

Background: TEM-1 β-lactamase hydrolyzes penicillins and early cephalosporins but not oxyimino-cephalosporins.
Results: A TEM-1 triple mutant, W165Y/E166Y/P167G, exhibits ceftazidime hydrolysis and a large active site conformational change.
Conclusion: The mutant has an enlarged active site to accommodate ceftazidime and an alternative catalytic residue, Tyr-166.
Significance: The study reveals plasticity in β-lactamase structure and mechanism in the evolution of altered substrate specificity.
Keywords: Antibiotic Resistance, Enzyme Catalysis, Enzyme Kinetics, Enzyme Structure, Protein Stability, Protein Structure, X-ray Crystallography, Beta-Lactamase, Enzyme Evolution
Abstract
β-Lactamases are bacterial enzymes that hydrolyze β-lactam antibiotics. TEM-1 is a prevalent plasmid-encoded β-lactamase in Gram-negative bacteria that efficiently catalyzes the hydrolysis of penicillins and early cephalosporins but not oxyimino-cephalosporins. A previous random mutagenesis study identified a W165Y/E166Y/P167G triple mutant that displays greatly altered substrate specificity with increased activity for the oxyimino-cephalosporin, ceftazidime, and decreased activity toward all other β-lactams tested. Surprisingly, this mutant lacks the conserved Glu-166 residue critical for enzyme function. Ceftazidime contains a large, bulky side chain that does not fit optimally in the wild-type TEM-1 active site. Therefore, it was hypothesized that the substitutions in the mutant expand the binding site in the enzyme. To investigate structural changes and address whether there is an enlargement in the active site, the crystal structure of the triple mutant was solved to 1.44 Å. The structure reveals a large conformational change of the active site Ω-loop structure to create additional space for the ceftazidime side chain. The position of the hydroxyl group of Tyr-166 and an observed shift in the pH profile of the triple mutant suggests that Tyr-166 participates in the hydrolytic mechanism of the enzyme. These findings indicate that the highly conserved Glu-166 residue can be substituted in the mechanism of serine β-lactamases. The results reveal that the robustness of the overall β-lactamase fold coupled with the plasticity of an active site loop facilitates the evolution of enzyme specificity and mechanism.
Introduction
An important mechanism of bacterial antibiotic resistance is the production of β-lactamases that catalyze the hydrolysis of β-lactam antibiotics (1). β-Lactamases are divided into four classes (A, B, C, and D) based on the sequence homology and mechanism of hydrolysis (2). Class A, C, and D β-lactamases utilize an active site serine residue to hydrolyze the antibiotics, whereas Class B includes metallo-β-lactamases that utilize one or two Zn2+ ions to stabilize and position an hydroxide ion to attack the β-lactam ring (2–5).
Class A β-lactamases share a common catalytic mechanism for the hydrolysis of β-lactam antibiotics that involves several highly conserved structural elements. Activation of the catalytic Ser-70 hydroxyl group initiates nucleophilic attack on the β-lactam carbonyl carbon and opening of the β-lactam ring to form a covalently linked acyl-enzyme intermediate. The subsequent nucleophilic attack by an activated catalytic water molecule leads to deacylation of the acyl-enzyme species, release of the inactivated antibiotic, and regeneration of the free enzyme (6, 7).
TEM-1 β-lactamase is a plasmid-encoded, class A enzyme with high catalytic efficiency (kcat/Km) for the hydrolysis of penicillins and early cephalosporins but not oxyimino-cephalosporins such as ceftazidime (8, 9). The extensive use of oxyimino-cephalosporins has led to the emergence of variants of TEM-1 that can more efficiently hydrolyze these drugs. These enzymes, termed extended spectrum β-lactamases, typically contain 1–5 amino acid substitutions that change the substrate profile of the enzyme. Over 200 unique variants of the TEM-1 enzyme have been identified (8–10).
TEM-1 β-lactamase is a 29-kDa enzyme and consists of one α and one α/β domain with the active site cavity located in a groove between the two domains (11, 12). The catalytic serine, Ser-70, is located on a helix (H2) along with Lys-73, which is proposed to participate, along with Glu-166 and an activated water molecule, to catalyze the initial acylation step (13, 14). A second active site serine residue, Ser-130, is located in the SDN loop and may participate in catalysis by shuttling a proton to the amide nitrogen during or after disruption of the amide bond (5, 15). Glu-166 is located in a Ω-loop structure forming the bottom edge of the active site (residues 164–179) and is considered essential as a general base in activating the catalytic water molecule in the deacylation step (11, 16, 17).
The active site Ω-loop is an important element of TEM-1 β-lactamase structure and function. Several different substitutions within the Ω-loop have been identified in TEM-1 variants that hydrolyze oxyimino-cephalosporins (18–20). These substitutions have been proposed to increase the flexibility of the Ω-loop, leading to enlargement of the active site pocket to accommodate newer β-lactams and their bulkier side groups (19, 20). In contrast to other residues in the Ω-loop, residue Glu-166 is conserved in all class A enzymes, and substitutions at this position result in slower rates of acylation and very slow rates of deacylation, leading to the accumulation of the acyl-enzyme intermediate (11, 16, 17).
Previous random mutagenesis studies of the Ω-loop region of TEM-1 identified a triple mutant, W165Y/E166Y/P167G, with a drastically altered substrate profile including increased activity for the oxyimino-cephalosporin, ceftazidime, and decreased activity for all other β-lactams tested (21). This mutant is particularly interesting because the conserved Glu-166 residue is substituted with tyrosine. In a subsequent study, the role of each individual position in the triple mutant was evaluated with respect to enzyme expression levels and in vivo activity (22). Increased antibiotic resistance levels conferred to Escherichia coli containing the mutant enzymes suggested that the individual substitutions act additively to increase hydrolysis of ceftazidime. Detailed biochemical characterization, however, was not performed (22).
To test the hypothesis that the active site is expanded in the triple mutant and that Tyr-166 is substituting for glutamate as a catalytic residue, biochemical and structural characterization was performed. The results indicate that the W165Y/E166Y/P167G enzyme exhibits a large change in the conformation of the Ω-loop compared with wild-type TEM-1, creating more space in the active site to facilitate ceftazidime hydrolysis. In addition, the results suggest that the hydroxyl group of Tyr-166 functions catalytically in the mechanism of hydrolysis by the triple mutant. The changes in conformation and mechanism of hydrolysis highlight the evolvability of the TEM-1 enzyme because of the robustness of the overall fold and the plasticity of the Ω-loop structure.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis PCR
The amino acid substitutions E166Y, P167G, E166Y/P167G, and W165Y/E166Y/P167G were introduced in the pET24a plasmid encoding TEM-1 β-lactamase by site-directed mutagenesis using the following primers: 165YYG167, 5′-GTAACTCGCCTTGATCGTTATTACGGGGAGCTGAATGAAGCATACC-3′; 166YG167, 5′-GTAACTCGCCTTGATCGTTGGTACGGGGAGCTGAATGAAGCCATACC-3′; E166Y, 5′-GTAACTCGCCTTGATCGTTGGTACCCGGAGCTGAATGAAGCCATACC-3′; P167G, 5′-GTAACTCGCCTTGATCGTTGGGAAGGGGAGCTGAATGAAGCCATACC-3′; S70G:165YYG167, 5′-CGTTTTCCAATGGGCACTTTTAAAGTTCTG-3′; and 165YFG167, 5′-GTAACTCGCCTTGATCGTTATTTCGGGGAGCTGAATGAAGCCATACC-3′. The primers were phosphorylated with T4 polynucleotide kinase, and QuikChange PCR was performed with Phusion® DNA polymerase (New England Biolabs, Ipswich, MA) according to the manufacturer's guidelines. DNA sequencing of the entire blaTEM gene of each mutant was performed to ensure the absence of extraneous mutations. Plasmid DNA of confirmed mutants was transformed into E. coli BL21(DE3) cells.
Protein Expression and Purification
Wild-type TEM-1 β-lactamase and designated mutants were expressed in E. coli BL21(DE3) cells as described previously (23). In brief, cells were grown in 250 ml of LB broth containing 300 mm sorbitol, 2.5 mm betaine, and 30 μg/ml kanamycin to an A600 of 0.6–0.8 before induction with 0.4 mm isopropyl β-d-thiogalactopyranoside. The culture was then grown at 23 °C for 18–20 h with shaking. Cell pellets were obtained by centrifugation, and the culture supernatant was dialyzed overnight at 4 °C against buffer containing 0.1 m sodium acetate and 0.4 m sodium chloride at pH 8.0. Proteins were purified to ∼95% homogeneity using a zinc-chelating column eluted with a pH gradient, as previously described (24–27). Purity was determined by SDS-PAGE, and protein fractions were concentrated with Amicon® centrifugal filters with a 10-kDa cutoff (Millipore Merck KGaA, Darmstadt, Germany). Protein concentrations were determined using the Bio-Rad Bradford protein assay reagent with a standard curve that was calibrated using wild-type TEM-1 β-lactamase whose concentration was determined by amino acid analysis. The TEM-1 W165Y/E166Y/P167G/L201P mutant was further purified to ≈99% homogeneity, judged by SDS-PAGE, using size exclusion chromatography in 25 mm HEPES buffer (pH 7.0) and 25 mm NaCl for crystallographic studies.
Enzyme Kinetics
Assays were performed at 30 °C in 50 mm sodium phosphate buffer, pH 7.2, as previously described (27). A DU800 spectrophotometer was utilized to monitor substrate hydrolysis at wavelengths 235 nm for ampicillin (Δϵ = −900 m−1 cm−1), 260 nm for ceftazidime (Δϵ = −10,500 m−1 cm−1), and cefotaxime (Δϵ = −7,600 m−1 cm−1), 262 nm for cephalothin (Δϵ = −7,960 m−1 cm−1), and 482 nm for nitrocefin (Δϵ = 20,500 m−1 cm−1). Enzyme kinetics data were analyzed with GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA) and fitted to the Michaelis-Menten equation. When Vmax determination was prevented by high Km values, the catalytic efficiency (kcat/Km) was estimated at [S] ≪ Km using Equation 1 (28).
 |
Progress curves that exhibited biphasic kinetics were fitted to the general integrated burst equation for a branched mechanism resulting in the steady-state accumulation of an inactive enzyme substrate/product complex (29),
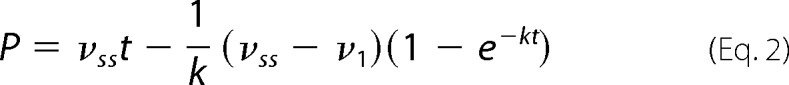 |
where P is the product concentration at time t, vi is the initial velocity in the absence of enzyme inactivation, vss is the steady-state velocity including the reversible accumulation of an inactive complex, and k is the rate constant characterizing the change. The burst amplitude was determined by extrapolation of the steady-state slope to time 0. The effect of ammonium sulfate concentration on the biphasic kinetics was also evaluated (30). All kinetic analysis experiments were performed at least in triplicate.
Determination of the pH Profile of TEM-1 and the W165Y/E166Y/P167G Mutant
The assay was performed as stated above (see “Enzyme Kinetics”) by monitoring initial velocities of nitrocefin hydrolysis at a range of substrate concentrations and pH conditions. The buffers used for the experiment were 50 mm sodium acetate (pH 5–6), 50 mm sodium phosphate (pH 6–7), 50 mm Tris (pH 7–9), and 50 mm CAPS (pH 9–10.5).3 Each buffer was supplemented with 150 mm NaCl to keep the ionic strength constant. Initial velocity data were analyzed with GraphPad Prism 6 and fitted to the Michaelis-Menten equation. The pH dependence of the steady-state parameters was fitted to double (kcat/Km and kcat) and single (Km) ionization model Equations 3 and 4 (13). The experiments were done in triplicate.
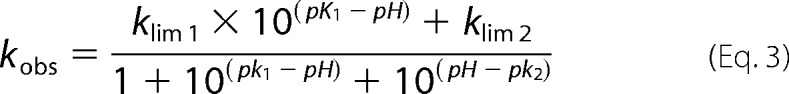 |
AND
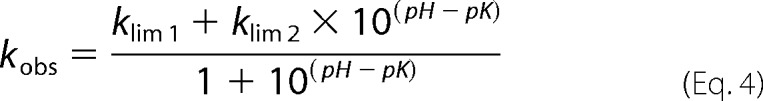 |
ThermoFluor Assay for Tm Determination
A thermal shift assay was performed using a Roche Applied Science Lightcycler 480 Real-Time PCR system (Roche Diagnostics). The assay was performed as follows: 10 μl of 0.4 mg/ml of enzyme in 50 mm sodium phosphate buffer at pH 7.2 was mixed with 10 μl of SYPRO dye (Invitrogen) (1:2500 dilution in 50 mm sodium phosphate buffer, pH 7.2). Data were collected using the manufacturer's software and following a standard protein denaturation program. The data were analyzed and fitted by a previously established method using the derivative of the melting curves (31).
Crystallization of the TEM-1 W165Y/E166Y/P167G Mutant
Crystal screening was performed using commercially available crystal screens from Hampton Research (Aliso Viejo, CA) and Qiagen. The hanging drop vapor diffusion method was used with an in-house TTP LabTech Mosquito instrument (TTP Labtech Ltd., Melbourn, UK). After extensive screening (pH, buffer, concentration), no crystallization hits were identified. An alternative approach was then employed whereby a previously identified stabilizing mutation, L201P, which is known to have no effect on the kinetic profile of the enzyme, was incorporated into the W165Y/E166Y/P167G mutant (26). The resulting enzyme was purified as described above and screened for crystallization. This approach yielded a suitable condition that was further optimized. Crystals were grown in 240 mm tri-sodium citrate with 25% (w/v) PEG 3350 and cryoprotected with peritone/paraffin oil (70:30). A 1.44 Å data set was collected at the Advanced Light Source synchrotron beam line in Berkeley, CA. The data set was processed using iMosflm (32) as implemented in the CCP4 program suite (33). The structure was determined by molecular replacement using MOLREP (34) with wild-type TEM-1 as a phasing model (PDB code 1BTL). Refinement was performed using phenix.refine program (35). In addition, another data set was collected from a crystal of the W165Y/E166Y/P167G triple mutant containing the M182T stabilizing mutation (36, 37). The crystallization condition contained 0.1 m HEPES at pH 6.5 and 30% (w/v) PEG 6,000. Data collection and processing was the same as for the W165Y/E166Y/P167G/L201P mutant. Refinement was performed for several cycles using the REFMAC5 program (38) and manually building the model using the molecular graphics program COOT (39).
Finally, an S70G mutation was introduced in the W165Y/E166Y/P167G mutant, and co-crystallization and soaking with ceftazidime were attempted. No crystals with bound substrate were obtained; however, apo-crystals of this mutant grew readily in 240 mm tri-sodium citrate and 25% PEG 4000 (w/v). A 1.39 Å data set was collected, and the structure was solved by molecular replacement using the same methods stated above.
The coordinates for the three structures were deposited in the Protein Data Bank (40) and have the following accession codes: 4RVA, 4RX2, and 4RX3. Superimpositions were done using the SSM procedure (41), which is part of the COOT program (39). All structural figures were generated with the UCSF Chimera program (42).
Modeling of Ceftazidime into the Active Site of W165Y/E166Y/P167G/L201P
The ceftazidime [CID481173] coordinates were retrieved from the PubChem compound database. The ceftazidime molecule was docked into the crystal structure of TEM-1 W165Y/E166Y/P167G/L201P and wild-type TEM-1 (PDB code 1XPB) using AutoDock Vina (Scripps Institute, La Jolla, CA) (43). Before docking, the proteins were processed by adding polar hydrogen atoms using AutoDockTools. The Lamarckian genetic algorithm was used to generate possible protein-ligand binding conformations (44). The receptor (β-lactamase) was treated as a rigid body, with all possible rotational angles in the substrate. The grid box was centered on the Ser-70 residue with the size (20 × 22 × 28 Å) of the box adjusted to cover the entire catalytic site. Docking was carried out with an exhaustiveness of eight.
RESULTS
Determination of Enzyme Kinetic Parameters
The TEM-1 β-lactamase triple mutant W165Y/E166Y/P167G had been identified previously from a random mutagenesis study of the plasticity of the Ω-loop region in altering the specificity of the enzyme (21). The mutant increases resistance to the oxyimino-cephalosporin, ceftazidime, compared with E. coli containing the wild-type TEM-1 enzyme. To assess the effects of each constituent single substitution, kinetic parameters for hydrolysis of ampicillin, cephalothin, cefotaxime, ceftazidime, and nitrocefin were evaluated for the TEM-1 mutants E166Y, P167G, E166Y/P167G, and W165Y/E166Y/P167G (Table 1).
TABLE 1.
Enzyme kinetic parameters for hydrolysis of β-lactam antibiotics by TEM-1 β-lactamase and mutants
| Enzyme | Substrate |
||||
|---|---|---|---|---|---|
| Ampicillin | Cephalothin | Nitrocefin | Cefotaxime | Ceftazidime | |
| Wild type | |||||
| kcat (s−1) | 1653 ± 370 | 142 ± 5 | 714 ± 80 | 6.5 ± 0.9 | NDa |
| Km (μm) | 63 ± 14 | 218 ± 15 | 30 ± 10 | 1469 ± 284 | >1000b |
| kcat/Km (μm−1s−1) | 26 | 0.65 | 24 | 0.004 | 0.0002b |
| E166Y | |||||
| kcat (s−1) | 1.1 ± 0.4 | 0.31 ± 0.02 | 0.55 ± 0.01 | 0.07 ± 0.01b | 0.09 ± 0.01c |
| Km (μm) | 6.6 ± 2 | 40 ± 11 | 2.9 ± 0.3 | 17 ± 5 | 32 ± 5 |
| kcat/Km (μm−1s−1) | 0.17 | 0.008 | 0.19 | 0.004 | 0.003 |
| P167G | |||||
| kcat (s−1) | 225 ± 34 | 552 ± 40 | 103 ± 5 | 1.3 ± 0.25 | 0.13 ± 0.02c |
| Km (μm) | 107 ± 20 | 208 ± 16 | 24 ± 2 | 776 ± 125 | 20 ± 3 |
| kcat/Km (μm−1s−1) | 2.1 | 2.7 | 4.3 | 0.002 | 0.007 |
| E166Y/P167G | |||||
| kcat (s−1) | 0.48 ± 0.02 | 0.14 ± 0.01 | 0.11 ± 0.01 | 0.03 ± 0.01 | 0.06 ± 0.01c |
| Km (μm) | 5.6 ± 3 | 19 ± 2 | 2.6 ± 0.5 | 3.2 ± 2 | 1.4 ± 0.2 |
| kcat/Km (μm−1s−1) | 0.09 | 0.007 | 0.04 | 0.01 | 0.04 |
| W165Y/E166Y/P167G | |||||
| kcat (s−1) | 3.4 ± 0.3 | 0.08 ± 0.01 | 2.6 ± 0.1 | 0.010 ± 0.001 | 0.2 ± 0.1c |
| Km (μm) | 68 ± 17 | 20 ± 5 | 45 ± 3 | 2.9 ± 2 | 2.4 ± 0.5 |
| kcat/Km (μm−1s−1) | 0.05 | 0.004 | 0.06 | 0.003 | 0.08 |
| W165Y/E166Y/P167G/L201P | |||||
| kcat (s−1) | 1.10 ± 0.03 | 0.04 ± 0.01 | 0.10 ± 0.02 | 0.005 ± 0.002 | 0.20 ± 0.01c |
| Km (μm) | 41 ± 5 | 5.6 ± 1 | 4 ± 1 | 1.9 ± 0.5 | 2.7 ± 0.3 |
| kcat/Km (μm−1s−1) | 0.03 | 0.007 | 0.03 | 0.003 | 0.07 |
a ND, not determined.
b The catalytic efficiency (kcat/Km) was determined using Equation 1 “Experimental Procedures,” because Vmax was not observed because of high Km value.
c The substrate hydrolysis followed branched-pathway; vi rates were determined using Equation 2 at varying substrate concentrations and fit to the Michaelis-Menton equation to determine kcat and Km.
The kinetic parameters of wild-type TEM-1 are consistent with previous observations (27) and revealed the penicillinase characteristics of the enzyme (Table 1). The oxyimino-cephalosporins, cefotaxime and ceftazidime, were hydrolyzed poorly because of very high Km values (>1000 μm) and low turnover rates resulting in a kcat/Km ratio of 0.004 μm−1 s−1 for cefotaxime, and 0.0002 μm−1 s−1 for ceftazidime. This is likely due to the bulky side chains of these drugs that cause steric hindrance in the active site of the enzyme (19).
The single mutant E166Y displayed significant decreases in catalytic efficiency for ampicillin, nitrocefin, and cephalothin hydrolysis (150-, 125-, and 80-fold) compared with the wild-type enzyme. The E166Y enzyme exhibited substantially reduced Km and kcat (85- and 90-fold) values for cefotaxime hydrolysis compared with wild type, resulting in an unchanged kcat/Km ratio (0.004 μm−1 s−1). In contrast to the other substrates, the catalytic efficiency for ceftazidime hydrolysis was 15-fold higher than that observed with wild-type TEM-1 because of a large decrease in the Km value (Table 1).
The P167G single mutant exhibited similar Km values for ampicillin and nitrocefin hydrolysis as wild-type TEM-1. The P167G enzyme, however, displayed a decrease in the kcat/Km ratio (12-fold for ampicillin and 5-fold for nitrocefin), because of significantly reduced kcat values. It is worth noting that P167G exhibited the highest catalytic efficiency for cephalothin hydrolysis (2.7 μm−1 s−1) out of all the enzymes tested, which was four times higher than wild type. Both Km and kcat of P167G for cefotaxime were modestly decreased, resulting in a similar catalytic efficiency for cefotaxime hydrolysis as wild-type TEM-1. The P167G enzyme, however, exhibited increased catalytic efficiency (35-fold) compared with wild type for ceftazidime hydrolysis because of a greatly reduced Km value. Thus, both the E166Y and P167G single substitutions increase catalytic efficiency for ceftazidime hydrolysis by over an order of magnitude by lowering Km.
The double mutant E166Y/P167G and E166Y single mutant exhibited similar kinetic parameters for the hydrolysis of ampicillin, cephalothin, nitrocefin, and cefotaxime (Table 1). These properties include low kcat and Km values relative to wild-type TEM-1 and indicate that the E166Y substitution dominates the catalytic properties of the E166Y/P167G double mutant for these substrates.
As noted above, both the E166Y and P167G single substitutions result in a greater than 10-fold increase in ceftazidime hydrolysis. If the two mutations were acting independently and additively, one would expect an even larger increase in kcat/Km (525-fold) in the double mutant compared with wild type. Combination of the substitutions into the E166Y/P167G enzyme results in a 13-fold increase in catalytic efficiency relative to E166Y, a 6-fold increase compared with P167G and a 200-fold increase over wild-type TEM-1. Thus, the favorable effects of the single mutants are observed in the double mutant, although the effects are not completely additive. The increased catalytic efficiency for ceftazidime hydrolysis by the double mutant is due to a >10-fold decrease in Km compared with either of the single mutants and a >700-fold decrease in Km relative to wild-type TEM-1.
The triple mutant W165Y/E166Y/P167G and the double mutant E166Y/P167G exhibit similar kinetic parameters for hydrolysis of cephalothin, nitrocefin, and cefotaxime (Table 1). The addition of the W165Y substitution, however, results in a small (2-fold) increase in turnover and catalytic efficiency for ceftazidime hydrolysis. The W165Y/E166Y/P167G triple mutant exhibits the highest catalytic efficiency for ceftazidime hydrolysis among all the mutants tested with a 400-fold increase in kcat/Km compared with wild-type TEM-1. The initial collection of random mutants was selected for increased ceftazidime resistance, and the high activity of the triple mutant explains why it was isolated in the original mutant selection experiment (21).
The previously identified L201P mutation that has been shown to stabilize TEM-1 β-lactamase was used to facilitate enzyme crystallization as described below (26). The W165Y/E166Y/P167G/L201P mutant had very similar catalytic efficiencies as the W165Y/E166Y/P167G triple mutant for all the substrates tested. Therefore, the L201P mutation does not affect the catalytic profile of the triple mutant. This is consistent with previous results where it was found that the L201P mutation has little effect on TEM-1 catalytic profile (26).
Overall, the steady-state kinetics of the mutant enzymes demonstrates a shift from the ability to hydrolyze early penicillins and cephalosporins such as ampicillin and cephalothin to more efficient hydrolysis of the oxyimino-cephalosporin ceftazidime. The E166Y substitution dominates the effect of the triple mutant for all substrates except ceftazidime. In the case of ceftazidime hydrolysis, the E166Y and P167G substitutions both contribute significantly to the catalytic efficiency of the triple mutant enzyme.
Characterization of Branched Pathway Kinetics for Ceftazidime Hydrolysis by Mutant Enzymes
It was noticed during the course of the kinetics experiments that ceftazidime hydrolysis by the mutant enzymes, as well as cefotaxime hydrolysis by the E166Y enzyme, followed biphasic progress curves suggesting branched kinetics (45). Substrate-induced reversible inactivation follows a branched pathway of substrate hydrolysis, in which a reversible, inactive enzyme-substrate complex accumulates with time. This behavior has been previously reported for several β-lactamases, particularly with large, bulky β-lactam antibiotic substrates (30, 46–52). The progress curves of the branched pathway are biphasic and start with an initial phase of substrate hydrolysis followed by a slower, linear, steady-state phase (Fig. 1) (29). Progress curves were fitted with Equation 2 (Experimental Procedures), and the values for the branched pathway parameters are listed in Table 2 under conditions of saturating concentrations of ceftazidime. The size of the burst is generally observed to be considerably greater than the concentration of the enzyme. However, a stoichiometric burst can be observed when the deacylation rate is slower than the reactivation rate (51). Previously, a stoichiometric burst was shown to be due to a branched pathway by adding ammonium sulfate to the reaction, which stabilizes and increases the fraction of active enzyme and leads to increase of the burst size (30, 51). This method enabled a pre-steady-state burst and branched pathway kinetics to be distinguished. To assess the nature of the burst, the presence of ammonium sulfate in the reaction was tested. The presence of ammonium sulfate increased the size of the burst in all cases (Table 2), which is indicative of the branched pathway of hydrolysis (30, 51).
FIGURE 1.
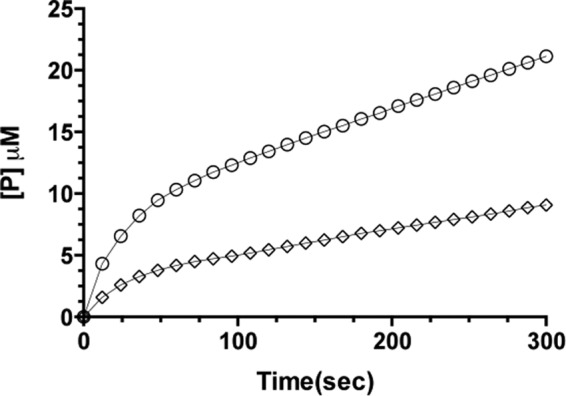
Progress curves of ceftazidime hydrolysis by the TEM-1 W165Y/E166Y/P167G enzyme. The experiments were performed in triplicate. The error bars are omitted for clarity (S.D. less than 5%). The line represents the fit of progress curve to the branched pathway equation for ceftazidime hydrolysis (diamonds) with the inclusion of 60% of saturated (NH4)2SO4 (circles). The increase of the burst in the presence of saturated (NH4)2SO4 is indicative of the branched pathway.
TABLE 2.
Branched pathway kinetics for cefotaxime and ceftazidime hydrolysis by TEM-1 β-lactamase mutants
CAZ, ceftazidime; CTX, cefotaxime; AS, ammonium sulfate.
| TEM-1 mutant | Substrate | Branched pathway parametersa |
Burstb | ||
|---|---|---|---|---|---|
| vi | vss | k | |||
| μmol/s | μmol/s | s−1 | |||
| E166Y | CAZ | ||||
| 0% AS | 0.090 | 0.015 | 0.009 | 5 | |
| 60% AS | 0.156 | 0.032 | 0.010 | 8 | |
| CTX | |||||
| 0% AS | 0.053 | 0.006 | 0.032 | 1 | |
| 60% AS | 0.517 | 0.003 | 0.034 | 7 | |
| P167G | CAZ | ||||
| 0% AS | 0.130 | 0.030 | 0.026 | 6 | |
| 60% AS | ND | ND | ND | NDc | |
| E166Y/P167G | CAZ | ||||
| 0% AS | 0.085 | 0.017 | 0.023 | 3 | |
| 60% AS | 0.138 | 0.031 | 0.021 | 5 | |
| W165Y/E166Y/P167G | CAZ | ||||
| 0% AS | 0.130 | 0.020 | 0.031 | 3 | |
| 60% AS | 0.670 | 0.090 | 0.037 | 16 | |
| W165Y/E166Y/P167G/L201P | CAZ | ||||
| 0% AS | 0.124 | 0.040 | 0.029 | 3 | |
| 60% AS | 0.620 | 0.070 | 0.034 | 15 | |
a The hydrolysis rate for ceftazidime was analyzed with the integrated branched pathway equation (Equation 2). Saturating concentrations of ceftazidime (>Vmax) were used (50 μm ceftazidime was used except in the case of E166Y and P167G, where 100 μm substrate was used because of their higher Km). The data are compiled from three separate experiments with an S.D. of less than 10%. vi is the initial velocity, vss is the steady-state velocity, and k is the rate constant representing the change. Presented rates are adjusted for enzyme concentration (1 μm enzyme for all mutants except 0.5 μm for W165Y/E166Y/P167G).
b The burst amplitude is calculated as the ratio of [P]/[E] by extrapolating the progress curve at the steady-state phase to time 0.
c ND, not determined.
The initial velocity (vi) from the biphasic curves using varying concentrations of ceftazidime was used to calculate kcat and Km in Table 1. The burst observed in the first part of the curve is the initial rate where almost the entire enzyme is in the active state. It is the phase in which the reversibly inactivated enzyme is present in negligible amount and therefore can be treated as a single species of active enzyme. When analyzing the progress curves, it is reasonable to compare the parameters calculated from the initial rate with their wild-type counterparts (this corresponds to the 400-fold increase in kcat/Km for the triple mutant compared with wild-type TEM-1). Furthermore, this can be interpreted as a lower limit of the initial rate because the concentration of active enzyme is decreasing over the course of the reaction until it reaches a steady state with the reversibly inactivated form (second part of the curve).
pH Profile of TEM-1 and the W165Y/E166Y/P167G Enzyme
In TEM-1, Glu-166 participates with Lys-73, Lys-234, and a bound water molecule in a catalytic network that activates Ser-70 during acylation and that activates a structural water molecule for attack on the carbonyl carbon of the acyl-enzyme intermediate in the deacylation reaction (13, 14). Glu-166 is substituted by tyrosine in the W165Y/E166Y/P167G triple mutant, suggesting that Tyr-166 may participate in the mechanism. The tyrosine hydroxyl group would be expected to have a higher pKa than the glutamate carboxyl and have a different pH-dependent effect on the pKa of Lys-73. Tyr-166 participation in the catalytic network would therefore be expected to have a significant effect on the pH dependence of the enzyme. This possibility was tested by examining the pH dependence of the wild-type and triple mutant enzymes for the hydrolysis of nitrocefin (“Experimental Procedures”). There are significant shifts in the pH profiles observed in kcat/Km, kcat, and Km (Fig. 2). The fitting of kcat/Km, and kcat in the double ionization model (Equation 3) produced a bell-shaped curve only for the wild-type kcat pH profile, which had values for pK1 = 6.1 ± 0.06 and pK2 = 8.6 ± 0.05. The other three curves (W165Y/E166Y/P167G kcat/Km and kcat, and wild-type kcat/Km) were not bell-shaped, and the pK values from these fits were ambiguous. Nevertheless, the curves can be described by the pH value of the peak and the width of the curve (50% of the peak value). Based on this analysis, the pH peak for the kcat of the triple mutant is pH 9.7 with a range of 9.1–10.3. The peak for kcat/Km of the wild type is pH 7.0, and the range is 6.2–7.8, and for the W165Y/E166Y/P167G triple mutant the pH peak is pH 9.1 with a range of 8.5–9.7. It is not possible to assign an identity to the various ionizing species, and the potential for a different rate-limiting step for the two enzymes makes a specific interpretation difficult. However, it is clear that there is a very significant effect of the substitution on catalytic residues, consistent with Tyr-166 participating in the active site catalytic network. Furthermore, a triple mutant in which Tyr-166 was substituted with phenylalanine was constructed and is catalytically inactive for all substrates. This suggests that the tyrosine hydroxyl group is essential for the function of the triple mutant.
FIGURE 2.
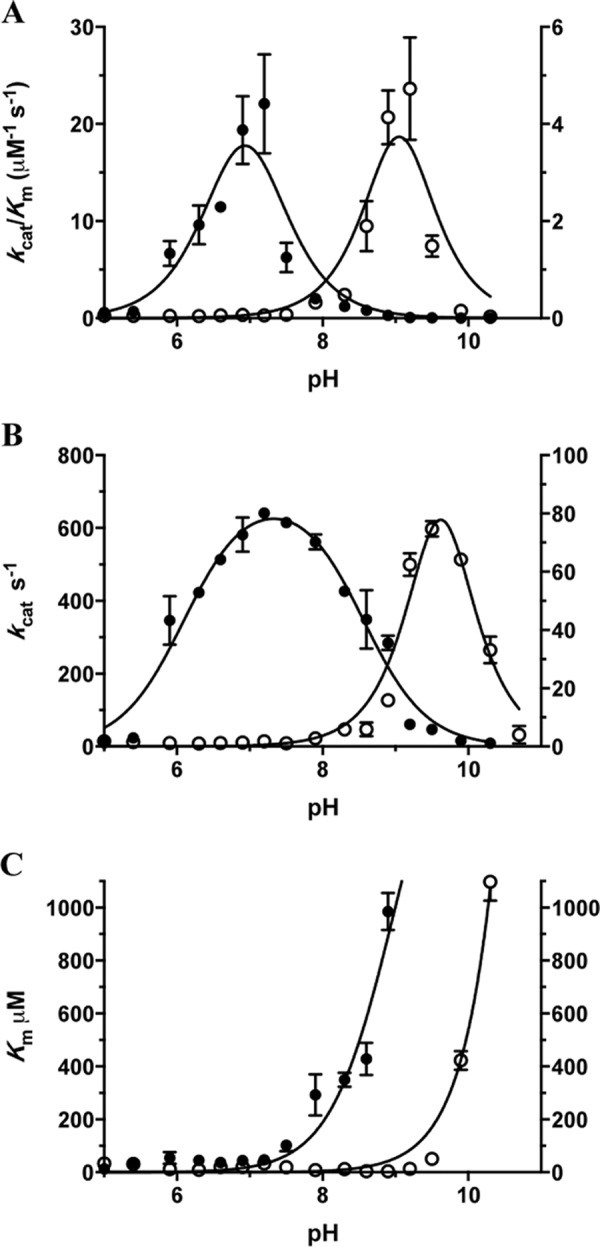
pH dependence of kcat/Km (A), kcat (B), and Km (C) for wild-type TEM-1 (filled circles, left y axis) and the W165Y/E166Y/P167G triple mutant (empty circles, right y axis) β-lactamases for hydrolysis of nitrocefin. The triple mutant exhibits a shift of two pH units in its optimal pH for kcat/Km and nearly two pH units for kcat compared with wild-type TEM-1 β-lactamase (A and B). The error bars in the plots represent standard deviations for each data point.
X-ray Crystal Structure Analysis
Previous studies of substitutions in TEM-1 that result in increased hydrolysis of oxyimino-cephalosporins such as ceftazidime indicate that the mutations lead to a widening of the active site to accommodate the bulky side chains of these drugs (19, 20). To investigate the structural changes resulting from the W165Y/E166Y/P167G mutations and address whether there is an enlargement in the active site of the triple mutant, x-ray crystallography was performed. Attempts to crystallize the W165Y/E166Y/P167G enzyme, however, were not successful.
A differential scanning fluorescence (ThermoFluor) assay was performed on purified wild-type TEM-1 and the triple mutant enzymes to examine their thermal stability. TEM-1 displayed a Tm of 55.0 °C, whereas the triple mutant was much less stable, with a Tm of 46.5 °C. Because the W165Y/E166Y/P167G enzyme is less stable than wild-type TEM-1, we reasoned that addition of a stabilizing mutation might facilitate crystallization. The L201P substitution has previously been shown to stabilize TEM-1 β-lactamase, and therefore it was introduced into the W165Y/E166Y/P167G triple mutant, and the resulting enzyme exhibited increased stability with a Tm of 48.3 °C (26). As noted above, the addition of L201P did not alter the kinetic profile of the W165Y/E166Y/P167G triple mutant (Table 1). The purified W165Y/E166Y/P167G/L201P enzyme was screened for suitable crystallization conditions. The mutant crystallized in the space group P212121 with one molecule in the asymmetric unit, and the structure was determined to 1.44 Å resolution by molecular replacement using the wild-type TEM-1 β-lactamase structure as a model (Table 3) (“Experimental Procedures”).
TABLE 3.
X-ray crystallography data collection and structure refinement statistics
| TEM mutant | W165Y/E166Y/P167G/L201P | W165Y/E166Y/P167G/M182T | S70G/W165Y/E166Y/P167G |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.997 | 0.997 | 0.997 |
| Resolution range (Å) | 34.9–1.44 (1.49–1.44) | 95.9–2.32 (2.40–2.32) | 30.5–1.39 (1.44–1.39) |
| Space group | P212121 | P1 | P212121 |
| Unit cell | |||
| a, b, c (Å) | 59.0, 59.5, 86.2 | 60.3, 83.3, 95.9 | 58.9, 60.1, 88.8 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.1, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Unique reflections | 55,671 (5467) | 80,979 (7943) | 63,941 (6283) |
| Multiplicity | 9.5 (7.4) | 7.8 (7.5) | 2.0 (2.0) |
| Completeness (%) | 99.9 (99.6) | 99.6 (97.7) | 99.6 (98.9) |
| Mean I/σ(I) | 18.2 (2.5) | 5.4 (3.1) | 18.1 (2.07) |
| Wilson B-factor (Å2) | 13.5 | 16.5 | 14.9 |
| Rmerge (%) | 6.5 (65) | 24 (48) | 1.8 (34.7) |
| Refinement | |||
| Rwork (Rfree) | 18.8 (20.3) | 23.5 (26.8) | 17.2 (19.4) |
| Number of non-hydrogen atoms | 2376 | 16,549 | 2487 |
| Protein | 1951 | 15,692 | 2023 |
| Ligands | 4 | 40 | 13 |
| Waters | 421 | 817 | 451 |
| Protein residues | 263 | 2036 | 263 |
| Root mean square deviation bond length (Å) | 0.006 | 0.013 | 0.007 |
| Root mean square deviation bond angle (°) | 1.08 | 1.49 | 1.11 |
| Ramachandran favored (%) | 98 | 97 | 98 |
| Ramachandran outliers (%) | 0 | 0.051 | 0 |
| Average B-factor (Å2) | 19.6 | 21.5 | 20.3 |
| Protein | 16.8 | 21.9 | 17.8 |
| Ligands | 25.2 | 22.3 | 13.8 |
| Waters | 32.4 | 12.9 | 31.8 |
With the exception of the Ω-loop, the W165Y/E166Y/P167G/L201P mutant structure is very similar to the wild-type TEM-1 structure (PDB code 1XPB) with a root mean square deviation of 0.287 Å for matching Cα atoms (Fig. 3, A and B). Compared with wild-type TEM-1, the Ω-loop of the mutant structure exhibits large positional and conformational changes, resulting in a significant expansion of the active site cavity (Fig. 3, A and C). The enlarged cavity provides extra space in the active site where the bulky side chain of ceftazidime would be expected to bind and presumably contributes to the much lower Km for ceftazidime hydrolysis observed with the W165Y/E166Y/P167G triple mutant compared with the wild-type enzyme.
FIGURE 3.

Crystal structure of the TEM-1 W165Y/E166Y/P167G/L201P mutant. A, top panel, TEM-1 in dark cyan (PDB code 1XPB, 1.9 Å resolution) aligned with the TEM-1 W165Y/E166Y/P167G/L201P (salmon) enzyme. The active-site Ser-70 is represented in both structures in stick model. The simulated annealing omit difference map contoured at ∼3 σ for residues 164–174 in the W165Y/E166Y/P167G/L201P structure is shown as a gray mesh and reveals the two different conformations that the Tyr-166 adopts in the W165Y/E166Y/P167G/L201P mutant structure. Bottom panels, detailed view of the catalytic apparatus of TEM-1 (left bottom panel) and proposed apparatus of W165Y/E166Y/P167G/L201P mutant (right bottom panel). In the mutant, the catalytic water molecule is not present because of the bulkier tyrosine residue and the movement of Asn-170. B, top panel, 120° rotation of the structure alignment with the dotted black box indicating the Ω-loop. Bottom panels, view of the Ω-loop electrostatic network. The Ω-loop structure of the mutant is maintained by the preserved salt bridges among the charged residues within the Ω-loop. C, surface representation of TEM-1 (dark cyan) (PDB code 1XPB) and W165Y/E166Y/P167G/L201P (salmon) structures. The active site Ser-70 and Asn-170 are labeled and represented in fluorescent green. The active site cavity of the mutant is enlarged and elongated, forming an L shape. In contrast, the TEM-1 active site is shallow and narrow.
The changes in the Ω-loop alter the catalytic apparatus of the mutant enzyme in several ways. First, the catalytic water molecule that interacts with Glu-166 in the wild-type structure and participates in the deacylation reaction is missing in the triple mutant structure (Fig. 3A). However, there is a water molecule hydrogen bonded (2.7 Å) to the Tyr-166 Oη at a shifted position in the active site. Second, the Asn-170 side chain that coordinates the catalytic water molecule along with Glu-166 in wild-type TEM-1 is shifted away from the active site. The Cα atom of Asn-170 is shifted by 8.4 Å in the mutant versus wild-type TEM-1. Finally, the hydroxyl group of the Tyr-166 residue in the W165Y/E166Y/P167G/L201P mutant is within hydrogen bond proximity of the Ser-70 Oγ and Lys-73 Nζ atoms, consistent with it participating in catalysis. Tyr-166 adopts two conformations in the crystal structure of the mutant (Fig. 3A). One of the rotamers points toward Ser-70 as described above, and the other points away from the active site toward the solvent.
Some aspects of the wild-type Ω-loop structure are preserved in the W165Y/E166Y/P167G/L201P mutant structure. The electrostatic interactions among the charged residues in the Ω-loop are the same as in wild-type TEM-1 in that Arg-161 participates in an ionic interaction with Asp-163, as does Arg-164 with Glu-171 and Asp-179 and also Arg-178 with Glu-171 and Asp-176 (Fig. 3B). However, the unraveling of the loop is apparent in the region of residues 165–170, which results in the large movement of Asn-170 and the creation of the active site cavity to better accommodate the bulky ceftazidime side chain (Fig. 3A).
There is also evidence of increased flexibility of the triple mutant Ω-loop region compared with wild-type TEM-1 in that there is a substantial increase of the B-factors for residues 167–172 (Fig. 4). The increase in the temperature factors suggests increased flexibility of these residues, which could facilitate ceftazidime binding and hydrolysis.
FIGURE 4.
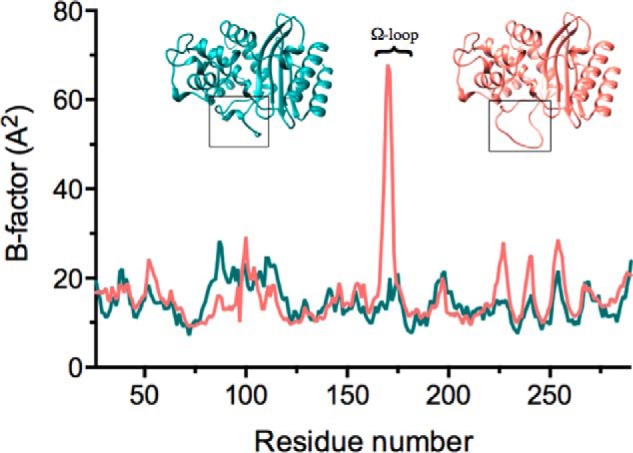
Temperature factors of the main chain of W165Y/E166Y/P167G/L201P (salmon) and TEM-1 (PDB code 1XPB) (dark cyan). The corresponding β-lactamase structures are shown above the temperature factor curves. On the left in dark cyan is TEM-1, and on the right is W165Y/E166Y/P167G/L201P in salmon. The Ω-loop is outlined in the black boxes. The peak in the temperature factor curve corresponds to an increase in the B-factors of residues 167–172 in W165Y/E166Y/P167G/L201P. The numbering follows the conventional numbering for class A β-lactamases.
It is unlikely that the positions of Tyr-166 and Asn-170 and the flexibility of the Ω-loop are an artifact of this crystallization condition and the crystal packing of the protein, because another crystallization condition was identified for a triple mutant containing the M182T stabilizing mutation (36, 37). A 2.32 Å data set was collected with P1 space group containing eight molecules per asymmetric unit (Table 3). The root mean square deviation of the two structures is 0.332 Å. After several refinement cycles, the position of the Ω-loop showed very little density for the 168–174 residue region, consistent with increased flexibility as observed in the temperature factors of the 1.44 Å structure. The electron density for Tyr-166 is observed clearly, however, and shows a single conformation pointing toward Ser-70. The alternative conformation of Tyr-166 that points toward the solvent was not observed. This further supports the hydrogen bond proximity of Tyr-166 to Ser-70. Additionally, a third structure of the triple mutant, this time containing an S70G substitution, was solved at a 1.39 Å resolution. The crystallization conditions and space group are the same as the L201P triple mutant structure (Table 3). This structure exhibits clear density in the Ω-loop and shows the highest similarity with the L201P triple mutant structure with a root mean square deviation of 0.175 Å. It is worth noting that only in the structure of the triple mutant containing L201P does Tyr-166 adopt two alternative conformations. An alignment of the Cα atoms of the three structures is shown in Fig. 5.
FIGURE 5.
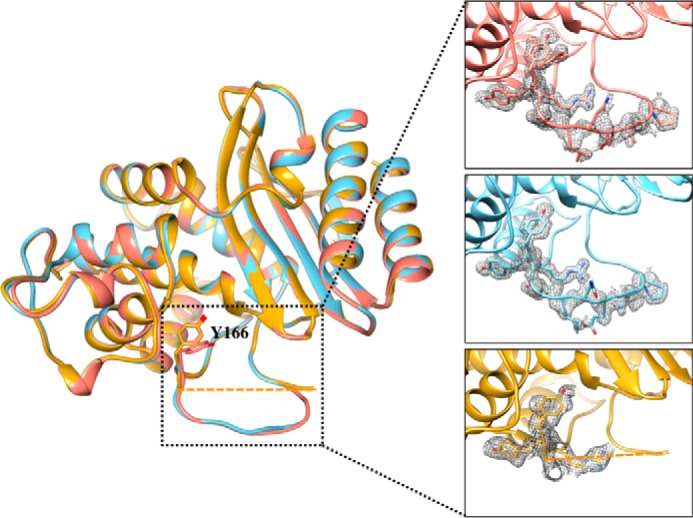
Alignment of the three structures of the TEM-1 W165Y/E166Y/P167G mutants solved in this study. In salmon is the structure of W165Y/E166Y/P167G/L201P. The structure of W165Y/E166Y/P167G/M182T is shown in gold. Residues 168–174 of the Ω-loop of W165Y/E166Y/P167G/M182T showed very little density, and were not modeled in the final structure (residues 167 and 175 are connected with a dashed line). The structure of S70G/W165Y/E166Y/P167G is shown in light blue. The Tyr-166 residue assumes the same conformation in the three structures; however, in the L201P structure, an addition conformation of Tyr-166 is observed. The inset shows a detailed view of the Ω-loop in the three structures, and the simulated annealing omit difference maps contoured at ∼3 σ are shown as a gray mesh for residues 164–174 in the W165Y/E166Y/P167G/L201P and S70G/W165Y/E166Y/P167G structures and for residues 164–167 in the W165Y/E166Y/P167G/M182T structure.
Docking of Ceftazidime into the Active Site of the W165Y/E166Y/P167G/L201P Structure
To assess the interactions between ceftazidime and the triple mutant, co-crystallization was attempted with the S70G/W165Y/E166Y/P167G mutant in which the catalytic Ser-70 residue was converted to glycine to avoid turnover of the substrate. Crystals of the complex, however, could not be obtained. As an alternative, substrate docking of ceftazidime was performed using Autodock Vina (43). The protein structures used for docking were wild-type TEM-1 (PDB code 1XPB) and TEM-1 W165Y/E166Y/P167G/L201P. The same constraints were used for all the docking experiments. Each docking round gave nine possible conformations, ranked from highest to lowest affinity. The highest ranked conformations for ceftazidime in the wild-type and mutant structures are discussed below.
In the triple mutant structure, the substrate is predicted to adopt a conformation where the imino side chain with the two methyl groups and the carboxylic acid moiety are accommodated by the cavity in the active site that is formed by the movement of the Ω-loop (Fig. 6). In wild-type TEM-1, this conformation would not be allowed because of the narrower active site constrained by the position of the Ω-loop. Residues Pro-167 and Asn-170 in the wild-type Ω-loop would sterically clash with the two methyl groups and the carboxylic acid moiety of ceftazidime if it were to adopt the conformation bound to the triple mutant. Instead, the docked structure has the thiazole ring flipped and the oxyimino region with its two methyl groups and the carboxylic acid point into the solvent. It is noteworthy that the orientation of the oxyimino-containing 7β side chain of ceftazidime in the wild-type docked structure is similar to that in a ceftazidime acyl-enzyme structure obtained previously by energy minimization modeling (19). This shallower binding would leave the ceftazidime exposed to the solvent and could contribute to the much higher Km of TEM-1 compared with W165Y/E166Y/P167G. The finding that the oxyanion hole formed by the main chain nitrogens of residues 70 and 237 hydrogen bonded to the carbonyl oxygen of the β-lactam ring of ceftazidime is present in both of the docked structures supports the accuracy of the docking experiment.
FIGURE 6.
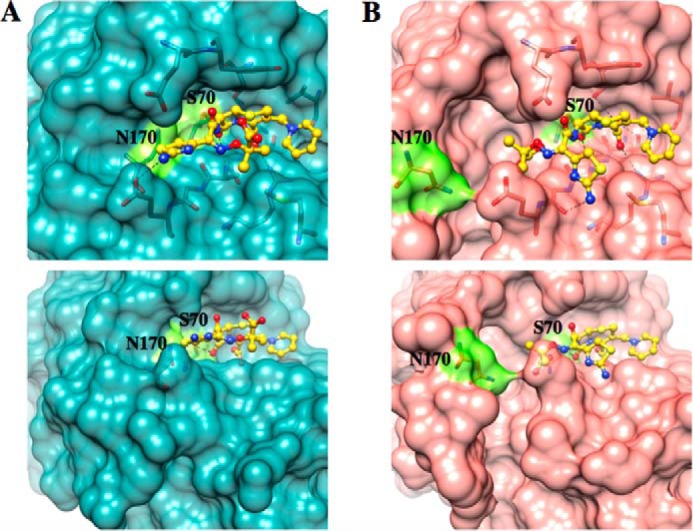
Docking results of ceftazidime with wild-type TEM-1 (A, dark cyan) and the W165Y/E166Y/P167G/L201P mutant (B, salmon). Ser-70 and Asn-170 are shown in fluorescent green in both structures. The same constraints were used for the docking parameters in Autodock Vina for both structures with an exhaustiveness of eight. The conformations with the lowest binding energy are shown. The movement of the Ω-loop in the triple mutant expands the active site, forming an L-shaped cavity (bottom panels) that provides more space to accommodate ceftazidime than in wild-type TEM-1.
DISCUSSION
The TEM-1 triple mutant, W165Y/E166Y/P167G, in which the conserved catalytic Glu-166 residue is substituted by tyrosine, is a functional enzyme. The kcat/Km value of this enzyme for ceftazidime hydrolysis is increased by 400-fold compared with wild-type TEM-1 β-lactamase. Coincident with improved ceftazidime hydrolysis, the triple mutant exhibits greatly reduced hydrolysis of penicillins and early cephalosporins, which are excellent substrates for the wild-type enzyme.
Over 200 TEM variants (extended spectrum β-lactamases) with increased activity toward oxyimino-cephalosporins or reduced susceptibility to inhibitors have been identified from clinical isolates of bacteria (8–10). Substitutions in the Ω-loop are often associated with increased catalysis of extended spectrum β-lactam hydrolysis. For example, R164S is an Ω-loop substitution that is commonly found among extended spectrum β-lactamase variants that exhibit increased catalytic efficiency for oxyimino-cephalosporin hydrolysis. The R164S substitution increases catalytic efficiency for ceftazidime hydrolysis by ∼100-fold and for cefotaxime by 10-fold (19, 53, 54). In addition, the substitution results in an ∼10- and 100-fold decrease in kcat/Km for benzylpenicillin and ampicillin hydrolysis, respectively (19, 53, 54). In comparison, the W165Y/E166Y/P167G mutant exhibits a 400-fold increase in ceftazidime hydrolysis, no change in kcat/Km for cefotaxime hydrolysis, and a 500-fold decrease in efficiency for ampicillin hydrolysis. Therefore, the change in substrate profile, with greatly increased ceftazidime activity, modest or no increase in cefotaxime catalysis, and a decrease in penicillin hydrolysis, is similar between the R164S extended spectrum β-lactamase mutation and the triple mutant, although the extent of the change is different.
Arg-164 is at the base of the Ω-loop and stabilizes the loop through ionic interactions with Glu-171 and Asp-179 (11, 55). The x-ray structure of the TEM-64 (E104K/R164S/M182T) variant has been solved in the presence of a boronic acid inhibitor (20). The structure shows that the short helical region containing Asn-170 unwinds and the Asn-170 Cα group moves by 4.5 Å, and the side chain flips conformation by 180° (20). These changes create a cavity in the active site that allows additional space for the bulky ceftazidime side chain (20). The authors also note that the enlarged site may be less optimal for the smaller side chains of penicillins, which fit optimally in the wild-type active site. A comparison of the conformation change of the Ω-loop in TEM-64 versus the triple mutant reveals Asn-170 is moved out of the active site in both; however, the Ω-loop conformation in the triple mutant is more drastically altered, and the cavity created is larger (Fig. 7). This could explain the greater catalytic efficiency of the triple mutant versus the R164S mutant enzyme for ceftazidime hydrolysis and the greater decrease in ampicillin hydrolysis by the triple mutant compared with R164S.
FIGURE 7.
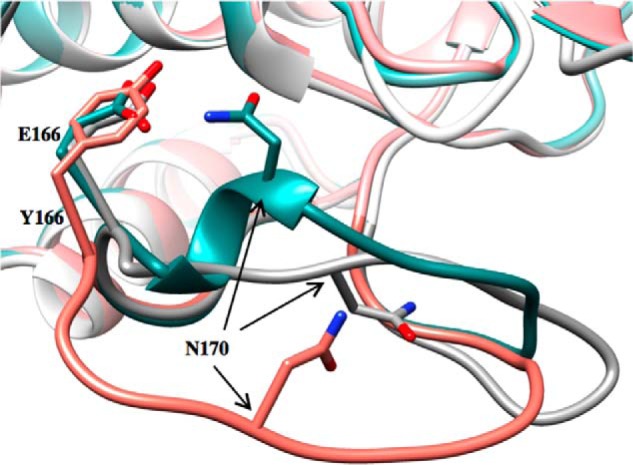
Ω-Loop structures in TEM-1, TEM-64, and TEM W165Y/E166Y/P167G/L201P. The structural conformation of the Ω-loop in TEM-1 (PDB code 1XPB) is shown in dark cyan, TEM-64 (PDB code 1JWZ) is in gray, and TEM W165Y/E166Y/P167G/L201P is in salmon. Represented in stick model are Glu-166 in TEM-1 and TEM-64, Tyr-166 in TEM W165Y/E166Y/P167G/L201P, and Asn-170 in all three enzymes. The position of the side chain at position 166 is very similar in all structures, but Asn-170 shows a great degree of variability. The three enzymes exhibit significant differences with respect to Ω-loop position and conformation. The TEM-1 W165Y/E166Y/P167G/L201P mutant enzyme has an Ω-loop conformation that results in wider active site pocket compared with both TEM-1 and TEM-64.
The E166Y substitution results in increased ceftazidime hydrolysis as well as greatly decreased ampicillin hydrolysis, and when combined with P167G, these effects are magnified (Table 1). The E166Y substitution in TEM-1 has been studied previously with respect to substrate specificity, pre-steady-state kinetics, and enzyme structure (18, 56). The results presented here are consistent with previous observations in that kcat/Km is decreased for penicillins and increased for ceftazidime hydrolysis. Previous pre-steady-state kinetics results also indicate that the E166Y mutant exhibits decreased hydrolysis of benzylpenicillin because of a greatly reduced rate of deacylation compared with wild-type TEM-1 (56).
The x-ray structure of E166Y revealed an enlargement of the active site because of movement of omega loop residues 167–170 by 0.2–1.0 Å (56). Of note, the Cα of Asn-170 is displaced by 0.5 Å, and the side chain moves by 1.0 Å in the E166Y structure (56). However, in contrast to the W165Y/E166Y/P167G structures, the main chain of the Ω-loop of the E166Y mutant retains a similar conformation as wild type. In addition, a rigid body movement of residues 85–142 creates additional space in the active site of the E166Y enzyme (56). The rigid body movement of residues 85–142 is not observed in the W165Y/E166Y/P167G structures. The fact that the TEM-1 E166Y structure does not display a large conformational change in the Ω-loop suggests that the addition of the P167G substitution to E166Y facilitates the conformational change observed in the W165Y/E166Y/P167G triple mutant. The Glu-166–Pro-167 peptide bond is cis in TEM-1, which contributes to the positioning of the Asn-170 region of the Ω-loop (11, 55, 57). Mutation of proline to glycine at position 167 allows a much wider range of dihedral angles, which likely contributes to the observed drastic change in Ω-loop conformation in the W165Y/E166Y/P167G structures.
Saturation-mutagenesis studies of the TEM-1 Glu-166 position have shown that mutations at this position, in addition to E166Y, can lead to increased ceftazidime resistance in E. coli (22, 58). Antunes et al. (58) showed that a TEM-1 E166R variant that also contains the M182T stabilizing substitution increases ceftazidime resistance by a covalent trapping mechanism. Stopped flow kinetics studies indicated that the rate of acylation (k2) of ceftazidime by the E166R/M182T enzyme is 16-fold faster than that catalyzed by wild-type TEM-1. However, deacylation (k3) was not detectable over a period of 20 h (58). Based on the concentration of E166R/M182T in the periplasm and known relationships between bacterial sensitivity to antibiotics and the production of β-lactamases, simulations showed that covalent trapping of ceftazidime as an acyl-enzyme could explain the observed ceftazidime resistance levels in E. coli (58). In contrast, enzyme kinetics analysis indicates the W165Y/E166Y/P167G triple mutant does not trap the substrate as an acyl-enzyme but rather hydrolyzes it via a branched pathway of substrate-induced reversible inactivation.
The TEM-1 W165Y/E166Y/P167G triple mutant is substantially less stable than wild-type TEM-1 with an 8.4 °C decrease in thermal stability. The large change in conformation observed in the Ω-loop in the triple mutant is therefore associated with a substantial loss in enzyme stability. This is not surprising because there are many changes in packing and hydrogen bonding interactions in the triple mutant compared with the wild-type enzyme. The low Tm may also explain why crystallization of the triple mutant enzyme was unsuccessful and why only after addition of the L201P stabilizing mutation were crystals obtained.
The pH rate profile and the crystal structure of the triple mutant indicate an altered catalytic apparatus. The optimal pH for the catalytic efficiency of the triple mutant enzyme is 9.1, which is two pH units higher than that for wild-type TEM-1. The increase in the pH optimum and the lack of detectable activity of the triple mutant in which Tyr-166 is substituted with Phe suggest that Tyr-166 participates in the catalytic mechanism. The inactivity of the Phe-166 mutant also supports a catalytic role for the tyrosine hydroxyl group, perhaps substituting mechanistically for Glu-166. The W165Y/E166Y/P167G crystal structures show that Tyr-166 is oriented toward and within hydrogen bond distance (3.1 Å) of the catalytic serine. Lys-73 and Lys-234 are also nearby so that Tyr-166 could participate in the activation of Ser-70 in the acylation step and/or the activation of water in the deacylation reaction. Wild-type TEM-1 contains a structural water that is positioned to bridge Glu-166 with Ser-70 and to serve as the nucleophile for deacylation (7). There is a water molecule within hydrogen bond distance (2.7 Å) from the Tyr-166 Oη in the triple mutant. This water is 4.3 Å from the Ser-70 Oγ; however, upon acylation, it may be positioned appropriately with the carbonyl carbon of the acyl-enzyme for nucleophilic attack.
The proposed mechanism of hydrolysis by the triple mutant resembles the mechanism for class C β-lactamases (5). In the active site of class C β-lactamases, the residue analogous to Glu-166 of class A is Tyr-150 (59, 60). The Tyr-150 residue is in proximity of two lysine residues that perturb the pKa of the hydroxyl of Tyr-150 and allow abstraction of a proton from the catalytic serine for nucleophilic attack on the β-lactam ring and formation of the acyl-enzyme complex (61). The covalent acyl-enzyme complex is then resolved by a catalytic water molecule activated by Tyr-150 (5). Therefore, the TEM-1 triple mutant has active site characteristics of both class A and C β-lactamases as previously noted for the TEM-1 E166Y enzyme (18).
Amino acid substitutions can result in a large movement of the polypeptide chain and a change of enzyme function. In the case of MutB trehalulose synthase, a sucrose isomerase from Rhizobium sp. that converts sucrose to trehalulose, a single substitution, R284C, causes a drastic conformational change resulting in an enlargement of the active site (62). The absence of the arginine in the R284C mutant disrupts several ionic interactions and subsequently leads to widening of the catalytic pocket. Additionally, this allows additional solvent molecules in the active site and reorganizes the hydrogen bond network of the enzyme. Ultimately, these changes lead to altered function and convert the sucrose isomerase into a more efficient sucrose hydrolase. Similarly, the triple mutant β-lactamase undergoes a large conformation change that alters catalytic function and substrate specificity.
The structural changes associated with the triple mutant reveal a remarkable robustness of the β-lactamase fold to allow large local conformational changes and yet maintain the overall folded enzyme. The substitutions associated with the triple mutant unravel a section of the Ω-loop to create a large cavity in the active site, and yet the enzyme remains stable enough to function. These findings suggest enzymes, or at least β-lactamases, possess a robust fold to allow extensive sampling of amino acid sequence space, which, coupled with selective pressure, can result in modified substrate specificity or new catalytic functions.
This work was supported, in whole or in part, by National Institutes of Health Grant AI32956 (to T. P.). This work was also supported by funds from NIGMS, National Institutes of Health and the Howard Hughes Medical Institute (to Berkeley Center for Structural Biology) and Contract DE-AC02-05CH11231 from the Director of the Office of Science, Office of Basic Energy Sciences, U.S. Department of Energy (to Advanced Light Source). V. S. was supported by NIH training Grant T32 AI55449.
The atomic coordinates and structure factors (codes 4RVA, 4RX2, and 4RX3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- PDB
- Protein Data Bank.
REFERENCES
- 1. Pfeifer Y., Cullik A., Witte W. (2010) Resistance to cephalosporins and carbapenems in Gram-negative bacterial pathogens. Int. J. Med. Microbiol. 300, 371–379 [DOI] [PubMed] [Google Scholar]
- 2. Ambler R. P. (1980) The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 289, 321–331 [DOI] [PubMed] [Google Scholar]
- 3. Massova I., Mobashery S. (1998) Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 42, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Page M. I. (1999) The reactivity of β-lactams, the mechanism of catalysis and the inhibition of β-lactamases. Curr. Pharm. Des. 5, 895–913 [PubMed] [Google Scholar]
- 5. Hata M., Fujii Y., Tanaka Y., Ishikawa H., Ishii M., Neya S., Tsuda M., Hoshino T. (2006) Substrate deacylation mechanisms of serine-β-lactamases. Biol. Pharm. Bull 29, 2151–2159 [DOI] [PubMed] [Google Scholar]
- 6. Fisher J. F., Meroueh S. O., Mobashery S. (2005) Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem Rev. 105, 395–424 [DOI] [PubMed] [Google Scholar]
- 7. Fisher J. F., Mobashery S. (2009) Three decades of the class A β-lactamase acyl-enzyme. Curr. Protein Pept. Sci. 10, 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salverda M. L., De Visser J. A., Barlow M. (2010) Natural evolution of TEM-1 β-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 34, 1015–1036 [DOI] [PubMed] [Google Scholar]
- 9. Pimenta A. C., Fernandes R., Moreira I. S. (2014) Evolution of drug resistance: insight on TEM β-lactamases structure and activity and β-lactam antibiotics. Mini Rev. Med. Chem. 14, 111–122 [DOI] [PubMed] [Google Scholar]
- 10. Bush K., Jacoby G. A. (2010) Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54, 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strynadka N. C., Adachi H., Jensen S. E., Johns K., Sielecki A., Betzel C., Sutoh K., James M. N. (1992) Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 A resolution. Nature 359, 700–705 [DOI] [PubMed] [Google Scholar]
- 12. Fisette O., Morin S., Savard P. Y., Lagüe P., Gagné S. M. (2010) TEM-1 backbone dynamics-insights from combined molecular dynamics and nuclear magnetic resonance. Biophys. J. 98, 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Golemi-Kotra D., Meroueh S. O., Kim C., Vakulenko S. B., Bulychev A., Stemmler A. J., Stemmler T. L., Mobashery S. (2004) The importance of a critical protonation state and the fate of the catalytic steps in class A β-lactamases and penicillin-binding proteins. J. Biol. Chem. 279, 34665–34673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meroueh S. O., Fisher J. F., Schlegel H. B., Mobashery S. (2005) Ab initio QM/MM study of class A β-lactamase acylation: dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J. Am. Chem. Soc. 127, 15397–15407 [DOI] [PubMed] [Google Scholar]
- 15. Atanasov B. P., Mustafi D., Makinen M. W. (2000) Protonation of the β-lactam nitrogen is the trigger event in the catalytic action of class A β-lactamases. Proc. Natl. Acad. Sci. U.S.A. 97, 3160–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adachi H., Ohta T., Matsuzawa H. (1991) Site-directed mutants, at position 166, of RTEM-1 β-lactamase that form a stable acyl-enzyme intermediate with penicillin. J. Biol. Chem. 266, 3186–3191 [PubMed] [Google Scholar]
- 17. Escobar W. A., Tan A. K., Fink A. L. (1991) Site-directed mutagenesis of β-lactamase leading to accumulation of a catalytic intermediate. Biochemistry 30, 10783–10787 [DOI] [PubMed] [Google Scholar]
- 18. Delaire M., Lenfant F., Labia R., Masson J. M. (1991) Site-directed mutagenesis on TEM-1 β-lactamase: role of Glu166 in catalysis and substrate binding. Protein Eng. 4, 805–810 [DOI] [PubMed] [Google Scholar]
- 19. Vakulenko S. B., Taibi-Tronche P., Tóth M., Massova I., Lerner S. A., Mobashery S. (1999) Effects on substrate profile by mutational substitutions at positions 164 and 179 of the class A TEM(pUC19) β-lactamase from Escherichia coli. J. Biol. Chem. 274, 23052–23060 [DOI] [PubMed] [Google Scholar]
- 20. Wang X., Minasov G., Shoichet B. K. (2002) Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95 [DOI] [PubMed] [Google Scholar]
- 21. Palzkill T., Le Q. Q., Venkatachalam K. V., LaRocco M., Ocera H. (1994) Evolution of antibiotic resistance: several different amino-acid substitutions in an active-site loop alter the substrate profile of β-lactamase. Mol. Microbiol. 12, 217–229 [DOI] [PubMed] [Google Scholar]
- 22. Petrosino J. F., Palzkill T. (1996) Systematic mutagenesis of the active-site omega-loop of TEM-1 β-lactamase. J. Bacteriol. 178, 1821–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sosa-Peinado A., Mustafi D., Makinen M. W. (2000) Overexpression and biosynthetic deuterium enrichment of TEM-1 β-lactamase for structural characterization by magnetic resonance methods. Protein Expr. Purif. 19, 235–245 [DOI] [PubMed] [Google Scholar]
- 24. Bowden G. A., Paredes A. M., Georgiou G. (1991) Structure and morphology of protein inclusion bodies in Escherichia coli. Biotechnology 9, 725–730 [DOI] [PubMed] [Google Scholar]
- 25. Cantu C., 3rd, Huang W., Palzkill T. (1997) Cephalosporin substrate specificity determinants of TEM-1 β-lactamase. J. Biol. Chem. 272, 29144–29150 [DOI] [PubMed] [Google Scholar]
- 26. Marciano D. C., Pennington J. M., Wang X., Wang J., Chen Y., Thomas V. L., Shoichet B. K., Palzkill T. (2008) Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 β-lactamase. J. Mol. Biol. 384, 151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brown N. G., Shanker S., Prasad B. V., Palzkill T. (2009) Structural and biochemical evidence that a TEM-1 β-lactamase N170G active-site mutant acts via substrate-assisted catalysis. J. Biol. Chem. 284, 33703–33712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wahl R. C. (1994) The calculation of initial velocity from product progress curves when [S] ≪ Km. Anal. Biochem. 219, 383–384 [DOI] [PubMed] [Google Scholar]
- 29. Waley S. G. (1991) The kinetics of substrate-induced inactivation. Biochem. J. 279, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen C. C., Herzberg O. (1999) Relocation of the catalytic carboxylate group in class A β-lactamase: the structure and function of the mutant enzyme Glu166 → Gln:Asn170 → Asp. Protein Eng. 12, 573–579 [DOI] [PubMed] [Google Scholar]
- 31. Phillips K., de la Pena A. H. (2011) The combined use of the Thermofluor assay and ThermoQ analytical software for the determination of protein stability and buffer optimization as an aid in protein crystallization. Curr. Protoc. Mol. Biol., Chapter 10, Unit 10.28 [DOI] [PubMed] [Google Scholar]
- 32. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 35. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang W., Palzkill T. (1997) A natural polymorphism in β-lactamase is a global suppressor. Proc. Natl. Acad. Sci. U.S.A. 94, 8801–8806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sideraki V., Huang W., Palzkill T., Gilbert H. F. (2001) A secondary drug resistance mutation of TEM-1 β-lactamase that suppresses misfolding and aggregation. Proc. Natl. Acad. Sci. U.S.A. 98, 283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 39. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 42. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 43. Trott O., Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. (1998) Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 [Google Scholar]
- 45. Page M. G. (2008) Extended-spectrum β-lactamases: structure and kinetic mechanism. Clin. Microbiol. Infect. 14, 63–74 [DOI] [PubMed] [Google Scholar]
- 46. Citri N., Samuni A., Zyk N. (1976) Acquisition of substrate-specific parameters during the catalytic reaction of penicillinase. Proc. Natl. Acad. Sci. U.S.A. 73, 1048–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kiener P. A., Knott-Hunziker V., Petursson S., Waley S. G. (1980) Mechanism of substrate-induced inactivation of β-lactamase I. Eur. J. Biochem. 109, 575–580 [DOI] [PubMed] [Google Scholar]
- 48. Charnas R. L., Knowles J. R. (1981) Inhibition of the RTEM β-lactamase from Escherichia coli: interaction of enzyme with derivatives of olivanic acid. Biochemistry 20, 2732–2737 [DOI] [PubMed] [Google Scholar]
- 49. Frère J. M. (1981) Interaction between serine β-lactamases and class A substrates: a kinetic analysis and a reaction pathway hypothesis. Biochem. Pharm. 30, 549–552 [DOI] [PubMed] [Google Scholar]
- 50. Persaud K. C., Pain R. H., Virden R. (1986) Reversible deactivation of β-lactamase by quinacillin. Extent of the conformational change in the isolated transitory complex. Biochem. J. 237, 723–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Escobar W. A., Tan A. K., Lewis E. R., Fink A. L. (1994) Site-directed mutagenesis of glutamate-166 in β-lactamase leads to a branched path mechanism. Biochemistry 33, 7619–7626 [DOI] [PubMed] [Google Scholar]
- 52. Zawadzke L. E., Chen C. C., Banerjee S., Li Z., Wäsch S., Kapadia G., Moult J., Herzberg O. (1996) Elimination of the hydrolytic water molecule in a class A β-lactamase mutant: crystal structure and kinetics. Biochemistry 35, 16475–16482 [DOI] [PubMed] [Google Scholar]
- 53. Sowek J. A., Singer S. B., Ohringer S., Malley M. F., Dougherty T. J., Gougoutas J. Z., Bush K. (1991) Substitution of lysine at position 104 or 240 of TEM-1pTZ18R β-lactamase enhances the effect of serine-164 substitution on hydrolysis or affinity for cephalosporins and the monobactam aztreonam. Biochemistry 30, 3179–3188 [DOI] [PubMed] [Google Scholar]
- 54. Raquet X., Lamotte-Brasseur J., Fonzé E., Goussard S., Courvalin P., Frère J. M. (1994) TEM β-lactamase mutants hydrolysing third-generation cephalosporins: a kinetic and molecular modelling analysis. J. Mol. Biol. 244, 625–639 [DOI] [PubMed] [Google Scholar]
- 55. Jelsch C., Mourey L., Masson J. M., Samama J. P. (1993) Crystal structure of Escherichia coli TEM1 β-lactamase at 1.8 A resolution. Proteins 16, 364–383 [DOI] [PubMed] [Google Scholar]
- 56. Maveyraud L., Saves I., Burlet-Schiltz O., Swarén P., Masson J. M., Delaire M., Mourey L., Promé J. C., Samama J. P. (1996) Structural basis of extended spectrum TEM β-lactamases: crystallographic, kinetic, and mass spectrometric investigations of enzyme mutants. J. Biol. Chem. 271, 10482–10489 [DOI] [PubMed] [Google Scholar]
- 57. Vanhove M., Raquet X., Palzkill T., Pain R. H., Frère J. M. (1996) The rate-limiting step in the folding of the cis-Pro167Thr mutant of TEM-1 β-lactamase is the trans to cis isomerization of a non-proline peptide bond. Proteins 25, 104–111 [DOI] [PubMed] [Google Scholar]
- 58. Antunes N. T., Frase H., Toth M., Mobashery S., Vakulenko S. B. (2011) Resistance to the third-generation cephalosporin ceftazidime by a deacylation-deficient mutant of the TEM β-lactamase by the uncommon covalent-trapping mechanism. Biochemistry 50, 6387–6395 [DOI] [PubMed] [Google Scholar]
- 59. Dubus A., Normark S., Kania M., Page M. G. (1994) The role of tyrosine 150 in catalysis of β-lactam hydrolysis by AmpC β-lactamase from Escherichia coli investigated by site-directed mutagenesis. Biochemistry 33, 8577–8586 [DOI] [PubMed] [Google Scholar]
- 60. Dubus A., Ledent P., Lamotte-Brasseur J., Frère J. M. (1996) The roles of residues Tyr150, Glu272, and His314 in class C β-lactamases. Proteins 25, 473–485 [DOI] [PubMed] [Google Scholar]
- 61. Hata M., Tanaka Y., Fujii Y., Neya S., Hoshino T. (2005) A theoretical study on the substrate deacylation mechanism of class C β-lactamase. J. Phys. Chem. B. 109, 16153–16160 [DOI] [PubMed] [Google Scholar]
- 62. Lipski A., Watzlawick H., Ravaud S., Robert X., Rhimi M., Haser R., Mattes R., Aghajari N. (2013) Mutations inducing an active-site aperture in Rhizobium sp. sucrose isomerase confer hydrolytic activity. Acta Crystallogr. D Biol. Crystallogr. 69, 298–307 [DOI] [PubMed] [Google Scholar]