

Background: Mechanisms underlying GPCR-mediated EGFR transactivation are not fully defined.
Results: Heterotrimeric G proteins directly regulate membrane-localized MMP14/MT1-MMP, resulting in HB-EGF release and EGFR transactivation.
Conclusion: These results define a previously unrecognized, membrane-delimited mechanism for EGFR transactivation.
Significance: This mechanism likely plays a role in settings that involve GPCR-RTK transactivation and may represent new therapeutic opportunities.
Keywords: Epidermal Growth Factor Receptor (EGFR), G Protein-coupled Receptor (GPCR), Matrix Metalloproteinase (MMP), Membrane Protein, Signal Transduction
Abstract
Agonist stimulation of G protein-coupled receptors (GPCRs) can transactivate epidermal growth factor receptors (EGFRs), but the precise mechanisms for this transactivation have not been defined. Key to this process is the protease-mediated “shedding” of membrane-tethered ligands, which then activate EGFRs. The specific proteases and the events involved in GPCR-EGFR transactivation are not fully understood. We have tested the hypothesis that transactivation can occur by a membrane-delimited process: direct increase in the activity of membrane type-1 matrix metalloprotease (MMP14, MT1-MMP) by heterotrimeric G proteins, and in turn, the generation of heparin-binding epidermal growth factor (HB-EGF) and activation of EGFR. Using membranes prepared from adult rat cardiac myocytes and fibroblasts, we found that MMP14 activity is increased by angiotensin II, phenylephrine, GTP, and guanosine 5′-O-[γ-thio]triphosphate (GTPγS). MMP14 activation by GTPγS occurs in a concentration- and time-dependent manner, does not occur in response to GMP or adenosine 5′-[γ-thio]triphosphate (ATPγS), and is not blunted by inhibitors of Src, PKC, phospholipase C (PLC), PI3K, or soluble MMPs. This activation is specific to MMP14 as it is inhibited by a specific MMP14 peptide inhibitor and siRNA knockdown. MMP14 activation by GTPγS is pertussis toxin-sensitive. A role for heterotrimeric G protein βγ subunits was shown by using the Gβγ inhibitor gallein and the direct activation of recombinant MMP14 by purified βγ subunits. GTPγS-stimulated activation of MMP14 also results in membrane release of HB-EGF and the activation of EGFR. These results define a previously unrecognized, membrane-delimited mechanism for EGFR transactivation via direct G protein activation of MMP14 and identify MMP14 as a heterotrimeric G protein-regulated effector.
Introduction
G protein-coupled receptors (GPCRs)2 are the largest family of membrane signaling receptors in the human genome and genomes of numerous other eukaryotes. The canonical mechanism for the alteration of cell function by GPCRs is their activation of membrane-localized heterotrimeric (αβγ) G proteins, subunits of which change the activity of membrane enzymes (e.g. adenylyl cyclases, phospholipase C-β (PLC-β), and certain ion channels). An additional, important aspect of cell regulation by GPCRs is their ability to transactivate receptor tyrosine kinases (RTKs), in particular epidermal growth factor receptors (EGFRs), thereby perturbing cell and tissue function via effects that include alterations in mitogenesis and differentiation (1, 2).
Matrix metalloproteases (MMPs) are zinc endopeptidases, most of which are cytoplasmic. Certain MMPs can localize to the plasma membrane, including MMP14, also known as membrane type-1 matrix metalloprotease (MT1-MMP) (3). MMP14 has been implicated in numerous physiological states and pathological events, including tissue development, angiogenesis, cardiac hypertrophy, and tumor invasion (4–12). The activation of MMPs can occur by various GPCRs depending on cell type, but precise mechanisms of this activation have not been elucidated (13–16).
The signaling mechanisms responsible for the transactivation of EGFRs by GPCRs are important for numerous physiological processes and disease-related responses. Current evidence suggests that transactivation depends not only on cell type, but also on the type and class of GPCRs and cellular environment (17, 18). GPCR-stimulated EGFR transactivation is mediated by both ligand-dependent and ligand-independent mechanisms. Ligand-independent activation has been shown to depend on G protein-coupled receptor kinase-dependent recruitment of β-arrestin and Src (19, 20) and results in Src-dependent activation of the intracellular tyrosine kinase domain of EGFR (21, 22). Ligand-dependent activation has been proposed to involve a triple membrane pass system wherein MMPs promote the cleavage and “shedding” of EGF-like ligands, including heparin-binding epidermal growth factor (HB-EGF), which in turn activate EGFRs in an autocrine/paracrine manner (23, 24). Although a number of intracellular molecules and pathways have been investigated for their role in the ligand-dependent triple membrane pass system, the possibility of a direct, membrane-delimited mechanism for ligand-dependent EGFR transactivation has not previously been assessed.
Based on the membrane localization of MMP14 and the ability of other MMPs to function as intermediates in GPCR-mediated EGFR transactivation, we reasoned that MMP14 might be akin to enzymes and ion channels that are activated by heterotrimeric G proteins in the plasma membrane. Using membrane preparations from various cell types, we show that the activation of GPCRs and heterotrimeric G proteins can activate MMP14. We find a role for pertussis toxin-sensitive G proteins and that Gβγ can activate MMP14 in the absence of intracellular mediators. This activation leads to the release of HB-EGF and the activation of EGFR. These results identify MMP14 as a membrane effector of heterotrimeric G proteins and define a membrane-delimited mechanism for GPCR-promoted transactivation of EGFRs, thus implying that EGFR transactivation need not require intracellular mediators.
EXPERIMENTAL PROCEDURES
Reagents
GTP, GTPγS, GMP, ATPγS, Gö6983, phenylephrine, AG1478, phosphatase inhibitor cocktail 3, EGF, and PP1/PP2 were all purchased from Sigma-Aldrich. Galardin, wortmannin, and MMP-2/9 inhibitor III were purchased from EMD Millipore (Billerica, MA). Angiotensin II, U73122, and gallein were purchased from Tocris Bioscience (Ellisville, MO). Peptide G (Ac-GACFSIAHCGA-NH2) was custom-synthesized from Biomatik (Wilmington, DE). 4-Aminophenylmercuric acetate (APMA) was purchased from AnaSpec (Fremont, CA). Recombinant MMP14 (rMMP14) was purchased from Abcam (Cambridge, MA), and active βγ subunits were generously donated by Dr. Alan Smrcka (University of Rochester Medical Center, Rochester, NY).
Cardiac Myocyte (CM) and Cardiac Fibroblast (CF) Isolation and Culture
CMs and CFs were isolated from adult (3–6-month-old) male Sprague-Dawley rats as described previously (25) using a modified reverse-Langendorff apparatus. The heart was minced and triturated in the presence of collagenase to separate cells from decellularized matrix. The CMs and CFs were separated by gravity separation and rinsed three times in ice-cold PBS before being used for membrane preparations.
Cell Membrane Preparation
Membranes were prepared from freshly isolated and PBS-rinsed primary CMs or CFs by Dounce homogenization in hypotonic buffer for 30 s followed by centrifugation at 300 × g for 5 min at 4 °C and then centrifugation of the supernatant at 5000 × g for 10 min at 4 °C. The resulting pellet was rinsed in high K+ buffer (500 mm) for 10 min at 4 °C and centrifuged again at 5000 × g for 10 min at 4 °C. The supernatant was discarded, and the pellet was resuspended in activity assay buffer for immediate use or resuspended in PBS and stored at −80 °C.
siRNA Transfection
NIH3T3 cells were plated in 10-cm plates and grown to 70% confluency in DMEM with 10% FBS plus penicillin-streptomycin. Cells were transfected with scrambled negative control siRNA (Ambion) or MMP14 Silencer Select siRNA (catalog number 4390771; Ambion) at 5 nm final concentration using Lipofectamine RNAiMAX (Invitrogen). After 4 h, the medium was replaced with serum-free DMEM; incubations were continued for 48 h before use for membrane preparations for MMP14 activity assays.
MMP14 Activity Assay
Membrane MMP14 activity was measured using a SensoLyte 520 MMP14 assay kit (AnaSpec, Fremont, CA) according to the manufacturer's instructions. Briefly, membranes were resuspended in assay buffer (final volume, 100 μl) and treated as indicated. Samples were then incubated for 2 h at 37 °C. 50 μl of sample was added to 50 μl of MMP14 substrate solution, and optical fluorescence intensity was measured at excitation/emission = 490 nm/520 nm every 30 min, or at a 3-h end-point, as indicated.
HB-EGF ELISA
Aliquots of the supernatant from stimulated CM membranes were collected and assessed for HB-EGF release using an ELISA kit (NovaTeinBio, Cambridge, MA) according to the manufacturer's instructions. Inhibitors were given as a 30-min pretreatment to membranes at 37 °C prior to a 60-min stimulation of the indicated activators at 37 °C. Optical density was measured at 450 nm, and results are normalized to control HB-EGF release.
EGFR Activation Assay
Phosphorylation of EGFR from CFs was measured using an EGFR (Tyr-845) In-Cell ELISA kit (Abcam, Cambridge, MA) according to the manufacturer's instructions. Primary rat CFs were seeded onto a 96-well tissue culture plate at 20,000 cells/well and incubated overnight at 37 °C, 5% CO2. Prior to treatment, cells were switched to serum-free DMEM that contained 0.1% Triton X-100 and phosphatase inhibitor. Inhibitors were added as a 30-min pretreatment to cells at 37 °C prior to a 60-min stimulation of the indicated activators at 37 °C. Optical density was measured at 450 nm.
Statistical Analysis
All data analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA). Data are presented as means ± S.E. Statistical calculations were done using one-way analysis of variance with either Dunnett's or Bonferroni's post tests. All experiments were conducted at least 3 times. Values of p ≤ 0.05 were considered significant.
RESULTS
MMP14 Is Directly Activated by G Protein βγ Subunits in a Membrane-delimited and Pertussis Toxin-sensitive Manner
Using membranes prepared from primary isolates of adult rat CMs, we found that GTP (Fig. 1A) and the non-hydrolyzable GTP analog GTPγS (Fig. 1B) increase the activity of MMP14 in a concentration-dependent manner. Enhancement of MMP14 activity did not occur with GMP (Fig. 1C) or ATPγS (Fig. 1D), thus showing specificity for the activation of G proteins. The action of GPCRs involves the formation of a receptor/heterotrimeric G protein complex and the exchange of GTP for Gα-bound GDP, leading to altered activity of downstream effector proteins by the Gα and Gβγ subunits. Binding and exchange of GTP for GDP are key steps in the activation of G proteins and proximal events following receptor activation. We used APMA, an organic mercury compound that directly activates MMPs, as a positive control for MMP14 activation in this and other experiments. APMA-promoted and GTPγS-mediated stimulations of MMP14 activity were inhibited by the non-selective MMP inhibitor galardin (Fig. 1, C–E). The stimulation of MMP14 activity by GTPγS continued for the duration of the 180-min time course (Fig. 1E). In membranes not preactivated by GTPγS, there was a lag in the increase in MMP14 activity, consistent with the time required to exchange GTPγS for GDP bound to the Gα subunit of heterotrimeric G proteins (Fig. 1F).
FIGURE 1.

MMP14 is directly activated by G protein βγ subunits in a membrane-delimited and pertussis toxin-sensitive manner. A–D, concentration-dependent stimulation of MMP14 activity in adult rat CM membranes by GTP (A) and GTPγS (B), but not GMP (C) or ATPγS (D). RFU, relative fluorescence units. VEH, vehicle. E, APMA (1 mm), an activator of MMPs, was used as a positive control in studies with GMP and ATPγS; the MMP inhibitor galardin (20 μm) blocked APMA-increased activity. Time-dependent stimulation of MMP14 activity by GTPγS (100 μm) is blocked by galardin (20 μm). F, lack of preactivation with reactants resulted in a lag for MMP14 activity. G, siRNA knockdown of MMP14, but not scrambled control siRNA, abolished GTPγS (100 μm)- and APMA (1 mm)-stimulated MMP14 activity in NIH3T3 cell membranes. H, a selective MMP14 peptide inhibitor, PG (250 μm), blocked GTPγS-stimulated (100 μm) MMP14 activity in CM membranes. I, Ang II (1 μm) stimulation of MMP14 activity is blocked by galardin (20 μm) and PG (250 μm). J, phenylephrine increased MMP14 activity. K, stimulation of MMP14 activity in adult rat CF membranes with APMA (1 mm) or GTPγS (100 μm) is blocked by PG (250 μm). L–P, GTPγS (100 μm)-stimulated MMP14 activity remained unchanged in the presence of the protein kinase C inhibitor Gö6983 (10 μm) (L), the PI3K inhibitor wortmannin (100 nm) (M), the PLC inhibitor U73122 (10 μm) (N), the Src inhibitors PP1 (1 μm) or PP2 (10 μm) (O), or an MMP-2/9 inhibitor (20 μm) (P).Q, the Gβγ subunit inhibitor gallein (10 μm) abolished the GTPγS (100 μm)-stimulated increase in MMP14 activity. R, stimulation of rMMP14 activity by APMA (1 mm) or active recombinant Gβ1γ2 subunits (10 μm) is inhibited by PG (250 μm). No activity was detected in the absence of rMMP14. S, APMA (1 mm) and GTPγS (100 μm) increased MMP14 activity in NIH3T3 cell membranes. T, GTPγS-promoted (100 μm) MMP14 activity was decreased in cell membranes prepared from NIH3T3 cells incubated with pertussis toxin for 16 h. The activation of MMP14 by APMA was unaltered by pertussis toxin treatment. *, p < 0.05, **, p < 0.01, ***, p < 0.005, ****, p < 0.001 as compared with control/vehicle; n ≥ 3 for all experiments.
Although the substrate used in the activity assay we employed is selective for MMP14, we wanted to confirm that the guanine nucleotide-stimulated activity was not attributable to the action of other MMPs. We thus used siRNA for MMP14 in NIH3T3 cells, as well as peptide G (PG), a specific MMP14 inhibitor (26), to inhibit MMP14 in CMs. siRNA knockdown of both MMP14 (Fig. 1G) and PG (Fig. 1H) abolished GTPγS-stimulated MMP14 activity; scrambled negative control siRNA had no effect (Fig. 1G).
To determine whether the activation of a GPCR could increase MMP14 activity in CM membranes, we tested the effect of angiotensin II (Ang II). Ang II, a peptide hormone, can promote cardiac hypertrophy, signaling via heterotrimeric G proteins to cause vasoconstriction and an increase in blood pressure (27). Ang II activated MMP14 activity in CM membranes; this stimulation was blocked by galardin and PG (Fig. 1I). The adrenergic receptor agonist phenylephrine also increased MMP14 activity (Fig. 1J).
We assessed the activation of MMP14 by GTPγS using membranes from adult rat primary CFs to test whether this effect was specific to CMs. Both APMA and GTPγS enhanced MMP14 activity in CF membranes, and the increases in activity by both were inhibited by PG (Fig. 1K), thus showing that this activation is not limited to CMs.
Previous research aimed at parsing the signaling network involved in GPCR ligand-dependent and ligand-independent transactivation of EGFR has shown a role for multiple entities depending on cell type, cellular environment, and the GPCRs that are activated (15, 28). We tested whether the increase in MMP14 activity produced by GTPγS in CM membranes might involve intracellular mediators (i.e. PKC, PLC, PI3K, and Src) or soluble MMPs (i.e. MMP-2/9). The PKC inhibitor Gö6983 (Fig. 1L), PI3K inhibitor wortmannin (Fig. 1M), PLC inhibitor U73122 (Fig. 1N), Src inhibitors PP1 and PP2 (Fig. 1O), and an MMP-2/9 inhibitor (Fig. 1P) failed to inhibit GTPγS-promoted MMP14 activity. All inhibitors were incubated with membranes for 30 min prior to the addition of GTPγS and were tested at concentrations that block their respective targets.
Both Gα and Gβγ subunits have been implicated in EGFR transactivation (29, 30). Because Gβγ can directly activate membrane-bound effectors such as ion channels (31), adenylyl cyclases, and PI3K (32), we tested the ability of the Gβγ inhibitor gallein to block GTPγS-stimulated MMP14 activity. We found that gallein inhibited stimulation of MMP14 activity by GTPγS in CM membranes (Fig. 1Q). As a further means to evaluate the role of Gβγ and determine whether the activation of MMP14 was due to a direct interaction of Gβγ and MMP14, we used an in vitro system with rMMP14 and purified Gβ1γ2 subunits. Both APMA and Gβ1γ2 subunits enhanced rMMP14 activity (Fig. 1R). This increase in activity was blocked by PG, a result consistent with the idea that the response was mediated by rMMP14. The addition of active Gβγ subunits alone was not able to cleave the FRET substrate (Fig. 1R). Thus, Gβγ subunits are able to activate MMP14.
To further characterize the role of G proteins in the activation of MMP14, we examined the effect of pertussis toxin on GTPγS-promoted MMP14 activity. EGFR transactivation has been shown to occur via both pertussis toxin-sensitive and pertussis toxin-insensitive pathways depending on the GPCR agonist and cell type involved (18, 33). We treated NIH3T3 cells for 16 h with pertussis toxin and then isolated membranes and assayed MMP14 activity. Control NIH3T3 cell membranes showed APMA- and GTPγS-promoted MMP14 activity (Fig. 1S). GTPγS-promoted MMP14 activity, but not activation by APMA, was lost in membranes prepared from cells treated with pertussis toxin (Fig. 1T). This decrease in GTPγS-stimulated MMP14 activity may be attributable to loss of activity of Gβγ or Gαi/o subunits (34). However, because Gβγ is able to directly activate rMMP14 in vitro, the loss in activity in response to pertussis toxin is consistent with a role for Gβγ subunits.
Membrane-delimited G Protein Activation of MMP14 Releases HB-EGF and Transactivates EGFR
MMPs are known to cleave EGF ligands, in particular HB-EGF, following activation by certain GPCRs (23, 35–37). We thus tested whether the membrane-delimited activation of MMP14 by G proteins could result in membrane shedding of HB-EGF and subsequent activation of EGFR.
We found that incubation of adult rat CM membranes with GTPγS for 60 min increased the release of HB-EGF. This release was blocked by the non-selective MMP inhibitor galardin, the MMP14 inhibitor PG, and the Gβγ subunit inhibitor gallein (Fig. 2A).
FIGURE 2.

Membrane-delimited G protein activation of MMP14 results in release of HB-EGF and transactivation of EGFR. A–C, GTPγS (100 μm)- (A), rMMP14 (100 nm)- (B), and APMA (1 mm) (C)-stimulated HB-EGF release from adult rat CM membranes in the presence of the MMP inhibitor galardin (20 μm), the selective MMP14 inhibitor PG (250 μm), or the Gβγ subunit inhibitor gallein (10 μm). RU, relative units. D, the adrenergic agonist phenylephrine (10 μm) increased HB-EGF release from CM membranes. E and F, EGF (100 nm) (E) and GTPγS (100 μm) (F) activate EGFR in adult rat CFs. EGF- and GTPγS-mediated activation of EGFR is inhibited by the selective EGFR antagonist AG1478 (2 μm). PG (250 μm), but not the selective MMP-2/9 inhibitor (inh, 10 μm), blocks GTPγS-mediated EGFR activation.*, p < 0.05, **, p < 0.01, as compared with control; n ≥ 3 for all experiments.
To evaluate whether the release of HB-EGF from CM membranes was due to the activity of MMP14, we tested whether rMMP14 could release HB-EGF and found that rMMP14 yielded results similar to those observed with GTPγS. This release of HB-EGF was blocked by galardin, but not by gallein (Fig. 2B).
In addition, the activation of MMPs by APMA released HB-EGF from CM membranes, and this release was blocked by pretreatment with either galardin or PG (Fig. 2C). Because APMA directly activates MMP14, HB-EGF release in response to APMA should not depend on Gβγ subunits, unlike release in response to GTPγS, but similar to rMMP14 treatment. Consistent with this prediction, HB-EGF release by APMA was not inhibited by gallein (Fig. 2C). The adrenergic agonist phenylephrine increased HB-EGF release from CM membranes (Fig. 2D), consistent with its ability to activate MMP14 (Fig. 1J).
To determine whether the membrane-delimited pathway of MMP14 activation by G proteins can transactivate EGFRs, we used adult rat CFs to assess EGFR Tyr-845 phosphorylation of Triton-permeabilized whole cells. EGF was used as a positive control for EGFR activation and was blocked by the EGFR inhibitor, AG1478 (Fig. 2E). GTPγS increased EGFR phosphorylation; this response was inhibited by AG1478 and by the MMP14 inhibitor PG, but not by the MMP-2/9 inhibitor (Fig. 2F), consistent with results in Fig. 1. Thus, the activation of MMP14 by G proteins can increase EGFR phosphorylation.
DISCUSSION
For almost two decades, it has been known that GPCRs can transactivate RTKs (e.g. EGFR) (17, 18), but a full understanding of the mechanisms involved has not been established. The current study provides a new mechanism for GPCR-RTK transactivation: the activation of a specific membrane-bound matrix metalloprotease (MMP14) acting as an effector whose activity is directly enhanced by activated G proteins (Gβγ and perhaps Gα), and in turn, an MMP14-promoted release of HB-EGF and the activation of EGFR (Fig. 3). We show here that this signaling pathway is mediated by MMP14 activation in response to GTP, GTPγS, and GPCR agonists and occurs in adult rat CM and CF membranes and 3T3 cell membranes.
FIGURE 3.
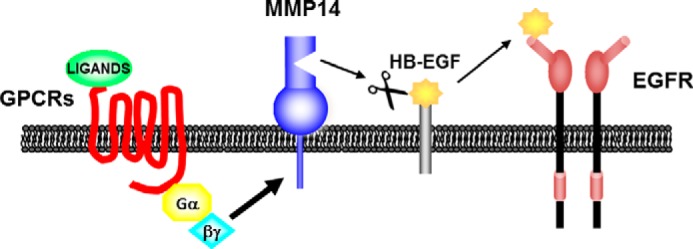
Model of GPCR/EGFR transactivation. GPCR agonist-promoted activation of a heterotrimeric G protein leads to direct activation of MMP14. MMP14 cleaves membrane-bound proteins, including HB-EGF, which following release occupies and activates EGFRs.
The kinetics of MMP14 activation are consistent with previous data regarding substrate cleavage by MMP (38). Several signaling entities can reportedly contribute to MMP activation by GPCRs, including PLC, PI3K, PKC, β-arrestin, and c-Src alone or in combination with reactive oxygen species (13, 14, 19, 39), but based on the use of inhibitors, we found that none of these nor MMP2 or MMP9 were required for the membrane-delimited, heterotrimeric G protein-mediated increase in MMP14 activity. These results thus support the conclusion that the increase in MMP14 activity occurs within the membrane and without the necessity of intracellular mediators or MMP2 or MMP9.
We also observed Ang II-promoted activation of MMP14 in CM membranes. Agonist activation of the type-1 Ang receptor is associated with the development of cardiac hypertrophy, which occurs in cultures of neonatal CM in an MMP/EGFR-dependent manner (40). Our results suggest that this may occur via direct activation of MMP14 by heterotrimeric G proteins.
It has been suggested that each of the four heterotrimeric G protein families (Gi/o, Gq/11, Gs, G12/13) could be involved in the transactivation of RTKs by GPCRs. Other studies have described RTK transactivation by the Gβγ subunits of Gi- and Gq-coupled GPCRs (30). Our findings that a Gβγ inhibitor blocks MMP14 activation by GTPγS and that purified Gβ1γ2 increases activity of purified rMMP14 imply a direct role for Gβγ in MMP14 activation. Such results are consistent with previous data that suggest Gβγ is involved in RTK transactivation (e.g. Ref. 30) but do not exclude a contribution by Gα subunits, including pertussis toxin-sensitive G proteins, such as Gαi/o, because incubation with pertussis toxin inhibits GTPγS-promoted activation of MMP14. Studies in COS-7 cells have shown pertussis toxin sensitivity of Gβγ-mediated EGFR transactivation, thus supporting our results implicating Gβγ in the activation of MMP14 (41).
A hallmark of GPCR-RTK transactivation is the release (shedding) of ligands for RTKs from the cell surface. Several ligands have been reported for EGFR transactivation, including HB-EGF, amphiregulin, and transforming growth factor-α (16). We find that phenylephrine- and GTPγS-stimulated MMP14 activation release HB-EGF (but not EGF (data not shown)) and that this shedding leads to tyrosine phosphorylation of EGFR. MMP14 is known to release other proteins from the plasma membrane (42). Perhaps GPCRs and heterotrimeric G proteins release other such entities. In addition, MMP14 and RTKs have been implicated in other cellular responses and disease settings (4, 43–45). We speculate that the activation of MMP14 by heterotrimeric G proteins may contribute to such responses and diseases and that therapeutic approaches designed to block heterotrimeric G protein-mediated activation of MMP14 may be useful in such settings.
Taken together, the current results identify MMP14 as an effector of heterotrimeric G proteins and a previously unappreciated membrane signaling module: GPCR/G protein/MMP14-mediated transactivation of EGFR (Fig. 3). This mechanism may contribute to settings of GPCR-RTK transactivation and may represent new therapeutic opportunities, especially as related to the activation and actions of MMP14.
Acknowledgments
We thank Dr. Alan Smrcka (University of Rochester Medical Center) for generously donating Gβ1γ2 subunits for these studies. We also thank Drs. Cicely Schramm, Aaron Snead, and Carrie Wade for helpful feedback while preparing this manuscript.
This work was supported by an American Heart Association postdoctoral fellowship (13POST14810015) (to A. C. O.).
- GPCR
- G protein-coupled receptor
- EGFR
- epidermal growth factor receptor
- MMP
- matrix metalloprotease
- rMMP14
- recombinant matrix metalloprotease 14
- HB-EGF
- heparin-binding epidermal growth factor
- ATPγS
- adenosine 5′-[γ-thio]triphosphate
- GTPγS
- guanosine 5′-O-[γ-thio]triphosphate
- PLC
- phospholipase C
- Src
- tyrosine-protein kinase CSK
- RTK
- receptor tyrosine kinase
- PG
- peptide G
- CM
- cardiac myocyte
- CF
- cardiac fibroblast
- APMA
- 4-aminophenylmercuric acetate
- Ang II
- angiotensin II.
REFERENCES
- 1. Gschwind A., Zwick E., Prenzel N., Leserer M., Ullrich A. (2001) Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 20, 1594–1600 [DOI] [PubMed] [Google Scholar]
- 2. Rozengurt E. (2007) Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 213, 589–602 [DOI] [PubMed] [Google Scholar]
- 3. Spinale F. G. (2007) Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol. Rev. 87, 1285–1342 [DOI] [PubMed] [Google Scholar]
- 4. Holmbeck K., Bianco P., Yamada S., Birkedal-Hansen H. (2004) MT1-MMP: a tethered collagenase. J. Cell. Physiol. 200, 11–19 [DOI] [PubMed] [Google Scholar]
- 5. Itoh Y. (2006) MT1-MMP: a key regulator of cell migration in tissue. IUBMB Life 58, 589–596 [DOI] [PubMed] [Google Scholar]
- 6. Arroyo A. G., Genís L., Gonzalo P., Matías-Román S., Pollán A., Gálvez B. G. (2007) Matrix metalloproteinases: new routes to the use of MT1-MMP as a therapeutic target in angiogenesis-related disease. Curr. Pharm. Des. 13, 1787–1802 [DOI] [PubMed] [Google Scholar]
- 7. Gingras D., Béliveau R. (2010) Emerging concepts in the regulation of membrane-type 1 matrix metalloproteinase activity. Biochim. Biophys. Acta 1803, 142–150 [DOI] [PubMed] [Google Scholar]
- 8. Strongin A. Y. (2010) Proteolytic and non-proteolytic roles of membrane type-1 matrix metalloproteinase in malignancy. Biochim. Biophys. Acta 1803, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sato H., Takino T. (2010) Coordinate action of membrane-type matrix metalloproteinase-1 (MT1-MMP) and MMP-2 enhances pericellular proteolysis and invasion. Cancer Sci. 101, 843–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sounni N. E., Paye A., Host L., Noël A. (2011) MT-MMPS as regulators of vessel stability associated with angiogenesis. Front. Pharmacol. 2, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Turner N. A., Porter K. E. (2012) Regulation of myocardial matrix metalloproteinase expression and activity by cardiac fibroblasts. IUBMB Life 64, 143–150 [DOI] [PubMed] [Google Scholar]
- 12. Spinale F. G., Janicki J. S., Zile M. R. (2013) Membrane-associated matrix proteolysis and heart failure. Circ. Res. 112, 195–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah B. H., Yesilkaya A., Olivares-Reyes J. A., Chen H. D., Hunyady L., Catt K. J. (2004) Differential pathways of angiotensin II-induced extracellularly regulated kinase 1/2 phosphorylation in specific cell types: role of heparin-binding epidermal growth factor. Mol. Endocrinol. 18, 2035–2048 [DOI] [PubMed] [Google Scholar]
- 14. Shah B. H., Shah F. B., Catt K. J. (2006) Role of metalloproteinase-dependent EGF receptor activation in α-adrenoceptor-stimulated MAP kinase phosphorylation in GT1–7 neurons. J. Neurochem. 96, 520–532 [DOI] [PubMed] [Google Scholar]
- 15. Belcheva M. M., Szùcs M., Wang D., Sadee W., Coscia C. J. (2001) μ-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J. Biol. Chem. 276, 33847–33853 [DOI] [PubMed] [Google Scholar]
- 16. Ohtsu H., Dempsey P. J., Eguchi S. (2006) ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am. J. Physiol. Cell Physiol. 291, C1–10 [DOI] [PubMed] [Google Scholar]
- 17. Daub H., Weiss F. U., Wallasch C., Ullrich A. (1996) Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379, 557–560 [DOI] [PubMed] [Google Scholar]
- 18. Daub H., Wallasch C., Lankenau A., Herrlich A., Ullrich A. (1997) Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 16, 7032–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Noma T., Lemaire A., Naga Prasad S. V., Barki-Harrington L., Tilley D. G., Chen J., Le Corvoisier P., Violin J. D., Wei H., Lefkowitz R. J., Rockman H. A. (2007) β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest. 117, 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim J., Ahn S., Rajagopal K., Lefkowitz R. J. (2009) Independent β-arrestin2 and Gq/protein kinase Cζ pathways for ERK stimulated by angiotensin type 1A receptors in vascular smooth muscle cells converge on transactivation of the epidermal growth factor receptor. J. Biol. Chem. 284, 11953–11962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bokemeyer D., Schmitz U., Kramer H. J. (2000) Angiotensin II-induced growth of vascular smooth muscle cells requires an Src-dependent activation of the epidermal growth factor receptor. Kidney Int. 58, 549–558 [DOI] [PubMed] [Google Scholar]
- 22. Amorino G. P., Deeble P. D., Parsons S. J. (2007) Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene 26, 745–756 [DOI] [PubMed] [Google Scholar]
- 23. Prenzel N., Zwick E., Daub H., Leserer M., Abraham R., Wallasch C., Ullrich A. (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402, 884–888 [DOI] [PubMed] [Google Scholar]
- 24. Miyamoto S., Hirata M., Yamazaki A., Kageyama T., Hasuwa H., Mizushima H., Tanaka Y., Yagi H., Sonoda K., Kai M., Kanoh H., Nakano H., Mekada E. (2004) Heparin-binding EGF-like growth factor is a promising target for ovarian cancer therapy. Cancer Res. 64, 5720–5727 [DOI] [PubMed] [Google Scholar]
- 25. Yokoyama U., Patel H. H., Lai N. C., Aroonsakool N., Roth D. M., Insel P. A. (2008) The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc. Natl. Acad. Sci. U.S.A. 105, 6386–6391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suojanen J., Salo T., Koivunen E., Sorsa T., Pirilä E. (2009) A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol. Ther. 8, 2362–2370 [DOI] [PubMed] [Google Scholar]
- 27. Smith N. J., Chan H. W., Osborne J. E., Thomas W. G., Hannan R. D. (2004) Hijacking epidermal growth factor receptors by angiotensin II: new possibilities for understanding and treating cardiac hypertrophy. Cell. Mol. Life Sci. 61, 2695–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prenzel N., Fischer O. M., Streit S., Hart S., Ullrich A. (2001) The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Relat. Cancer 8, 11–31 [DOI] [PubMed] [Google Scholar]
- 29. Daaka Y., Luttrell L. M., Lefkowitz R. J. (1997) Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91 [DOI] [PubMed] [Google Scholar]
- 30. Luttrell L. M., Della Rocca G. J., van Biesen T., Luttrell D. K., Lefkowitz R. J. (1997) Gβγ subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor: a scaffold for G protein-coupled receptor-mediated Ras activation. J. Biol. Chem. 272, 4637–4644 [DOI] [PubMed] [Google Scholar]
- 31. Mahajan R., Ha J., Zhang M., Kawano T., Kozasa T., Logothetis D. E. (2013) A computational model predicts that Gβγ acts at a cleft between channel subunits to activate GIRK1 channels. Sci. Signal. 6, ra69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwindinger W. F., Robishaw J. D. (2001) Heterotrimeric G-protein βγ-dimers in growth and differentiation. Oncogene 20, 1653–1660 [DOI] [PubMed] [Google Scholar]
- 33. Castagliuolo I., Valenick L., Liu J., Pothoulakis C. (2000) Epidermal growth factor receptor transactivation mediates substance P-induced mitogenic responses in U-373 MG cells. J. Biol. Chem. 275, 26545–26550 [DOI] [PubMed] [Google Scholar]
- 34. Rusinova R., Mirshahi T., Logothetis D. E. (2007) Specificity of Gβγ signaling to Kir3 channels depends on the helical domain of pertussis toxin-sensitive Gα subunits. J. Biol. Chem. 282, 34019–34030 [DOI] [PubMed] [Google Scholar]
- 35. Arribas J., Coodly L., Vollmer P., Kishimoto T. K., Rose-John S., Massagué J. (1996) Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J. Biol. Chem. 271, 11376–11382 [DOI] [PubMed] [Google Scholar]
- 36. Dempsey P. J., Meise K. S., Yoshitake Y., Nishikawa K., Coffey R. J. (1997) Apical enrichment of human EGF precursor in Madin-Darby canine kidney cells involves preferential basolateral ectodomain cleavage sensitive to a metalloprotease inhibitor. J. Cell Biol. 138, 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown C. L., Meise K. S., Plowman G. D., Coffey R. J., Dempsey P. J. (1998) Cell surface ectodomain cleavage of human amphiregulin precursor is sensitive to a metalloprotease inhibitor. Release of a predominant N-glycosylated 43-kDa soluble form. J. Biol. Chem. 273, 17258–17268 [DOI] [PubMed] [Google Scholar]
- 38. Ouyang M., Huang H., Shaner N. C., Remacle A. G., Shiryaev S. A., Strongin A. Y., Tsien R. Y., Wang Y. (2010) Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer Res. 70, 2204–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uchiyama-Tanaka Y., Matsubara H., Mori Y., Kosaki A., Kishimoto N., Amano K., Higashiyama S., Iwasaka T. (2002) Involvement of HB-EGF and EGF receptor transactivation in TGF-β-mediated fibronectin expression in mesangial cells. Kidney Int. 62, 799–808 [DOI] [PubMed] [Google Scholar]
- 40. Thomas W. G., Brandenburger Y., Autelitano D. J., Pham T., Qian H., Hannan R. D. (2002) Adenoviral-directed expression of the type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via transactivation of the epidermal growth factor receptor. Circ. Res. 90, 135–142 [DOI] [PubMed] [Google Scholar]
- 41. Luttrell L. M., Hawes B. E., van Biesen T., Luttrell D. K., Lansing T. J., Lefkowitz R. J. (1996) Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gβγ subunit-mediated activation of mitogen-activated protein kinases. J. Biol. Chem. 271, 19443–19450 [DOI] [PubMed] [Google Scholar]
- 42. Nishihara T., Remacle A. G., Angert M., Shubayev I., Shiryaev S. A., Liu H., Dolkas J., Chernov A. V., Strongin A. Y., Shubayev V. I. (2015) Matrix Metalloproteinase-14 both sheds cell surface neuronal glial antigen 2 (NG2) proteoglycan on macrophages and governs the response to peripheral nerve injury. J. Biol. Chem. 290, 3693–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang S. M., Harari P. M. (1999) Epidermal growth factor receptor inhibition in cancer therapy: biology, rationale and preliminary clinical results. Invest. New Drugs 17, 259–269 [DOI] [PubMed] [Google Scholar]
- 44. Olayioye M. A., Neve R. M., Lane H. A., Hynes N. E. (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19, 3159–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rossé C., Lodillinsky C., Fuhrmann L., Nourieh M., Monteiro P., Irondelle M., Lagoutte E., Vacher S., Waharte F., Paul-Gilloteaux P., Romao M., Sengmanivong L., Linch M., van Lint J., Raposo G., Vincent-Salomon A., Bièche I., Parker P. J., Chavrier P. (2014) Control of MT1-MMP transport by atypical PKC during breast-cancer progression. Proc. Natl. Acad. Sci. U.S.A. 111, E1872–E1879 [DOI] [PMC free article] [PubMed] [Google Scholar]