgG, coimmunoprecipitated endogenous Rnd3 and p190RhoGAP. B, PLA demonstrating a proximity of <40 nm of Rnd3 and p190RhoGAP in EMCs as evidenced by each bright dot. C, control for PLA illustrating that under the same conditions no signal was observed for p190RhoGAP antibody and ROCK antibody. The presence of cells is indicated by the nuclear stain DAP1. D and E, control panels for the PLA assay showing the immunofluorescence of p190 RhoGAP and Rnd3 antibodies in these cells. G, p190RhoGAP activity, as indicated by Tyr-1105 phosphorylation, significantly decreases in the presence of 250 pm TGF-β1 (36 h) in EECs; n = 8. H, scheme of signaling pathways contributing to the maintenance of the proepicardial progenitor phenotype. EMT initiated by growth factors leads to activation of p63RhoGEF and GEF-H1 to exchange GTP for GDP and activate RhoA to stimulate the transcription of SM marker proteins such as SMA, SM myosin, and SM22 and transformation of the proepicardial cells into coronary vascular SM cells. RhoA activity is suppressed and kept in check in the proepicardial cells by the increased activity of p190RhoGAP and Rnd proteins as well as through signaling from cAMP to Epac, the GTP exchange factor for the small GTPase, Rap1, to further down-regulate RhoA activity.
DISCUSSION
In this study we identified signaling pathways that play key roles in the formation and maintenance of epicardial cells, a source of progenitors for coronary SMCs. We have confirmed and focused on the RhoA signaling pathway as it has been shown to play a critical role in epicardial EMT and serum response factor-dependent transcription of SM marker genes (14, 41–43), although upstream RhoGEFs responsible for its activation are unknown.
p63RhoGEF and GEF-H1 Drive RhoA-mediated EMT
Our study provides the first identification of GEF-H1 and p63RhoGEF as RhoGEFs that drive RhoA-mediated epicardial EMT and differentiation into coronary SM cells (Fig. 3). RhoA is known to up-regulate transcription of SM marker genes such as SMA (44), and our results indicate that p63RhoGEF and GEF-H1 are upstream regulators of these promotors. We also provide evidence that signaling through Epac/Rap1, p190RhoGAP, and Rnds, the RhoA antagonists, contribute to the maintenance of the progenitor state through their suppression of RhoA activity and of EMT (Fig. 7H). Using GST-RhoAG17A and Western blotting analysis to sediment active GEFs, we showed that the activity of GEF-H1 increased significantly with a consistent trend for an increase in p63RhoGEF but not VAV2, consistent with the changes in the expression level of GEF proteins with TGF-β1-induced EMT. The small increase in p63RhoGEF activity likely reflects processes that inactivate it during the 60-min incubation needed for the activity assay. These processes may be different for p63RhoGEF and GEF-H1. It is also possible that p63RhoGEF and GEF-H1 affinities for the RhoA mutant are different. Importantly, silencing of either p63RhoGEF or GEF-H1 using shRNAs suppressed the expression of TGF-β1-induced SMA expression, a marker for differentiation into SM cells, in further support for a significant role for p63RhoGEF. p63RhoGEF couples selectively and directly to the heterotrimeric G-protein Gαq/11 by agonists selective for this Gα subunit. Interaction of the activated Gαq/11 subunit with p63RhoGEF relieves its autoinhibited state (45). Although we detected Gαq/11 in epicardial cells, how TGF-β1 activates p63RhoGEF remains to be determined. One possibility is through a process called receptor transactivation, where G protein-coupled receptors are activated by growth factor receptor-tyrosine kinases (46). Interestingly, GEF-H1 has been shown in fibroblasts to be activated by stretch through integrins (47), and this is a potential mechanism for GEF-H1 in epicardial cells as they undergo cyclical stretch when attached to the beating embryonic heart. GEF-H1 has also been identified in retinal pigment epithelium where TGF-β1-induced dedifferentiation results in RhoA-mediated SMA expression that is suggested to occur in diseased epithelium (48), further linking GEF-H1 to TGF-β1 stimulation. Apart from our identification for the first time of RhoGEFs mediating the increased RhoA activity, a sine qua non of EMT in the PE, our findings also provide another example in support of the concept that different RhoGEFs are selectively activated to target discrete RhoA-mediated pathways such as has been shown for salt-induced hypertension or basal blood pressure (28, 49, 50). This may reflect the fact that RhoGEFs have multiple domains in addition to their characteristic tandem Dbl and pleckstrin homology domains that catalyze nucleotide exchange on RhoA, allowing these enzymes to activate additional signaling pathways. Understanding the function of domains in p63RhoGEF and GEF-H1 is an important next step.
Epac Activation Suppresses and Partial Silencing of Epac Enhances EMT and Cell Migration in Epicardial Cells
cAMP activity is known to decrease with TGF-β1-induced EMT, which in turn can be inhibited by treatments that increase cyclic nucleotides as shown in other cell types (16, 18). Increased expression and activity of cAMP PDEs with a fall in cAMP levels occur during EMT (18). Of the 11 cyclic nucleotide PDE family members, only PDE4A and PDE4D mRNA levels were increased in human alveolar epithelial cells after TGF-β1-induced EMT, resulting in a 2-fold decrease in cAMP due to PDE activity (18). Furthermore, these authors also demonstrated that inhibition of PDE4D attenuated EMT in these cells. Reduced PDE expression and activity have also been implicated in the vascular smooth muscle synthetic phenotype associated with vascular damage (51). In keeping with these findings, we now find that PDE4D mRNA significantly increased with TGF-β1 treatment of epicardial cells, and SM22 expression was suppressed in the presence of the PDE inhibitor IBMX (data not shown). Furthermore, the TGF-β1-induced loss of localization of the epithelial marker ZO1 at cell-cell contacts is reversed with the general PDE inhibitor IBMX (Fig. 5, K versus L) as well as the PDE4-specific inhibitor, Rolipram. Thus, cAMP also plays a critical role in the suppression of EMT in epicardial progenitor cells.
Although there is good evidence for a significant role of cAMP activity in EMT, which is assumed to be entirely through activation of PKA signaling, the direct involvement of the cAMP-activated Rap1 GEF, Epac, in the mechanism of EMT has not been reported. We find that Epac and Rap1 are present in the PE and EMCs and that partial silencing of Epac in EECs accelerated EMT as evidenced by increased expression of SM22 at both the progenitor state and after TGF-β1 treatment. Our findings demonstrating that the Epac-selective cAMP analogue, 8-pCPT-2′-O-Me-cAMP (007), increased Rap1-GTP and inhibited TGF-β1-induced expression of SM22 and SMA mRNA and protein as well as inhibited TGF-β1-induced dissociation of cell-cell adhesions and morphological changes, also strongly support a role for Epac in contributing to maintenance of the epicardial epithelial phenotype. The decrease in Rap1A-GTP during EMT is consistent with a loss of Epac activity with TGF-β1-induced EMT. In addition, a role for Epac/Rap1 in maintenance of the epicardial progenitor state is further supported by our demonstration that increased cell migration and development of stress fibers upon growth factor stimulation are both inhibited by treatment with 007 and enhanced by partial silencing of Epac in epicardial cells growing out of E10.5 heart explants. We previously showed in SM tissues and cultured SMCs that activation of Epac results in a PKA-independent, Rap1-dependent Ca2+ desensitization of force through down-regulation of RhoA activity (19). An increase in Rap1-GTP after activation of cAMP also results in a decrease in RhoA activity in endothelial cells and neurons (52–54). The decreased RhoA activity was suggested to reflect a Rap1-activated RhoGAP (19, 53). In view of the increased RhoA activity during transformation of epicardial cells (14), we suggest that a Rap1-activated RhoGAP may also operate in the untransformed epicardial cells to maintain the low RhoA activity observed in the epicardial progenitor state (Fig. 2B), yet the molecular mechanism remains to be determined in future studies. Active Rap1 has also been shown in cultured epithelial cells to localize to and regulate the formation of E-cadherin-based cell-cell contacts (55). Thus, our observed Epac-activated Rap1 localized to cell membrane regions in the epicardial cells may also contribute to maintaining cell-cell contacts before EMT. In conclusion, our findings provide evidence for a significant role for cAMP signaling through the Epac pathway in the maintenance of the epicardial progenitor state and for down-regulation of cAMP in EMT via an increase in PDE expression and activity leading to lower cAMP and Epac/Rap1 levels resulting in an increase RhoA activity to promote EMT.
Rnd Proteins and p190RhoGAP Activity Inhibit RhoA Activity and EMT
Rnd proteins, known to be potent RhoA antagonists, unlike other small GTPases, are always bound to GTP and are regulated by their expression level and by post-translational modification mechanisms such as phosphorylation, subcellular localization, and proteasomal degradation (21, 56, 57) rather than by a GDP/GTP molecular switch. We show for the first time that Rnd proteins are present in epicardial cells and that they are regulated through changes in expression level, being expressed in the progenitor state and decreased with EMT. We suggest that they play a significant role in antagonizing RhoA activity to maintain the epicardial cell progenitor state. Their ability to abolish stress fibers and adhesions leading to rounding of cells gave rise to the terminology, Rnds (58). Indeed, the round cells in the PE (Fig. 1) and their lack of stress fibers before transformation are consistent with the Rnd phenotype. Rnd proteins have been shown to function as RhoA antagonists by interacting with and activating p190RhoGAP to reduce cellular levels of RhoA-GTP (22). p190RhoGAP is activated through Tyr-1105 phosphorylation upon treatment with the α4β3 integrin ligand, VCAM-1, in adult rat epicardial cells to inhibit EMT (20). However, the role of p190RhoGAP and Rnd proteins in epicardial progenitor cells has been largely unknown. We now demonstrate in embryonic epicardial cells through co-immunoprecipitation and co-localization that Rnd3 interacts and co-localizes with p190RhoGAP in progenitor epicardial cells. This is expected to lead to stabilization of Rnds, preventing their degradation (59). On the other hand, the fall in Rnd expression associated with the increase in RhoA/ROCK activity in EMT is likely due to the known ability of ROCK to phosphorylate Rnd3 and increase its turnover (59) and to phosphorylate and inhibit p190RhoGAP (60). We found Rnds and p190RhoGAP distributed in both the cytosol and at the cell plasma membrane region, although we cannot say how much is in a complex. We suggest that ROCK inhibitory phosphorylation of Rnds and p190RhoGAP serves as a feedback mechanism to silence Rnd/p190RhoGAP antagonism of RhoA activity upon EMT as has been suggested for other cells (59). In explanted epicardium, EMT is progressive and not reversible, consistent with such a feedback mechanism (14). The catalytic activity of p190RhoGAP is up- and down-regulated, respectively, by phosphorylation and dephosphorylation at Tyr-1105 (41, 61) through tyrosine kinases such as c-Src and cAbl or tyrosine phosphatases such as SHP2, PTP-PEST, or LMW-PTP (62, 63). We report significant phosphorylation of p190RhoGAP Tyr-1105 in EECs consistent with p190RhoGAP activity contributing to the suppression of RhoA activity in untransformed epicardial cells. In SMCs angiotensin II has been reported to increase RhoA activity through SHP2-mediated dephosphorylation of p190RhoGAP Tyr-1105 in SM (62). However, we do not know whether a Tyr kinase, phosphatase, or both regulate p190RhoGAP activity in untransformed epicardial cells, an area of future study. Rnd3 is reported to localize to both membrane and cytosolic fractions (64). Rnds are farnesylated, accounting for their association with cell membranes (65). Our membrane fractionation data showed Rnds in both the cytosolic- and detergent-soluble fractions, in agreement with our immunolocalization findings, and that more Rnd proteins were localized to the Triton-soluble membrane fraction in the control cells compared with TGF-β1-treated cells. When SM is stimulated, RhoA translocates to the membrane (27). These findings are consistent with Rnds-antagonizing membrane-associated RhoA activity and conserving the progenitor state of epicardial cells through their binding and activation of p190RhoGAP. Altogether, we propose that RhoA activity is kept in check in the epicardial cell progenitor state through both expression of Rnds 1 and 3, which in turn activate p190RhoGAP, as well as through activation of GAP activity through p190RhoGAP Tyr-1105 phosphorylation.
In conclusion, we have identified new signaling molecules, p63RhoGEF, GEF-H1, Epac, Rap1, Rnds, and p190RhoGAP, that act together to control RhoA activity and play key roles in the maintenance of vascular smooth muscle progenitor cells in the embryonic epicardium. Although it has been suggested that adult epicardial cells lose their ability to undergo EMT during later stages of embryogenesis (20), considerable evidence supports the possibility that the epicardium can serve as a source of vascular progenitors for cell based therapies (1, 6). Altogether, if these pathways also regulate progenitor cells in the adult epicardium, understanding the signaling molecules regulating RhoA activity that function to maintain the progenitor phenotype of the embryonic epicardium might eventually lead to their manipulation in order to facilitate a robust and stable neovascularization or myocardial regeneration of the injured adult heart (7).
Footnotes
- SMC
- smooth muscle (SM) cell
- EMT
- mesenchymal transition
- GEF
- GTPase exchange factor
- GAP
- GTP activating protein
- ROCK
- Rho kinase
- EMC
- epicardial/mesothelial cell
- EEC
- embryonic epicardial cell
- PE
- proepicardium
- PDE
- phosphodiesterase
- IBMX
- isobutylmethylxanthine
- PLA
- protein ligation assay
- WB
- Western blot
- PRG
- PDZ-RhoGEF
- LARG
- leukemia-associated RhoGEF
- bFGF
- bovine FGF.
REFERENCES
- 1. Tang X. L., Rokosh D. G., Guo Y., Bolli R. (2010) Cardiac progenitor cells and bone marrow-derived very small embryonic-like stem cells for cardiac repair after myocardial infarction. Circ. J. 74, 390–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou B., Ma Q., Rajagopal S., Wu S. M., Domian I., Rivera-Feliciano J., Jiang D., von Gise A., Ikeda S., Chien K. R., Pu W. T. (2008) Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Winter E. M., Gittenberger-de Groot A. C. (2007) Epicardium-derived cells in cardiogenesis and cardiac regeneration. Cell. Mol. Life Sci. 64, 692–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muñoz-Chápuli R., González-Iriarte M., Carmona R., Atencia G., Macías D., Pérez-Pomares J. M. (2002) Cellular precursors of the coronary arteries. Tex. Heart Inst. J. 29, 243–249 [PMC free article] [PubMed] [Google Scholar]
- 5. Cai C. L., Martin J. C., Sun Y., Cui L., Wang L., Ouyang K., Yang L., Bu L., Liang X., Zhang X., Stallcup W. B., Denton C. P., McCulloch A., Chen J., Evans S. M. (2008) A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou B., Pu W. T. (2008) More than a cover: epicardium as a novel source of cardiac progenitor cells. Regen. Med. 3, 633–635 [DOI] [PubMed] [Google Scholar]
- 7. Smart N., Risebro C. A., Melville A. A., Moses K., Schwartz R. J., Chien K. R., Riley P. R. (2007) Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature 445, 177–182 [DOI] [PubMed] [Google Scholar]
- 8. Merki E., Zamora M., Raya A., Kawakami Y., Wang J., Zhang X., Burch J., Kubalak S. W., Kaliman P., Izpisua Belmonte J. C., Chien K. R., Ruiz-Lozano P. (2005) Epicardial retinoid X receptor α is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. U.S.A. 102, 18455–18460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morabito C. J., Dettman R. W., Kattan J., Collier J. M., Bristow J. (2001) Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev. Biol. 234, 204–215 [DOI] [PubMed] [Google Scholar]
- 10. Tevosian S. G., Deconinck A. E., Tanaka M., Schinke M., Litovsky S. H., Izumo S., Fujiwara Y., Orkin S. H. (2000) FOG-2, a cofactor for GATA transcription factors, is essential for heart morphogenesis and development of coronary vessels from epicardium. Cell 101, 729–739 [DOI] [PubMed] [Google Scholar]
- 11. Dettman R. W., Denetclaw W., Jr., Ordahl C. P., Bristow J. (1998) Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 193, 169–181 [DOI] [PubMed] [Google Scholar]
- 12. Gittenberger-de Groot A. C., Vrancken Peeters M. P., Mentink M. M., Gourdie R. G., Poelmann R. E. (1998) Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 82, 1043–1052 [DOI] [PubMed] [Google Scholar]
- 13. Pérez-Pomares J. M., Macías D., García-Garrido L., Muñoz-Chápuli R. (1997) Contribution of the primitive epicardium to the subepicardial mesenchyme in hamster and chick embryos. Dev. Dyn. 210, 96–105 [DOI] [PubMed] [Google Scholar]
- 14. Lu J., Landerholm T. E., Wei J. S., Dong X. R., Wu S. P., Liu X., Nagata K., Inagaki M., Majesky M. W. (2001) Coronary smooth muscle differentiation from proepicardial cells requires rhoA-mediated actin reorganization and p160 rho-kinase activity. Dev. Biol. 240, 404–418 [DOI] [PubMed] [Google Scholar]
- 15. Landerholm T. E., Dong X. R., Lu J., Belaguli N. S., Schwartz R. J., Majesky M. W. (1999) A role for serum response factor in coronary smooth muscle differentiation from proepicardial cells. Development 126, 2053–2062 [DOI] [PubMed] [Google Scholar]
- 16. Zhang A., Dong Z., Yang T. (2006) Prostaglandin D2 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in MDCK cells. Am. J. Physiol. Renal Physiol. 291, F1332–F1342 [DOI] [PubMed] [Google Scholar]
- 17. Santibáñez J. F., Olivares D., Guerrero J., Martínez J. (2003) Cyclic AMP inhibits TGFβ1-induced cell-scattering and invasiveness in murine-transformed keratinocytes. Int. J. Cancer 107, 715–720 [DOI] [PubMed] [Google Scholar]
- 18. Kolosionek E., Savai R., Ghofrani H. A., Weissmann N., Guenther A., Grimminger F., Seeger W., Banat G. A., Schermuly R. T., Pullamsetti S. S. (2009) Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: the role of phosphodiesterase 4. Mol. Biol. Cell 20, 4751–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zieba B. J., Artamonov M. V., Jin L., Momotani K., Ho R., Franke A. S., Neppl R. L., Stevenson A. S., Khromov A. S., Chrzanowska-Wodnicka M., Somlyo A. V. (2011) The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J. Biol. Chem. 286, 16681–16692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dokic D., Dettman R. W. (2006) VCAM-1 inhibits TGFβ stimulated epithelial-mesenchymal transformation by modulating Rho activity and stabilizing intercellular adhesion in epicardial mesothelial cells. Dev. Biol. 299, 489–504 [DOI] [PubMed] [Google Scholar]
- 21. Chardin P. (2006) Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 7, 54–62 [DOI] [PubMed] [Google Scholar]
- 22. Wennerberg K., Forget M. A., Ellerbroek S. M., Arthur W. T., Burridge K., Settleman J., Der C. J., Hansen S. H. (2003) Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr. Biol. 13, 1106–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cario-Toumaniantz C., Reillaudoux G., Sauzeau V., Heutte F., Vaillant N., Finet M., Chardin P., Loirand G., Pacaud P. (2003) Modulation of RhoA-Rho kinase-mediated Ca2+ sensitization of rabbit myometrium during pregnancy: role of Rnd3. J. Physiol. 552, 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wada A. M., Smith T. K., Osler M. E., Reese D. E., Bader D. M. (2003) Epicardial/mesothelial cell line retains vasculogenic potential of embryonic epicardium. Circ. Res. 92, 525–531 [DOI] [PubMed] [Google Scholar]
- 25. Austin A. F., Compton L. A., Love J. D., Brown C. B., Barnett J. V. (2008) Primary and immortalized mouse epicardial cells undergo differentiation in response to TGFβ. Dev. Dyn 237, 366–376 [DOI] [PubMed] [Google Scholar]
- 26. Momotani K., Khromov A. S., Miyake T., Stukenberg P. T., Somlyo A. V. (2008) Cep57, a multidomain protein with unique microtubule and centrosomal localization domains. Biochem. J. 412, 265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gong M. C., Fujihara H., Somlyo A. V., Somlyo A. P. (1997) Translocation of RhoA associated with Ca2+ sensitization of smooth muscle. J. Biol. Chem. 272, 10704–10709 [DOI] [PubMed] [Google Scholar]
- 28. Momotani K., Artamonov M. V., Utepbergenov D., Derewenda U., Derewenda Z. S., Somlyo A. V. (2011) p63RhoGEF couples Gαq/11-mediated signaling to Ca2+ sensitization of vascular smooth muscle contractility. Circ. Res. 109, 993–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neppl R. L., Lubomirov L. T., Momotani K., Pfitzer G., Eto M., Somlyo A. V. (2009) Thromboxane A2-induced bi-directional regulation of cerebral arterial tone. J. Biol. Chem. 284, 6348–6360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren X. D., Schwartz M. A. (2000) Determination of GTP loading on Rho. Methods Enzymol. 325, 264–272 [DOI] [PubMed] [Google Scholar]
- 31. van Triest M., Bos J. L. (2004) Pulldown assays for guanoside 5′-triphosphate-bound Ras-like guanosine 5′-triphosphatases. Methods Mol. Biol. 250, 97–102 [DOI] [PubMed] [Google Scholar]
- 32. García-Mata R., Wennerberg K., Arthur W. T., Noren N. K., Ellerbroek S. M., Burridge K. (2006) Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 406, 425–437 [DOI] [PubMed] [Google Scholar]
- 33. Szulc J., Wiznerowicz M., Sauvain M. O., Trono D., Aebischer P. (2006) A versatile tool for conditional gene expression and knockdown. Nat. Methods 3, 109–116 [DOI] [PubMed] [Google Scholar]
- 34. Artamonov M. V., Momotani K., Stevenson A., Trentham D. R., Derewenda U., Derewenda Z. S., Read P. W., Gutkind J. S., Somlyo A. V. (2013) Agonist-induced Ca2+ sensitization in smooth muscle: redundancy of Rho guanine nucleotide exchange factors (RhoGEFs) and response kinetics, a caged compound study. J. Biol. Chem. 288, 34030–34040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Momotani K., Somlyo A. V. (2012) p63RhoGEF: A New switch for Gq-mediated activation of smooth muscle. Trends Cardiovasc. Med. 22, 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shankaranarayanan A., Boguth C. A., Lutz S., Vettel C., Uhlemann F., Aittaleb M., Wieland T., Tesmer J. J. (2010) Gαq allosterically activates and relieves autoinhibition of p63RhoGEF. Cell. Signal. 22, 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Christensen A. E., Selheim F., de Rooij J., Dremier S., Schwede F., Dao K. K., Martinez A., Maenhaut C., Bos J. L., Genieser H. G., Døskeland S. O. (2003) cAMP analog mapping of Epac1 and cAMP kinase: discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J. Biol. Chem. 278, 35394–35402 [DOI] [PubMed] [Google Scholar]
- 38. Rehmann H., Schwede F., Døskeland S. O., Wittinghofer A., Bos J. L. (2003) Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J. Biol. Chem. 278, 38548–38556 [DOI] [PubMed] [Google Scholar]
- 39. Roscioni S. S., Maarsingh H., Elzinga C. R., Schuur J., Menzen M., Halayko A. J., Meurs H., Schmidt M. (2011) Epac as a novel effector of airway smooth muscle relaxation. J. Cell Mol. Med. 15, 1551–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Somlyo A. P., Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 41. Bernards A., Settleman J. (2004) GAP control: regulating the regulators of small GTPases. Trends Cell Biol. 14, 377–385 [DOI] [PubMed] [Google Scholar]
- 42. Mack C. P., Somlyo A. V., Hautmann M., Somlyo A. P., Owens G. K. (2001) Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 276, 341–347 [DOI] [PubMed] [Google Scholar]
- 43. Wamhoff B. R., Bowles D. K., McDonald O. G., Sinha S., Somlyo A. P., Somlyo A. V., Owens G. K. (2004) L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ. Res. 95, 406–414 [DOI] [PubMed] [Google Scholar]
- 44. Mack C. P., Owens G. K. (1999) Regulation of smooth muscle α-actin expression in vivo is dependent on CArG elements within the 5′ and first intron promoter regions. Circ. Res. 84, 852–861 [DOI] [PubMed] [Google Scholar]
- 45. Rossman K. L., Cheng L., Mahon G. M., Rojas R. J., Snyder J. T., Whitehead I. P., Sondek J. (2003) Multifunctional roles for the PH domain of Dbs in regulating Rho GTPase activation. J. Biol. Chem. 278, 18393–18400 [DOI] [PubMed] [Google Scholar]
- 46. Patel T. B. (2004) Single transmembrane spanning heterotrimeric g protein-coupled receptors and their signaling cascades. Pharmacol. Rev. 56, 371–385 [DOI] [PubMed] [Google Scholar]
- 47. Guilluy C., Swaminathan V., Garcia-Mata R., O'Brien E. T., Superfine R., Burridge K. (2011) The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 13, 722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsapara A., Luthert P., Greenwood J., Hill C. S., Matter K., Balda M. S. (2010) The RhoA activator GEF-H1/Lfc is a transforming growth factor-β target gene and effector that regulates α-smooth muscle actin expression and cell migration. Mol. Biol. Cell 21, 860–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wirth A., Benyó Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Orsy P., Horváth B., Maser-Gluth C., Greiner E., Lemmer B., Schütz G., Gutkind J. S., Offermanns S. (2008) G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 14, 64–68 [DOI] [PubMed] [Google Scholar]
- 50. Wuertz C. M., Lorincz A., Vettel C., Thomas M. A., Wieland T., Lutz S. (2010) p63RhoGEF: a key mediator of angiotensin II-dependent signaling and processes in vascular smooth muscle cells. FASEB J. 24, 4865–4876 [DOI] [PubMed] [Google Scholar]
- 51. Dunkerley H. A., Tilley D. G., Palmer D., Liu H., Jimmo S. L., Maurice D. H. (2002) Reduced phosphodiesterase 3 activity and phosphodiesterase 3A level in synthetic vascular smooth muscle cells: implications for use of phosphodiesterase 3 inhibitors in cardiovascular tissues. Mol. Pharmacol. 61, 1033–1040 [DOI] [PubMed] [Google Scholar]
- 52. Cullere X., Shaw S. K., Andersson L., Hirahashi J., Luscinskas F. W., Mayadas T. N. (2005) Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105, 1950–1955 [DOI] [PubMed] [Google Scholar]
- 53. Yamada T., Sakisaka T., Hisata S., Baba T., Takai Y. (2005) RA-RhoGAP, Rap-activated Rho GTPase-activating protein implicated in neurite outgrowth through Rho. J. Biol. Chem. 280, 33026–33034 [DOI] [PubMed] [Google Scholar]
- 54. Birukova A. A., Zagranichnaya T., Alekseeva E., Bokoch G. M., Birukov K. G. (2008) Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J. Cell Physiol. 215, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hogan C., Serpente N., Cogram P., Hosking C. R., Bialucha C. U., Feller S. M., Braga V. M., Birchmeier W., Fujita Y. (2004) Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol. Cell. Biol. 24, 6690–6700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Madigan J. P., Bodemann B. O., Brady D. C., Dewar B. J., Keller P. J., Leitges M., Philips M. R., Ridley A. J., Der C. J., Cox A. D. (2009) Regulation of Rnd3 localization and function by protein kinase C α-mediated phosphorylation. Biochem. J. 424, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lonjedo M., Poch E., Mocholí E., Hernández-Sánchez M., Ivorra C., Franke T. F., Guasch R. M., Pérez-Roger I. (2013) The Rho family member RhoE interacts with Skp2 and is degraded at the proteasome during cell cycle progression. J. Biol. Chem. 288, 30872–30882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nobes C. D., Lauritzen I., Mattei M. G., Paris S., Hall A., Chardin P. (1998) A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J. Cell Biol. 141, 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goh L. L., Manser E. (2012) The GTPase-deficient Rnd proteins are stabilized by their effectors. J. Biol. Chem. 287, 31311–31320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mori K., Amano M., Takefuji M., Kato K., Morita Y., Nishioka T., Matsuura Y., Murohara T., Kaibuchi K. (2009) Rho-kinase contributes to sustained RhoA activation through phosphorylation of p190A RhoGAP. J. Biol. Chem. 284, 5067–5076 [DOI] [PubMed] [Google Scholar]
- 61. Roof R. W., Haskell M. D., Dukes B. D., Sherman N., Kinter M., Parsons S. J. (1998) Phosphotyrosine (p-Tyr)-dependent and -independent mechanisms of p190 RhoGAP-p120 RasGAP interaction: Tyr-1105 of p190, a substrate for c-Src, is the sole p-Tyr mediator of complex formation. Mol. Cell. Biol. 18, 7052–7063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bregeon J., Loirand G., Pacaud P., Rolli-Derkinderen M. (2009) Angiotensin II induces RhoA activation through SHP2-dependent dephosphorylation of the RhoGAP p190A in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 297, C1062–C1070 [DOI] [PubMed] [Google Scholar]
- 63. Sastry S. K., Rajfur Z., Liu B. P., Cote J. F., Tremblay M. L., Burridge K. (2006) PTP-PEST couples membrane protrusion and tail retraction via VAV2 and p190RhoGAP. J. Biol. Chem. 281, 11627–11636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Riento K., Guasch R. M., Garg R., Jin B., Ridley A. J. (2003) RhoE binds to ROCK I and inhibits downstream signaling. Mol. Cell. Biol. 23, 4219–4229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Foster R., Hu K. Q., Lu Y., Nolan K. M., Thissen J., Settleman J. (1996) Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol. Cell. Biol. 16, 2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]