

Background: The Escherichia coli Dgt enzyme hydrolyzes the important DNA building block dGTP, but the cellular role of this activity is still unclear.
Results: The enzyme is shown to be a hexameric structure containing regulatory DNA molecules.
Conclusion: DNA mediates the regulation of dGTPase activity in the cell.
Significance: This is the first demonstration of regulation of dNTPase by DNA.
Keywords: Allosteric Regulation, Cooperativity, Crystal Structure, DNA-binding Protein, Escherichia coli (E. coli), dGTPase, Mutator Phenotype
Abstract
The Escherichia coli dgt gene encodes a dGTP triphosphohydrolase whose detailed role still remains to be determined. Deletion of dgt creates a mutator phenotype, indicating that the dGTPase has a fidelity role, possibly by affecting the cellular dNTP pool. In the present study, we have investigated the structure of the Dgt protein at 3.1-Å resolution. One of the obtained structures revealed a protein hexamer that contained two molecules of single-stranded DNA. The presence of DNA caused significant conformational changes in the enzyme, including in the catalytic site of the enzyme. Dgt preparations lacking DNA were able to bind single-stranded DNA with high affinity (Kd ∼ 50 nm). DNA binding positively affected the activity of the enzyme: dGTPase activity displayed sigmoidal (cooperative) behavior without DNA but hyperbolic (Michaelis-Menten) kinetics in its presence, consistent with a specific lowering of the apparent Km for dGTP. A mutant Dgt enzyme was also created containing residue changes in the DNA binding cleft. This mutant enzyme, whereas still active, was incapable of DNA binding and could no longer be stimulated by addition of DNA. We also created an E. coli strain containing the mutant dgt gene on the chromosome replacing the wild-type gene. The mutant also displayed a mutator phenotype. Our results provide insight into the allosteric regulation of the enzyme and support a physiologically important role of DNA binding.
Introduction
dNTP triphosphohydrolases represent an intriguing class of enzymes capable of hydrolyzing deoxynucleoside 5′-triphosphates (dNTPs) into the corresponding deoxynucleosides and tripolyphosphate (PPPi) (1–3). The first discovered enzyme in this group is the product of the Escherichia coli dgt gene (3). This enzyme (Dgt) has a strong preference for hydrolyzing dGTP, and is generally referred to a dGTPase (2). With respect to their substrate specificity, the triphosphohydrolases can be divided into two major groups. One group includes E. coli dGTPase and Pseudomonas aeruginosa PA1124, both of which have a strong preference for dGTP. A second group includes Thermus thermophilus TT1383, Enterococccus faecalis EF1143, and P. aeruginosa PA3043, which have a broader specificity and readily hydrolyze multiple dNTPs (4–6). In humans, a dGTP-dependent triphosphohydrolase has been identified as SAMHD1, which likewise hydrolyzes dNTPs into the corresponding deoxynucleoside and tripolyphosphate (7, 8). All these enzymes belong to the HD superfamily (9), as they have conserved His and Asp residues that are coordinated to a metal in a diverse group of phosphohydrolases.
The focus of the current work is on the dGTPase of E. coli encoded by the dgt gene. Early studies had shown that deletion of the dgt gene led to a 2-fold increase in the cellular dGTP pool (10), whereas its overexpression (optA1 strain) led to a 5-fold decrease in the dGTP level (11). Despite these early findings, no cellular function of the enzyme could be described (2). More recently, our laboratory discovered that inactivation of the dgt gene produced a novel mutator phenotype (12). This effect, which suggested a fidelity function for the dgt gene, may be attributed to an increase in the cellular dGTP concentration resulting from the lack of Dgt function. Our laboratory has also demonstrated the involvement of the dgt gene in the thymine-salvage pathway by way of its role in producing deoxyguanosine, a compound critical for thymine salvage (13). Severe depletion of the dGTP pool by Dgt overproduction can actively kill E. coli (14) in a manner similar to the well known phenomenon of thymineless death (15).
Biochemical studies on E. coli dGTPase (Dgt) revealed that the enzyme has a strong single-stranded DNA-binding (ssDNA)2 activity (16). However, the role of this DNA-binding activity has not been defined. For human SAMHD1, DNA binding is required for the oligomerization of the protein, which in turn is required for its dNTPase activity (17, 18). There is no information available on the DNA-binding properties of other bacterial dNTPases.
Crystal structures of other bacterial dNTP triphosphohydrolases (TT1383, PA1124, PA3043, and EF1143) have revealed a domain architecture where the dNTPase exists as a hexamer, except for EF1143, which is a tetramer (4, 6, 19). The crystal structure of the complex of EF1143 with its substrate (dATP bound at the active site) and activator (dGTP bound at the regulatory site) revealed an allosteric mechanism in which a dNTP in one site (regulatory site) promotes binding of a dNTP substrate molecule in the catalytic site (6). However, no structure of any protein-DNA complex is available that would elucidate the mode of DNA binding and its possible role in affecting the dNTPase activity.
In the present work, we have obtained the first crystal structure of E. coli Dgt. Surprisingly, this crystal structure showed the presence of DNA in complex with the enzyme. In the structure, Dgt exists as a hexamer, containing two DNA units per hexamer. Mutation of the Dgt residues interacting with DNA lead to a complete loss of the DNA-binding activity. Genetic studies on an E. coli dgt mutant defective in DNA binding showed a modest mutator phenotype for A·T → G·C transition mutations. This is the first time that the DNA-binding activity of triphosphohydrolases has been explored. Our analysis of the DNA binding leads us to postulate a possible allosteric mechanism that links the catalytic site and the DNA-binding site of the dNTPase.
EXPERIMENTAL PROCEDURES
Cloning of E. coli dgt
The dgt gene was cloned under expression of the T7 promoter in plasmid pET30-Ek/LIC (pET30-dgt). The pET30-Ek/LIC vector introduced an N-terminal hexahistidine tag, which can be cleaved off due to the presence of an enterokinase protease site (Ek) just before the start of the gene sequence. Primers for amplification were designed to contain overhangs at the 5′- and 3′-termini (underlined in DNA sequence, see below) that allowed for the directional cloning of the gene into the vector. The entire gene from its ATG start codon to its TAA stop codon was amplified from genomic DNA using the TaqDNA polymerase kit (Invitrogen) using 5′-GACGACGACAAGATGGCACAGATTGATTTCCGA-3′ (forward primer) and 5′-CTCCTCTTCGGGCCTTATTGTTCTACGGCCATATCCCA-3′ (reverse primer) (Dgt start and stop codon in bold). After gel purification of the amplification product, a LIC (ligation independent cloning) reaction was performed using the pET-30 Ek/LIC Vector kit (Novagen). The insertion of the gene into the vector was confirmed by a restriction digestion reaction using BglII and BamHI, sites for which are present in the vector on either side of the dgt insert. The plasmid was then transformed into DH5α cells selecting for kanamycin-resistant (kanR) colonies. To confirm that there were no mutations created within the dgt gene during its amplification, DNA sequencing was performed using the Big Dye Terminator kit (Life Technologies) and 5 sequencing primers covering the entire dgt sequence. For Dgt overexpression studies, the plasmid, named pET30-dgt, was transformed into strain BL21(DE3).
Protein Purification
Three-liter cultures of BL21(DE3) containing pET30-dgt were grown at 37 °C in LB to A600 nm ∼ 0.5, at which point the temperature was reduced to 18 °C and expression was induced by addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. After overnight incubation at 18 °C, the cells were harvested by centrifugation. Three slightly different Dgt purification protocols were used throughout the study, as detailed below.
Protocol 1
Cell lysis was performed in the presence of Bugbuster (Novagen), lysozyme (Sigma), and Benzonase. After high-speed centrifugation, the lysate was purified over a Ni-NTA column at room temperature, as further described in protocol 2. The His6 tag at the N-terminal of Dgt was cleaved off using the protease enterokinase (Ek). This particular protocol led to a Dgt preparation containing enzyme-bound DNA. The salt concentration in the lysis solution was 25 mm, as per the manufacturer's instructions.
Protocol 2
This modified protocol was developed to minimize DNA contamination of purified Dgt. Cells were grown as described in protocol 1, but lysis was performed by addition of Bugbuster, lysozyme, and 2 m NaCl, thus creating high-salt conditions and omitting the Benzonase. After centrifugation at 30,000 × g for 30 min, 6% (w/v) PEG 6000, and 5% (w/v) dextran 500 were added to the clear supernatant to precipitate out any remaining DNA. The clear upper phase obtained after a high-speed spin was then loaded on the Ni-NTA column and the purification was performed at room temperature. The Ni-NTA resin packed in a column was equilibrated with 5–10 column volumes of equilibration buffer from the HisTALON buffer set (Clontech). The lysate containing Dgt was loaded on the column and the resin was further washed with a wash buffer containing 10 mm imidazole. Finally the protein was eluted using the elution buffer (Clontech). The purity of protein was analyzed on an SDS-PAGE gel, and after cleaving off the histidine tag the protein was concentrated to 7 mg/ml and stored at −80 °C. This protocol yielded an essentially DNA-free enzyme preparation, which was used for crystallography of the DNA-free enzyme.
Protocol 3
Cells were grown as described in protocol 1. Lysis and purification were performed at 4 °C. For lysis, cell pellets were resuspended and sonicated in 25 mm Tris-HCl, pH 8.0, 1 m NaCl, 10% (w/v) sucrose, 5 mm EDTA, 1 mm β-mercaptoethanol, 2–3 tablets of EDTA-free protease inhibitor mixture (Roche Applied Science), and lysozyme (Sigma). Polyethyleneimine to a final concentration of 0.3% (v/v) was added to the lysate to further reduce DNA contamination. A clear supernatant containing the protein was obtained by high-speed centrifugation at 16,000 × g for 30 min. The protein was fractionated from the supernatant by the addition of solid ammonium sulfate to 50% (w/v), over a period of 10 min on ice. The precipitated protein was collected using high-speed centrifugation, and the pellet was resuspended in 25 mm Tris-HCl, pH 8.0, 2 mm EDTA, 15% (v/v) glycerol, and 1 mm β-mercaptoethanol. Before purification the protein was dialyzed overnight against 25 mm Tris-HCl, pH 8.0, 300 mm NaCl. The dialyzed protein was purified using Ni-NTA-agarose resin (Qiagen) using a batch procedure. The resin was equilibrated with 25 mm sodium/potassium phosphate, pH 8.0, 300 mm NaCl. The protein was incubated with the equilibrated resin for 45 min and then washed with a solution of 25 mm sodium/potassium phosphate, pH 8.0, 2 m NaCl, and 5 mm imidazole, pH 8.0. Finally the protein was eluted from the resin in a step gradient using 25 mm sodium/potassium phosphate, pH 8.0, 300 mm NaCl, and 200 mm imidazole. After cleaving off the histidine tag, the purified protein was dialyzed overnight against 25 mm Tris·HCl, pH 8.0, 10 mm MgCl2, and 75 mm sodium citrate (buffer A). Dgt was concentrated using a 10,000 MWCO concentrator (Millipore) before freezing the enzyme in small aliquots at −80 °C at a concentration of 15 mg/ml. Dgt purified using protocol 3 was used for DNA binding studies as well as activity assays in the presence and absence of DNA. In all cases, the histidine tag was removed by enterokinase.
Analytical Gel Filtration
Dgt protein was analyzed on a Superdex 200 16/60 size exclusion column to investigate its oligomerization state in solution. Protein elution was measured at 280 nm. The Dgt molecular weight was determined by comparing its elution volume to molecular weight standards (thyroglobulin, γ-globulin, ovalbumin, myoglobin, and vitamin B12) and by SEC-MALS (size exclusion chromatography multi-angle light scattering) using the miniDAWNTM TREOS® connected to a Superdex 200 10/30 column.
Crystallization, X-ray Diffraction, Phasing, and Refinement
Crystals of Dgt protein were grown by the sitting-drop vapor diffusion method by mixing 2 μl of protein solution (10 mg/ml of dGTPase) with 2 μl of well solution 1 (0.1 m sodium acetate, 8% (w/v) polyethylene glycol 4000, pH 4.6) and incubation at room temperature (20 °C). Crystals appeared in a few days. For data collection, crystals were transferred to a cryoprotectant solution (crystallization solution plus 25% (v/v) ethylene glycol) and flash cooled in liquid nitrogen. Crystals of Dgt without any bound DNA (see protocol 2) were produced similarly with a protein solution containing 1 mm deoxy-CCC and a well solution of 0.5 m ammonium sulfate, 0.1 m sodium citrate, 1.0 m lithium sulfate, pH 5.6. X-ray data were collected at beamline 22-ID of the Advanced Photon Source at Argonne National Laboratory in Chicago. All data reduction and scaling was performed using the HKL2000 data processing software (20). The structure was phased by molecular replacement using the structure of dGTPase from Pseudomonas syringae (PDB accession 2PGS). Maps were improved using solvent flattening and 6-fold non-crystallographic averaging using density modifications (21, 22). The model was fit manually using O (23), and refined using PHENIX and CNS (24, 25). Structure refinement was done using NCS restraints on Dgt monomers each divided into six rigid subdomains, with some loops excluded. Final refinement of the DNA-bound structure restrained chain A with B, and grouped chains C through F. The final model displays reasonable Ramachandran geometry (favored 93.6%, outliers 1.2%).
Construction of a Dgt Mutant (S34D/G37E) Defective in DNA Binding
The S34D/G37E double mutation was generated in plasmid pET30-dgt using the QuikChange site-directed mutagenesis kit (Stratagene). The oligonucleotides containing the double mutations (underlined) were 5′-CGGATCTTCGAGGATGATCGCGAACGTATCATCAACTCTCCG-3′ and 5′-CGGAGAGTTGATGATACGTTCGCGATCATCCTCGAAGATCCG-3′. The presence of both mutations within dgt was confirmed using DNA sequencing.
Modeling of dGTP in the Active Site of Dgt
The dGTP substrate was modeled by superposition of the active site region with the structure of SAMHD1 bound to dGTP (Protein Data Bank 4MZ7) (26). In the reference structure, the triphosphate is shifted away from the active site due to the use of the non-catalytic metal Zn+2. Therefore, the triphosphate group was modeled to approximate a catalytically competent orientation with Pα near the active site metal, and the remaining two phosphates near residues Lys-211 and Lys-232.
Fluorescence Anisotropy for Assessing DNA Binding
Single-stranded DNA or RNA (22-mer) containing a fluorescein label at the 5′ end (HPLC purified, purchased from Sigma) of the following sequence was used for binding studies to Dgt: 5′-TTGTACATTTCTCTCCTTGTCT-3′. The DNA or RNA was resuspended in 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, and 0.1 mm EDTA at a concentration of 35 nm. An Olis RSM1000 spectrofluorometer (Bogart, GA) was used to measure the steady-state fluorescence anisotropy at 22 °C. The excitation wavelength was 480 nm and the emission wavelength was 520 nm. The protein concentration was varied from 0 to 1000 nm, and the changes in the fluorescence anisotropy (r) were measured in response to the stepwise addition of the protein. The data were fit to a quadratic equation (27) via nonlinear regression using Kaleidagraph version 3.6 software to obtain the dissociation constant (Kd) values.
dGTP Triphosphohydrolase Activity Assay in Absence and Presence of DNA
dGTPase activity of the Dgt protein was measured using an enzyme-coupled assay in which deoxyguanosine (the product of dGTP hydrolysis by Dgt) is converted into 8-oxoguanine by the action of purine nucleoside phosphorylase and xanthine oxidase. The details of this assay will be described elsewhere.3 Briefly, the reaction mixture contained 100 mm Tris-HCl, pH 8.0, 5 mm MgCl2, 50 milliunits/ml of purine nucleoside phosphorylase (Sigma), 500 milliunits/ml of xanthine oxidase (Sigma), 5 mm sodium phosphate, and a desired dGTP concentration. To start the reaction, Dgt enzyme was added (4 nm, final concentration in 100 mm Tris-HCl, pH 8.0, 5 mm MgCl2). The reaction was performed in a 1-cm pathway quartz cuvette inside a Beckman Coulter DU-640 spectrophotometer. The formation of 8-oxoguanine was monitored continuously at 297 nm, and the rate of the reaction was determined from the linear slope of the absorbance increase. To analyze enzyme activity in the presence of DNA, a single-stranded DNA 40-mer (5′-ATGCTAGCTTGGCTGTGACTTTAAACCTGTCGTGCCAGCT-3′) was included in the reaction mixture. Prior to initiation of the reaction, Dgt enzyme was preincubated at 37 °C for 20 min (2, 3). The rate data were fit to the Hill equation (28) using nonlinear regression on Kaleidagraph software version 3.6.
Construction of an E. coli Mutant Carrying the S34D/G37E dgt Mutation on the Chromosome
To achieve replacement of the wild-type dgt gene on the chromosome with the DNA-binding defective S34D/G37E variant, we used the splicing by overlap extension-LRed method, which uses both splicing by overlap extension PCR and the λRed system (29). Using this method, the wild-type and mutant dgt gene were fused to a kanamycin-resistant (kan) gene serving as selectable marker. Three separate PCR were used to amplify the wild-type dgt gene (from pET30-dgt), dgt mutant gene (from pET30-dgt S34D/G37E), and the kan gene (from plasmid pKD4) (30). Primers (listed below) were designed so that 50-bp extensions (capital letters) were added at the 5′ and 3′ ends of each amplification product. The four primers used were: forward primer (FP1) and reverse primer (RP1) for amplifying the dgt gene (lowercase letters) along with its promoter region (underlined), and FP2 and RP2 to amplify the kan gene (lowercase letters) from plasmid pKD4 (30). Primers RP1 and FP2 had a common linker region (italicized) that could be used to fuse the dgt gene to the kanR gene in a subsequent step.
The primer sequences are as follows: FP1, 5′-GCATAGTTTACCATGCGCTTACGGGGAAGCGTATTTCTCACGCGGGAGAGGACatggcacagattgatttccg-3′; RP1, 5′-CTCTATGCGTGGGATGAATACCGACGTCTGATGGCCGTAGAACAAtaaccaggcttttgtaaagacgaac-3′; FP2, 5′-GGGATGAATACCGACGTCTGATGGCCGTAGAACAAtaaCCccggaattgccagctggggcgccctctggt-3′; and RP2, 5′-gccttcttgacgagttcttctgaAGGCTTTTGTAAAGACGAACAATAAATTTTTACCTTTTGCAGAAACTTTAGTTCGGAACT-3′.
In the next step, the dgt and dgt S34D/G37E PCR products were each separately amplified together with the amplified kan gene with primers FP1 and RP2, leading to a PCR product in which the kan and dgt genes were joined. The fusion PCR products were electroporated into BW25113 cells containing plasmid pKD46, which expresses the phage λRed proteins under control of an arabinose-inducible promoter (30). For efficient recovery of the desired recombinants, the dgt gene in BW25113 was first replaced with a chloramphenicol-resistant variant (Δdgt::cam) (12) by P1 transduction. Cells were plated on LB plates containing 25 μg/ml of kanamycin and incubated at 37 °C for 24 h. Colonies were purified and tested for loss of the camR marker. Genomic DNA of several kanRcamS colonies was isolated, and a PCR was performed to confirm the replacement of dgt with the joint dgt-kan product. DNA sequencing using the Big Dye kit was used to confirm the presence of both the wild-type and the S34D/E37E dgt gene. P1 transduction was used to transfer the constructs to tester strain NR10836 (12) using a NR10836 Δdgt::cam strain as recipient. kanRcamS colonies were selected for DNA sequencing to confirm the presence of the desired dgt allele in the NR10836 background.
LacZ Papillation Assay and Mutant Frequency Determination
Derivatives of strain NR10836 (12) containing dgt+, Δdgt::cam, or dgt S34D/E37E were streaked on XPG plates (12) and incubated at 37 °C for up to 8 days. The plates were inspected at regular intervals for the number of blue (lac+) papillae appearing within individual colonies indicating a reversion of the lac gene to lac+. For mutant frequencies, for each strain, 28–30 independent LB cultures were grown overnight in 1 ml of LB medium at 37 °C with shaking. The total cell count in the resulting cultures was determined by plating 0.1 ml of a 106 dilution on MM-glucose plates (12). The number of lac+ mutants in each culture was determined by plating 0.1 ml of either undiluted culture (MMR-deficient strains) or 10-fold concentrated cultures (MMR-proficient strains) on MM-lactose plates (12). The resulting number of lac+ revertants per plate was divided by the number of total cells to calculate the mutant frequency.
RESULTS
Purification and Properties of E. coli dGTPase
The Dgt protein was overexpressed from plasmid pET30-dgt in good yield (15–20 mg/liter) and high purity after nickel chromatography and removal of the histidine tag. Denaturing SDS electrophoresis revealed a single polypeptide near 60 kDa (see Fig. 1, inset), consistent with the predicted 59.4 kDa from the amino acid sequence (504 residues) and a previous experimental result (58 kDa) (2). Size exclusion chromatography of the nondenatured protein revealed the protein to elute as a high molecular weight assembly (Fig. 1, 2nd peak). Subsequent analysis by SEC-MALS revealed a protein hexamer (330 kDa). This hexameric mass contrasts to previous reports that Dgt is a tetramer (2). As shown below, the crystal structure confirmed the hexameric composition of the protein. The purified protein was fully active in hydrolyzing its preferred substrate, dGTP (see below).
FIGURE 1.

Gel filtration of Dgt on a Superdex 200 16/60 column. The 1st peak (fractions 40–47) represents the aggregated protein and the 2nd peak (fractions 51–60) the hexameric Dgt. The inset shows a denaturing SDS-PAGE electrophoresis gel indicating the purity of Dgt as present in the 2nd peak. Lane 1 = molecular weight marker; lane 2 = Dgt.
Crystal Structure of E. coli Dgt
Crystals of Dgt (irregular diamond-shaped) were obtained by vapor diffusion, diffracting to 3.1 Å, permitting determination of the protein structure (PDB code 4X9E) (see Table 1 for the collected crystallographic data and refinement results). The structure and various close-ups are presented in Figs. 2, 3, and 4. As shown in Fig. 2, A, B, and D, the structure contains six molecules of Dgt per asymmetric unit, arranged as a hexamer with approximate D3 molecular symmetry. The hexameric composition is consistent with the SEC-MALS result described above. The structure is all α-helical, with 21 helices per chain (Table 2 and Fig. 2C). The structure may be viewed as two stacked trimer rings. However, the largest subunit-subunit interactions, as defined by buried surface areas (Table 3), are found within the Dgt dimers that are perpendicular to the two trimer rings (a-b, c-d, and e-f pairs in the side view of Fig. 2D). Hence, the protein is best described as a set of three dimers. The importance of the dimers is further supported by the functional characteristics discussed below. Each Dgt dimer consists of two antiparallel (head-to-head) monomers (2-fold symmetry) with an extensive interaction area of ∼2000 Å2. A noteworthy feature of the hexameric Dgt structure is the intrusion of a fingerlike structural element formed by helices 17 and 18 and an intervening loop (indicated by an arrow in Fig. 2, A and B) into the adjacent, clockwise monomer belonging to a different Dgt dimer. This finger extends toward the active sites (marked A) and is likely catalytically important (see below).
TABLE 1.
Data collection and refinement statistics for Dgt crystals with and without DNA
| With DNA (4X9E) | Without DNA (4XDS) | |
|---|---|---|
| Data collectiona | ||
| Space group | P212121 | P43212 |
| Cell dimensions | ||
| a, b, c (Å) | 136.8, 159.8, 190.0 | 189.3, 189.3, 296.7 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 3.1 (3.21-3.1)b | 3.35 (3.47-3.35)b |
| Rsym (%)c | 11.0 (72.5) | 14.7 (69.9) |
| I/σI | 10.3 (3.3) | 8.1 (2.1) |
| Completeness (%) | 99.9 (99.9) | 98.6 (98.7) |
| Redundancy | 6.0 (6.1) | 6.2 (5.0) |
| Refinement | ||
| Resolution (Å) | 3.1 | 3.3 |
| No. reflections | 75,861 | 66,200 |
| Rwork/Rfree (%)c | 16.7/22.2 | 17.2/20.9 |
| Number of atoms | ||
| Protein | 24,358 | 24,119 |
| Ligand (DNA or Ni+2)d | 113 | 6 |
| Average B-factors | ||
| Protein | 108.3 | 95.49 |
| Ligand | 130.8 | 96.33 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.010 | 0.014 |
| Bond angles (°) | 1.17 | 1.24 |
a Each data set was obtained from a single crystal.
b Values in parentheses represent statistics from the highest resolution shell (10%).
c R-factor values according to standard equations.
d The crystal without DNA obtained from Dgt purified by protocol 2 (see “Experimental Procedures”) revealed Ni2+ density at the metal-binding site.
FIGURE 2.

Views of the Dgt hexameric crystal structure. A, top view of the hexameric organization showing Dgt as a stack of two trimers. The top trimer consists of monomers labeled a (yellow), c (blue), and e (hot pink). The bottom three monomers are labeled b (orange), d (cyan), and f (magenta). The two DNA molecules (modeled as a CCC trinucleotide) bound at the interface of monomers a and b are shown as solid green spheres. The active site region of monomers a, c, and e is indicated with a circled capital A. The arrow is pointing toward a small loop in monomer e (in hot pink) that reaches out into the active site of monomer a (in yellow). See text for details. B, surface representation of structure as shown in panel A. The putative active sites of top monomers (a, c, and e) are indicated with capital A. The long arrow points toward one example of a small loop in each monomer that reaches out into the active site of the next clockwise monomer; see text and Fig. 4D for details. C, ribbon diagram of monomer a (in yellow) with individual helices numbered α1 to α21. The two DNA molecules are in green. D, side view of the Dgt hexamer with the bound DNA (green) at the interface of a and b. The 5′ → 3′ orientations of the DNA are also indicated.
FIGURE 3.
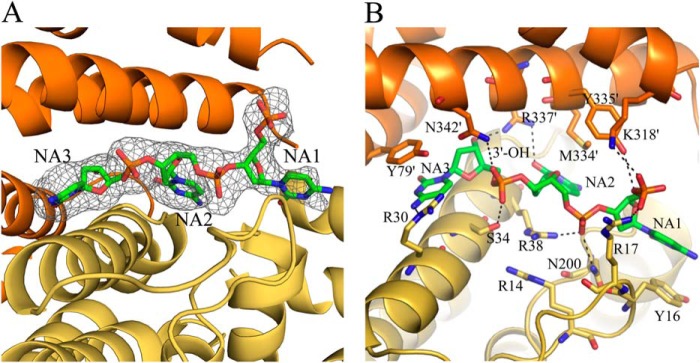
Close-up views of the DNA-binding site of Dgt. A, simulated annealing Fo − Fc omit map of a DNA molecule bound at one of the two interfaces of monomers a (yellow) and b (orange). B, DNA-binding site showing potential hydrogen-bonding interactions. Hydrogen bonds (3.2 Å or less) are shown in dashed black lines. Additional ambiguous H-bond interactions are also shown, and measure 3.3–3.8 Å.
FIGURE 4.

Conformational changes within Dgt due to DNA binding. A, superposition of DNA-bound and non-DNA bound Dgt dimers. The DNA-bound dimer is represented in yellow and orange (chains a and b, respectively) and the unliganded dimer in gray (as in monomers c and d). Superposition is achieved by aligning the three central helices (α1, α3, α4) of monomer b with the corresponding helices of an unliganded monomer. The resulting shift is most clearly seen for the bottom monomer (yellow) as indicated by the red arrow (3 Å). The expansion of the two individual DNA-binding clefts is indicated by blue arrows. B, effect of DNA binding on helices α10 and α13, and the movement of residue Tyr-272 relative to the dGTP substrate. C, effects of DNA binding on the Dgt active site. Superposition was as shown in A with gray representing the unliganded state. The movement of the Tyr-272 side chain of helix 10 away from the dGTP substrate upon DNA binding is clearly seen. D, additional residue shifts in the Dgt active site upon DNA binding. Note the rotation of His-126 and shift in helix α′18 of the cyan-colored monomer (chain d), which intrudes into the active site. Also shown are the nearby His-69, His-117, Asp-118, and Asp-268 residues that define the HD family and coordinate metal binding for catalysis (9).
TABLE 2.
Dgt helices
The total length of Dgt protein is 505 residues.
| Helix | Start | End |
|---|---|---|
| 1 | 24 | 41 |
| 2 | 42 | 50 |
| 3 | 65 | 90 |
| 4 | 102 | 116 |
| 5 | 125 | 141 |
| 6 | 166 | 181 |
| 7 | 184 | 197 |
| 8 | 202 | 208 |
| 9 | 239 | 250 |
| 10 | 260 | 283 |
| 11 | 287 | 299 |
| 12 | 306 | 320 |
| 13 | 325 | 355 |
| 14 | 355 | 362 |
| 15 | 373 | 390 |
| 16 | 393 | 414 |
| 17 | 421 | 432 |
| 18 | 437 | 447 |
| 19 | 448 | 460 |
| 20 | 468 | 486 |
| 21 | 489 | 501 |
TABLE 3.
Contact surface areas within the Dgt hexamer
Interestingly, the structure also contains density consistent with the presence of two single-stranded nucleic acid fragments of three nucleotides long. At the 3.1-Å resolution of the crystal, no direct identification of this nucleic acid as either DNA or RNA could be made. However, experiments described later in this report strongly indicate that this nucleic acid is ssDNA, which is also consistent with the previously described high affinity of Dgt for ssDNA (2, 16). The DNA molecules occupy two separate monomer-monomer interfaces within one single Dgt dimer. Thus, only one of the three dimers contained the DNA. The DNA is shown in Fig. 2, A, C, and D, as the green inserts between monomers a (yellow) and b (orange). Note that this DNA was not added in the crystallization process, but was either retained or acquired during the lysis or purification procedures (see protocol 1 under “Experimental Procedures”). The bound DNA imparts a clear asymmetry within the hexameric structure when comparing the DNA bound dimers to the non-DNA containing dimers.
Mode of DNA Binding
As seen in Fig. 2D, the head-to-head monomer-monomer configuration within each Dgt dimer provides for two symmetrically located DNA-binding sites per dimer. A close-up view of one of the DNA molecules in its binding site between two Dgt monomers is shown in Fig. 3, A and B. The electron density is fit optimally using DNA bases the size of pyrimidines (in particular, cytosines) as shown in Fig. 3A, but would also be consistent with a pyrimidine/purine mixture, as each of the three base-occupied sites appears to have sufficient room to accommodate a purine base. The first (5′) base (NA1) is less well ordered, with its 5′-phosphate extending into solution. The 3′ end of the DNA is bound deep in the binding pocket, possibly indicating a preference for binding a 3′ terminus at that location, and suggesting that binding to the middle of a DNA strand would require a different ligand or protein conformation. The 3′-hydroxyl forms a hydrogen bond with Arg-337 of one of the monomers (Arg-337′ in Fig. 3B). Phosphate groups are bound to residues Arg-17, Ser-34, Arg-38, Asn-200, and Lys-318′. The DNA bases are bound primarily by hydrophobic interactions along their face, involving residues Tyr-16, Tyr-79′, and Met-334′. The hydrogen bonding edges of the bases have few interactions, with a single hydrogen bond between the 2nd base (NA2) and Arg-337′. The binding pocket appears to have sufficient room for purines, and has no obvious features to confer sequence specificity. Ionic hydrogen bonds are observed between DNA phosphates and side chains Arg-17, Arg-38, and Lys-318′. The phosphate groups of the DNA are solvent exposed.
DNA Binding-induced Conformational Changes
When the DNA-bound dimer (chains a and b) was superimposed on the unliganded dimers (c-d or e-f), and a structural shift due to DNA binding was observed. When comparing the DNA-bound monomers a and b to the non-DNA-bound monomers, the average root mean square deviation for the 479 Cα positions range from 0.38 to 0.41, whereas the values for comparison of the non-DNA-bound monomers are significantly lower (0.12–0.15). The various shifts within the protein due to DNA binding are shown in Fig. 4, A–D. DNA binding results in an expansion of the two binding pockets within the DNA-bound dimer (indicated by the blue arrows in Fig. 4A), also illustrated by the 3-Å shift (red arrow) of monomer a (yellow) relative to an unliganded monomer in gray (see legend for details of the superposition). Importantly, the shift also affects residues in the putative active site of the enzyme (see below).
The Dgt Active Site and DNA Binding
Although no substrate was present in the structure to identify the Dgt active site, this site could be readily identified based on published data on the active sites of other HD nucleases, such as EF1143 from E. faecalis (PDB 3IRH) (6) or human SAMHD1 (26), which both include a bound dNTP substrate. The modeled dGTP substrate (see “Experimental Procedures” for details) is shown in Fig. 4, B–D. Fig. 4, B and C, clearly shows how the binding of DNA leads to changes in the active site. In particular, there are notable shifts of helices α13 and α10. The most significant shift is in the center of helix α10, which along with other helices forms the active site of the protein. In the absence of DNA, this helix is kinked near Tyr-272. With DNA bound, the helix is straightened and there is a shift of Tyr-272, as well as the adjacent Cys-273 (2.4 Å shift of the Cα positions). As a result, Tyr-272 is moved away from the dGTP substrate, in particular the deoxyribose moiety, which may have catalytic consequences.
The view of Fig. 4D shows another interesting feature of the DNA-bound Dgt structure, in particular as it relates to residue Arg-442′ on helix 18′ (monomer d, cyan). This arginine is part of the fingerlike structure (α17-loop-α18) from the adjacent clockwise monomer that intrudes toward the active site. Due to the expansion of the DNA binding cleft and the associated movement of the two DNA-binding monomers, Arg-442′ encroaches further into the active site of the DNA-bound monomer b (orange), and this is likely catalytically relevant. Fig. 4D also shows the close proximity of the presumed metal-binding ligands His-69 (α3), His-117, and Asp-118 (just outside the C terminus of α4), and Asp-268 (α10), the four defining residues (H—HD—D) of the HD family of nucleases (9). Indeed, density was found at this location (see Fig. 5A) consistent with the presence of a metal, most likely Mg2+.
FIGURE 5.
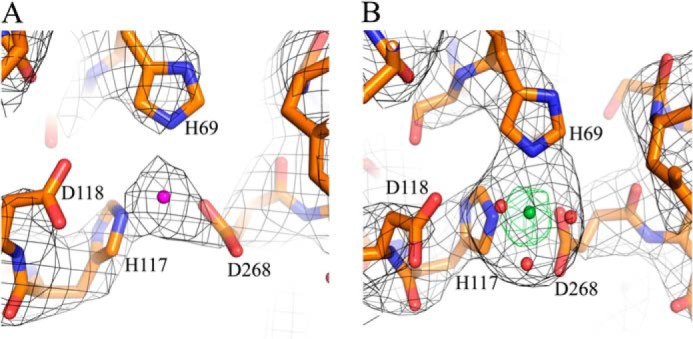
Observed density at the Dgt metal-binding site. The density shown is at the metal-binding site defined by the His-69, His-117, Asp-118, and Asp-268 residues, as also shown in Fig. 4D. The density is most consistent with the presence of Mg2+ for structure 4X9E (purple, panel A) and Ni2+ for structure 4XDS (green, panel B). The small red spheres in panel B indicate water molecules.
A Dgt Crystal Structure without DNA
A modified Dgt purification protocol was used to obtain an enzyme preparation not containing any DNA (see “Experimental Procedures”). Experiments to co-crystallize this enzyme with a DNA of defined length and composition were not successful. Instead, we obtained Dgt crystals lacking any DNA (see Table 1 for crystallographic data). In this apo-structure (PDB code 4XDS), all six monomers within the asymmetric unit were structurally similar. The average root mean square deviation values for 479 Cα positions between individual monomers ranges from 0.13 to 0.27. This structure also showed defined metal-binding sites with the density for Ni2+ ion, as verified by anomalous diffraction (Fig. 5B). This finding was also consistent with the positioning of the metal density (likely Mg2+) noted in the DNA-bound structure (Fig. 5A).
DNA- and RNA-binding Properties of Dgt
Dgt protein purified by a slightly modified procedure (protocol 3, see “Experimental Procedures”) and lacking DNA was used to study the interaction of Dgt with added ssDNA or ssRNA using fluorescence polarization (27). Binding of the fluorescent nucleic acid to protein produces an increase in the fluorescence anisotropy due to its decreased rotational mobility. Titration of increasing amounts of Dgt into a solution containing the labeled DNA yielded a Dgt-dependent increase in anisotropy, reflecting binding of the DNA by Dgt (Fig. 6). The data were fit to a binding curve using a quadratic equation (27), allowing calculation of Kd (dissociation constant) values. Dgt bound to the 22-mer ssDNA with a Kd of 52 nm, indicative of a tight protein-DNA complex. In contrast, no change in anisotropy was seen for a fluorescein-labeled ssRNA of the same length and sequence (Fig. 6), indicating that Dgt has no affinity for the RNA molecule.
FIGURE 6.

Nucleic acid binding to Dgt measured by fluorescence anisotropy. The fluorescence anisotropy (r) of a 22-mer fluorescein-labeled DNA substrate (see “Experimental Procedures”) present at 35 nm was measured. Error bars are standard deviations of triplicate measurements. The dissociation constant (Kd) of 52 ± 5 nm for binding of Dgt to DNA was obtained after fitting the data to a quadratic equation (27). The graph also shows the lack of Dgt binding to the RNA 22-mer of identical sequence, as well as the lack of DNA binding to the Dgt S34D/G37E mutant protein.
dGTP Triphosphohydrolase Activity of Dgt in the Absence and Presence of DNA
E. coli Dgt activity has been described to have a strong preference for dGTP among the four canonical dNTP substrates (1–3, 31). To further investigate the effects of DNA binding on the properties of E. coli Dgt, we analyzed dGTPase activity in the absence and presence of ssDNA. For this experiment we used a 40-mer oligonucleotide (see “Experimental Procedures”). The results are shown in Fig. 7. In the absence of DNA, the dGTP response curve is sigmoidal with an apparent Km of 33 ± 1.5 μm. Upon DNA addition, the curve is hyperbolic with a Km of 10 ± 1.5 μm. Little or no effect of DNA on the Vmax was observed. Hence, the presence of DNA leads to an activation of the dGTPase activity specifically at low substrate concentrations (Km effect) and this likely reflects improved substrate binding.
FIGURE 7.

dGTPase activity of Dgt in absence and presence of DNA. Dgt concentration was 4 nm, and the added DNA was a 40-mer oligomer (see “Experimental Procedures” for sequence) at 50 nm. The curve was fitted using the Hill equation. In the absence of DNA, the Km was calculated at 33 ± 1.5 μm and the Vmax at 4.2 ± 0.2 s−1. In the presence of DNA, the Km was 10 ± 1.5 μm and the Vmax 3 ± 0.3 s−1 (average of three repeated experiments).
A Dgt Mutant Lacking DNA-binding Activity
To further explore the role of the DNA-binding properties of Dgt, a putative DNA binding-defective mutant was created. Using site-directed mutagenesis we generated double S34D and G37E substitutions in the DNA-binding cleft (Fig. 3B), which were expected to introduce significant electrostatic repulsion toward the DNA backbone. The mutant protein eluted as a hexamer, similar to the wild-type protein (data not shown). In the DNA binding analysis using fluorescence anisotropy, no increase in anisotropy was observed for this mutant protein, as shown in Fig. 6. Thus, as expected, the S34D and G37E mutations abolish the ability of Dgt to bind DNA. To investigate the catalytic properties of mutant protein, we assayed its dGTPase activity. Fig. 8A compares the wild-type (WT) and mutant protein in the absence of DNA. Interestingly, the mutant protein is active and, in fact, displays increased activity compared with the WT enzyme at low substrate concentrations. The apparent Km for the mutant was 16 μm, which is 2-fold lower than the value for the wild-type enzyme (33 μm). This suggests that the mutant enzyme has an increased affinity for dGTP in comparison with the WT Dgt. However, as expected, when testing the effect of added DNA to the S34D/G37E enzyme, no stimulation of dGTPase was observed (Fig. 8B).
FIGURE 8.

dGTPase activity of the DNA binding-deficient Dgt mutant (S34D/G37E). A, dGTPase activity of mutant and wild-type Dgt enzyme in the absence of DNA. The Vmax and Km of the mutant enzyme were 3.85 ± 0.65 s−1 and 16 ± 4 μm, respectively, compared with 4.2 ± 0.2 s−1 and 33 ± 1.5 μm for the wild-type enzyme. Conditions were as described in the legend to Fig. 6. B, dGTPase activity of the S34D/G37E enzyme with or without DNA. The DNA concentration (ss 40-mer) was 50 nm, and the enzyme concentration 4 nm. Results are average of three repeated experiments.
Mutator Effect of dgt Mutants
Previous studies showed that deletion of the dgt gene leads to a mutator effect (12). For example, a 40-fold increase in the frequency of A·T → G·C base pair substitution mutations was observed (12). In view of the DNA-binding properties of Dgt, we wished to see whether the loss of DNA binding capacity also has mutational consequences. We constructed an E. coli strain that has the wild-type dgt gene replaced by the double S34D/G37E mutant (see “Experimental Procedures”). In Fig. 9, we show the results of a papillation assay for scoring mutations in the lacZ gene. In this assay the strain carries a lacZ missense mutation that can revert to lac+ by specifically A·T → G·C transition (32). Reversion to lac+ in growing colonies restores the ability to hydrolyze X-Gal and gives rise to a blue dot (mini-colony or papilla). Thus, the number of blue papillae provides an easy visual indicator for enhanced mutagenesis. In the dgt+ strain only very few papillae appear, representing the background of the assay. Significantly increased papillation is seen for the Δdgt strain, confirming the previously reported mutator activity (12). However, a modest increase in papillation was also observed for the dgt S34D/G37E mutant, indicating a mutator effect upon loss of DNA binding capacity.
FIGURE 9.
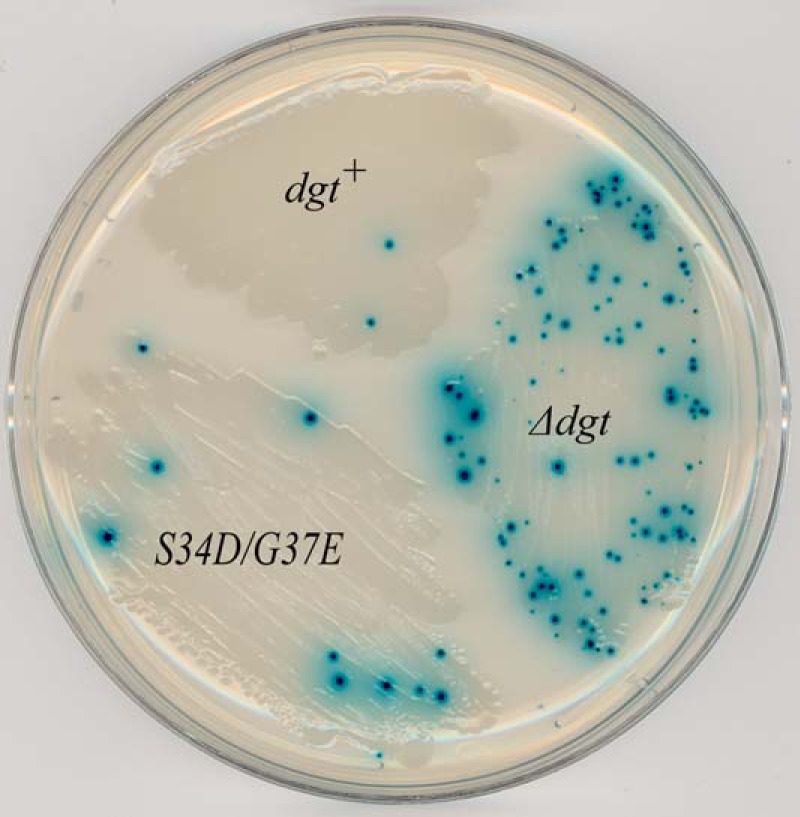
Mutator activity of Δdgt and dgt S34D/G37E E. coli strains. Strongly increased papillation (mutator phenotype) is observed for Δdgt strain and moderate papillation for the DNA-binding defective S34D/G37E strain, indicating a modest mutator phenotype for this strain. The assay scores for the production of lacZ A·T → G·C transition mutations (12). The strains in this experiment were mismatch-repair proficient.
Direct measurements of lac reversion frequencies were also conducted. In this case both mismatch repair (MMR)-proficient and mismatch repair-defective (mutL) backgrounds were investigated. The MMR-defective strains are often used to facilitate scoring of replication errors without interference of MMR. Table 4 shows that no significant increase in lac A·T → G·C transitions could be demonstrated for the DNA-binding mutant in the MMR-proficient background. Nevertheless, in the MMR-defective background a noticeable (2.5-fold) mutator effect was seen for this mutant, compared with the 5-fold mutator effect for the Δdgt mutant in this background. Thus, it follows that loss of the DNA binding capacity in vivo affects the functioning of the protein in the cell, signaling the importance of Dgt activity in the cell.
TABLE 4.
Mutability of dgt strains
Mutability of wild-type (dgt+), Δdgt, and dgt S34D/G37E strains in MMR-proficient (wt) and MMR-deficient (mutL) backgrounds. Mutant frequencies were determined for the lac A·T → G·C strain as described under “Experimental Procedures.” The average values for 28–30 independent cultures in three different experiments are shown with standard deviation.
| Mismatch repair status |
lac+ revertants per 108 cells in indicated dgt strain |
||
|---|---|---|---|
| dgt+ | Δdgt | dgt S34D/G37E | |
| Wild-type | 0.05 ± 0.02 | 0.8 ± 0.02 | 0.06 ± 0.04 |
| mutL | 7 ± 0.6 | 34 ± 3 | 17 ± 4 |
DISCUSSION
E. coli Dgt Triphosphohydrolase Is a Hexamer Capable of Binding DNA
In this work we report on the crystal structure of the dGTP triphosphohydrolase from E. coli (dgt gene product), the first described member of the group of the dNTP triphosphohydrolases (3). Although structures for a total of five dNTPases from distant species, bacteria to humans, have been described (4, 6, 19, 26, 33, 34), the current E. coli structure is unique, as it is the first dNTPase described to contain DNA as an allosteric factor. The presence of ssDNA in the Dgt structure was not expected, but is fully consistent with earlier reports of a strong binding affinity of Dgt to single-stranded DNA (2, 16). Our results showed the enzyme to exist as a hexamer. This finding contrasts with an earlier characterization based on gel filtration and sucrose gradient data supporting a tetrameric structure (2). Within the hexameric structure, the strongest interactions are within the individual Dgt dimers, containing two Dgt chains in 2-fold symmetry with an extensive interaction area of about 2000 Å2.
Mode of DNA Binding
The binding of DNA imparts a clear asymmetry to the overall structure, as the two single-stranded DNAs are found within only one Dgt dimer. Further inspection reveals a plausible explanation as to why only one dimer was found to contain DNA. At the DNA-binding sites, the presence of DNA causes a significant expansion of the binding cleft by about 3.0 Å (1.5 Å in either direction). Due to the hexamer arrangement, this expansion is in the direction of the other two dimers and is likely to restrict expansion of the cleft in the other dimers, such that DNA can no longer be accommodated. From a functional perspective, binding to only one dimer may make sense, as binding of more than two DNA stretches of the same DNA strand would create a topologically complicated situation. Also, note that if the two DNA sites were bound by the same single-stranded DNA molecule, the strand would have to be looped around to enter each cleft in the 5′ to 3′ orientation. Such looping could occur across the outside surface of the protein, although no evidence for this has been obtained. Possibly, the DNAs found in our crystals are small oligomeric stretches left over from the Benzonase treatment of longer DNA during protein isolation. Nevertheless, looped binding to a continuous DNA strand may be a physiologically relevant mode. Earlier studies describing DNA binding to Dgt have suggested cooperative binding to long single-stranded (i.e. bacteriophage M13) DNA, but not small oligomeric DNA, suggesting that Dgt can potentially form a chain of hexamer molecules along a strand of DNA (16).
Within the DNA-bound dimer several important changes are observed influencing the Dgt active site. Notably, the conformational shift involves the important helices α13, closest to the DNA, and the adjacent α10, closest to the active site (see Fig. 4B). Helix α10 is of interest, as without the DNA it contains a significant kink near residue Tyr-272. This residue is well conserved among the dNTP triphosphohydrolases (4, 5, 9) and likely plays an important role. Upon DNA binding, the helix straightens and Tyr-272 moves by 2.4 Å relative to the modeled dGTP in the active site. This shift moves Tyr-272 away from the dGTP substrate, in particular the deoxyribose moiety, which may facilitate substrate binding.
A second consequence of DNA binding that may affect the active site concerns the movement of Arg-442′ that is part of the intruding fingerlike structure from an adjacent monomer (Fig. 2, A and B). Upon DNA binding, Arg-442′ is moved closer to the dGTP substrate, which is likely to affect the positioning of the substrate. This arginine is an attractive candidate for further mutational studies and for probing the precise catalytic mechanism. Note that involvement of residues from the adjacent monomers is a unique feature of the Dgt enzyme that is not seen in any other of the triphosphohydrolases.
Allosteric Regulation and Role of DNA Binding
Measurements of dGTPase activity at low dGTP concentrations yield a sigmoidal curve in the range of 5–10 μm, indicative of positive cooperative behavior. Thus, binding of a dGTP molecule to the active site in one monomer may cause a conformational change that stabilizes binding of dGTP to a second active site, most logically within the same Dgt dimer.
Activity studies in the presence of DNA indicated a modest but notable stimulative effect of DNA. This effect was primarily on the Km (∼3-fold lowering) and did not significantly affect the Vmax (Fig. 7). Thus, like dGTP binding, DNA also has an allosteric effect. We hypothesize that this effect is similar to the cooperative effect of increasing dGTP concentrations. Thus, binding of DNA to the allosteric cleft promotes increased substrate binding in the two active sites of the affected Dgt dimer. Indeed, no cooperativity is noted when DNA is present (hyperbolic curve instead of a sigmoidal curve). This allosteric effect of DNA resembles the stimulative effect of dNTP binding to a second site in the E. faecalis EF1143 enzyme (35).
Further information was obtained from the S34D/G37E mutant carrying negative charges in the DNA binding groove. As expected, the mutant enzyme was defective in DNA binding and could not stimulate dGTP hydrolysis (Fig. 8B). Interestingly, the S34D/G37E substitution by itself altered the kinetic behavior of the enzyme in the absence of DNA (Fig. 8A). The lowering of the Km and loss of sigmoidality suggest that the mutant enzyme is constitutively activated, perhaps due to a widening of the DNA binding cleft due to the amino acid substitutions, which may resemble the DNA-mediated widening of the cleft.
Other dNTP triphosphohydrolases have been reported to be regulated by allosteric factors as well. In particular, several including hSAMHD1 (7, 33, 34), T. thermophilus TT1383 (5), and E. faecalis EF1143 (6) have been reported to exhibit allosteric activation due to dNTP binding at secondary sites distinct from the catalytic site. SAMHD1 is activated by either GTP or dGTP (7, 33, 36), whereas the T. thermophilus TT1383 dNTPase requires two dNTPs as activator/inhibitors (5). Clearly, among these enzymes, E. coli Dgt is unique in its ability to be affected by a ssDNA cofactor. hSAMHD1 has been reported to require DNA or RNA for tetramer stabilization, but no structural evidence with regard to DNA binding is available (17), and for its enzymatic reaction, the dNTP cofactors are clearly the primary regulators. It appears that during evolution multiple options for the allosteric site have developed at the interface of the individual monomers in both tetrameric (EF1143, SAMHD1) and hexameric enzymes (E. coli, TT1383).
In Vivo Role of dGTPase
Understanding the in vivo role of E. coli dGTPase is an intriguing question, and this is similarly true for the other dNTPases of the triphosphohydrolase family. Recent studies on the human SAMHD1 enzyme have indicated a potentially dual role in protection against viruses (33) and in controlling the cellular dNTP pools and cell cycle regulation (37). Bacterial triphosphohydrolases could likewise have multiple functions.
A possible antiviral role in the bacterial systems is indicated by observations in E. coli that overproduction of the enzyme (optA1 strain) is strongly inhibitory to certain bacteriophage mutants (38). Of further significance is that at least one bacteriophage (T7) is known to carry an inhibitor of dGTPase (gp1.2) (39). The possibility that E. coli Dgt functions in control of cellular dNTP levels for fidelity purposes has been addressed (12). We show here that both the loss of Dgt function and its constitutive activation can lead to a mutator phenotype, consistent with the notion that both increases and decreases in individual dNTP levels can promote infidelity (12). The ability of dGTPase to control the in vivo dGTP concentration was also demonstrated by experiments in which E. coli could be killed under conditions of dGTP starvation in a manner resembling the well known phenomenon of thymineless death (13–15). Thus, dNTP triphosphohydrolases could be used for multiple purposes, and may in fact serve different roles in different organisms.
What Is the Specific in Vivo Role of DNA Binding for Dgt?
It is also an interesting question to ask how much of the Dgt protein in an actively growing cell is actually DNA-bound. Generally, single-stranded DNA is of limited availability. Most of it is at the replication fork, where any protein that wishes to bind needs to compete with single-stranded DNA-binding protein. The Kd for Dgt for binding to DNA was reported to be 7-fold higher than for single-stranded DNA-binding protein (16), which binds very cooperatively (40). So, perhaps not much Dgt might be bound to ssDNA under normal conditions. On the other hand, Dgt could get access to the genome under conditions of DNA damage and replication stress, and this is one of the issues that needs to be investigated.
Certain in vivo results with the DNA binding-defective E. coli mutant strain also point to the physiologically important role of DNA binding. The mutant displays a (modest) mutator effect in a mismatch-repair-defective background (Table 4). Although this mutator effect is less than the effect for the Δdgt deletion, one must conclude that the mutant enzyme is defective in vivo in one or more ways, and this constitutes evidence for a role for DNA binding in vivo. Second, we have noted that expression of the S34D/G37E mutant protein from a pET vector, even under conditions of modest overexpression (low isopropyl 1-thio-β-d-galactopyranoside concentration), is clearly toxic to the cells (data not shown). This is an interesting observation that requires further investigation. Possibly, in the mutant, critical changes in the dNTP levels occur that interfere with viability. As a 50-fold overexpression of Dgt in the optA1 strain is not toxic (11, 14), this suggests that the rate-limiting step for Dgt activity in the cell may not be the amount of protein, but rather its activation status. Further studies will be necessary to address this question as well as the broader role of the dNTP triphosphohydrolases in bacterial metabolism.
Acknowledgments
We thank Drs. K. Bebenek and L. Pedersen, NIEHS, for helpful comments on the manuscript. Crystallographic data were collected at the Southeast Regional Collaborative Access Team 22-ID beam line at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract W-31-109-Eng-38.
Note Added in Proof
Some of the values in the version of Table 1 that was published as a Paper in Press on February 18, 2015 have been corrected based on the latest wwPDB validation reports.
This work was supported, in whole or in part, by project number Z01 ES101905 of the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
The atomic coordinates and structure factors (codes 4X9E and 4XDS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
A. Hochkoeppler, D. Singh, and R. M. Schaaper, manuscript in preparation.
- ssDNA
- single-stranded DNA
- LIC
- ligation independent cloning
- Ni-NTA
- nickel-nitrilotriacetic acid
- Ek
- enterokinase
- SEC-MALS
- size exclusion chromatography multi-angle light scattering
- MMR
- mismatch repair.
REFERENCES
- 1. Beauchamp B. B., Richardson C. C. (1988) A unique deoxyguanosine triphosphatase is responsible for the optA1 phenotype of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 85, 2563–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seto D., Bhatnagar S. K., Bessman M. J. (1988) The purification and properties of deoxyguanosine triphosphate triphosphohydrolase from Escherichia coli. J. Biol. Chem. 263, 1494–1499 [PubMed] [Google Scholar]
- 3. Kornberg S. R., Lehman I. R., Bessman M. J., Simms E. S., Kornberg A. (1958) Enzymatic cleavage of deoxyguanosine triphosphate to deoxyguanosine and tripolyphosphate. J. Biol. Chem. 233, 159–162 [PubMed] [Google Scholar]
- 4. Mega R., Kondo N., Nakagawa N., Kuramitsu S., Masui R. (2009) Two dNTP triphosphohydrolases from Pseudomonas aeruginosa possess diverse substrate specificities. FEBS J. 276, 3211–3221 [DOI] [PubMed] [Google Scholar]
- 5. Kondo N., Kuramitsu S., Masui R. (2004) Biochemical characterization of TT1383 from Thermus thermophilus identifies a novel dNTP triphosphohydrolase activity stimulated by dATP and dTTP. J. Biochem. 136, 221–231 [DOI] [PubMed] [Google Scholar]
- 6. Vorontsov I. I., Minasov G., Kiryukhina O., Brunzelle J. S., Shuvalova L., Anderson W. F. (2011) Characterization of the deoxynucleotide triphosphate triphosphohydrolase (dNTPase) activity of the EF1143 protein from Enterococcus faecalis and crystal structure of the activator-substrate complex. J. Biol. Chem. 286, 33158–33166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Powell R. D., Holland P. J., Hollis T., Perrino F. W. (2011) Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 286, 43596–43600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. (2011) SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aravind L., Koonin E. V. (1998) The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 23, 469–472 [DOI] [PubMed] [Google Scholar]
- 10. Quirk S., Bhatnagar S. K., Bessman M. J. (1990) Primary structure of the deoxyguanosine triphosphate triphosphohydrolase-encoding gene (dgt) of Escherichia coli. Gene 89, 13–18 [DOI] [PubMed] [Google Scholar]
- 11. Myers J. A., Beauchamp B. B., Richardson C. C. (1987) Gene 1.2 protein of bacteriophage T7: effect on deoxyribonucleotide pools. J. Biol. Chem. 262, 5288–5292 [PubMed] [Google Scholar]
- 12. Gawel D., Hamilton M. D., Schaaper R. M. (2008) A novel mutator of Escherichia coli carrying a defect in the dgt gene, encoding a dGTP triphosphohydrolase. J. Bacteriol. 190, 6931–6939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Itsko M., Schaaper R. M. (2011) The dgt gene of Escherichia coli facilitates thymine utilization in thymine-requiring strains. Mol. Microbiol. 81, 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Itsko M., Schaaper R. M. (2014) dGTP starvation in Escherichia coli provides new insights into the thymineless-death phenomenon. PLoS Genet. 10, e1004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cohen S. S., Barner H. D. (1954) Studies on unbalanced growth in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 40, 885–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wurgler S. M., Richardson C. C. (1993) DNA binding properties of the deoxyguanosine triphosphate triphosphohydrolase of Escherichia coli. J. Biol. Chem. 268, 20046–20054 [PubMed] [Google Scholar]
- 17. Tüngler V., Staroske W., Kind B., Dobrick M., Kretschmer S., Schmidt F., Krug C., Lorenz M., Chara O., Schwille P., Lee-Kirsch M. A. (2013) Single-stranded nucleic acids promote SAMHD1 complex formation. J. Mol. Med. 91, 759–770 [DOI] [PubMed] [Google Scholar]
- 18. Yan J., Kaur S., DeLucia M., Hao C., Mehrens J., Wang C., Golczak M., Palczewski K., Gronenborn A. M., Ahn J., Skowronski J. (2013) Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J. Biol. Chem. 288, 10406–10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kondo N., Nakagawa N., Ebihara A., Chen L., Liu Z. J., Wang B. C., Yokoyama S., Kuramitsu S., Masui R. (2007) Structure of dNTP-inducible dNTP triphosphohydrolase: insight into broad specificity for dNTPs and triphosphohydrolase-type hydrolysis. Acta Crystallogr. D Biol. Crystallogr. 63, 230–239 [DOI] [PubMed] [Google Scholar]
- 20. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 21. Cowtan K. D., Zhang K. Y. (1999) Density modification for macromolecular phase improvement. Prog. Biophys. Mol. Biol. 72, 245–270 [DOI] [PubMed] [Google Scholar]
- 22. Kleywegt G. J., Read R. J. (1997) Not your average density. Structure 5, 1557–1569 [DOI] [PubMed] [Google Scholar]
- 23. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 26. Zhu C., Gao W., Zhao K., Qin X., Zhang Y., Peng X., Zhang L., Dong Y., Zhang W., Li P., Wei W., Gong Y., Yu X. F. (2013) Structural insight into dGTP-dependent activation of tetrameric SAMHD1 deoxynucleoside triphosphate triphosphohydrolase. Nat. Commun. 4, 2722. [DOI] [PubMed] [Google Scholar]
- 27. Heyduk T., Lee J. C. (1990) Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl. Acad. Sci. U.S.A. 87, 1744–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hill A. V. (1910) The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. 40, iv-viii [Google Scholar]
- 29. Benson R. W., Cafarelli T. M., Godoy V. G. (2011) SOE-LRed: A simple and time-efficient method to localize genes with point mutations onto the Escherichia coli chromosome. J. Microbiol. Methods 84, 479–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quirk S., Seto D., Bhatnagar S. K., Gauss P., Gold L., Bessman M. J. (1989) Location and molecular cloning of the structural gene for the deoxyguanosine triphosphate triphosphohydrolase of Escherichia coli. Mol. Microbiol. 3, 1391–1395 [DOI] [PubMed] [Google Scholar]
- 32. Cupples C. G., Miller J. H. (1989) A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl. Acad. Sci. U.S.A. 86, 5345–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goldstone D. C., Ennis-Adeniran V., Hedden J. J., Groom H. C., Rice G. I., Christodoulou E., Walker P. A., Kelly G., Haire L. F., Yap M. W., de Carvalho L. P., Stoye J. P., Crow Y. J., Taylor I. A., Webb M. (2011) HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382 [DOI] [PubMed] [Google Scholar]
- 34. Ji X., Wu Y., Yan J., Mehrens J., Yang H., DeLucia M., Hao C., Gronenborn A. M., Skowronski J., Ahn J., Xiong Y. (2013) Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol. 20, 1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vorontsov I. I., Wu Y., DeLucia M., Minasov G., Mehrens J., Shuvalova L., Anderson W. F., Ahn J. (2014) Mechanisms of allosteric activation and inhibition of the deoxyribonucleoside triphosphate triphosphohydrolase from Enterococcus faecalis. J. Biol. Chem. 289, 2815–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amie S. M., Bambara R. A., Kim B. (2013) GTP is the primary activator of the anti-HIV restriction factor SAMHD1. J. Biol. Chem. 288, 25001–25006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franzolin E., Pontarin G., Rampazzo C., Miazzi C., Ferraro P., Palumbo E., Reichard P., Bianchi V. (2013) The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, 14272–14277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saito H., Richardson C. C. (1981) Genetic analysis of gene 1.2 of bacteriophage T7: isolation of a mutant of Escherichia coli unable to support the growth of T7 gene 1.2 mutants. J. Virol. 37, 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huber H. E., Beauchamp B. B., Richardson C. C. (1988) Escherichia coli dGTP triphosphohydrolase is inhibited by gene 1.2 protein of bacteriophage T7. J. Biol. Chem. 263, 13549–13556 [PubMed] [Google Scholar]
- 40. Williams K. R., Spicer E. K., LoPresti M. B., Guggenheimer R. A., Chase J. W. (1983) Limited proteolysis studies on the Escherichia coli single-stranded DNA binding protein. Evidence for a functionally homologous domain in both the Escherichia coli and T4 DNA binding proteins. J. Biol. Chem. 258, 3346–3355 [PubMed] [Google Scholar]