

Background: Alcohol (EtOH) exposure has detrimental effects on fracture healing.
Results: EtOH inhibits TGF-β1 protein expression via interference with the transcription factor myeloid zinc finger 1.
Conclusion: EtOH-induced deficient fracture healing may be related to reduced TGF-β1.
Significance: Further understanding of the mechanisms responsible for the effects of EtOH are crucial for the development of interventions aimed at averting morbidities related to EtOH consumption.
Keywords: Gene Expression, Gene Transcription, Mesenchymal Stem Cells (MSCs), Osteopontin, Promoter, Protein Kinase A (PKA), Transforming Growth Factor β (TGF-β), Zinc Finger, Alcohol (EtOH)
Abstract
Alcohol (EtOH) intoxication is a risk factor for increased morbidity and mortality with traumatic injuries, in part through inhibition of bone fracture healing. Animal models have shown that EtOH decreases fracture callus volume, diameter, and biomechanical strength. Transforming growth factor β1 (TGF-β1) and osteopontin (OPN) play important roles in bone remodeling and fracture healing. Mesenchymal stem cells (MSC) reside in bone and are recruited to fracture sites for the healing process. Resident MSC are critical for fracture healing and function as a source of TGF-β1 induced by local OPN, which acts through the transcription factor myeloid zinc finger 1 (MZF1). The molecular mechanisms responsible for the effect of EtOH on fracture healing are still incompletely understood, and this study investigated the role of EtOH in affecting OPN-dependent TGF-β1 expression in MSC. We have demonstrated that EtOH inhibits OPN-induced TGF-β1 protein expression, decreases MZF1-dependent TGF-β1 transcription and MZF1 transcription, and blocks OPN-induced MZF1 phosphorylation. We also found that PKA signaling enhances OPN-induced TGF-β1 expression. Last, we showed that EtOH exposure reduces the TGF-β1 protein levels in mouse fracture callus. We conclude that EtOH acts in a novel mechanism by interfering directly with the OPN-MZF1-TGF-β1 signaling pathway in MSC.
Introduction
Alcohol intoxication is a significant risk factor associated with traumatic injury. Nearly half of patients with traumatic injuries who present to emergency departments test positive for blood alcohol content (1), and intoxication is associated with an increased risk of mortality among trauma patients (2). Among the numerous physiological effects of alcohol exposure, it has been widely shown that alcohol interferes with bone formation and fracture healing. Patients who abuse alcohol have prolonged healing time following transverse tibial fractures (3). The link between alcohol use and bone healing has serious implications for intoxicated patients suffering from fractures.
Much has been discovered regarding the effects of alcohol on fracture healing in animal models. Alcohol exposure in rodents prior to fracture injury results in inhibition of bone repair, decreased fracture callus volume, and reduced biomechanical strength (4–6). Although some of the signaling pathways involved in alcohol-induced deficient bone repair have been implicated, such as Wnt signaling (4), the molecular mechanisms and signaling pathways responsible for the physical manifestations remain to be elucidated.
Central to the process of bone healing is the role of mesenchymal stem cells (MSC).2 MSC are vital to bone healing through their ability to migrate to fracture sites in response to chemokines, secrete cytokines, and differentiate into multiple cell types that constitute mesenchymal tissues. Differentiation of MSC into osteoblasts and chondrocytes forms the fracture callus, which serves as the scaffolding for bone repair (7, 8). Interestingly, exogenous intravenous administration of MSC to alcohol-exposed mice can partly reverse the deficits in fracture healing attributed to alcohol exposure (9). This raises the possibility of MSC use as a treatment modality. Any direct interference in MSC function due to alcohol would clearly disrupt normal fracture healing.
One of the important signaling molecules that affect MSC function during fracture healing is osteopontin (OPN). Animal models show OPN mRNA increases during the mineralization phase of fracture healing (10), and in vitro studies show that OPN functionally stimulates MSC migration and attachment to fracture sites (11). OPN-deficient mice display smaller fracture calluses, reduced bone strength, decreased fracture neovascularization, and altered osteoclast functionality (12). OPN has also been implicated in human studies, as patients show a marked increase in serum OPN within a few days following long bone fractures (13).
The transforming growth factor-β (TGF-β) superfamily, including bone morphogenetic proteins, is widely implicated in fracture healing (14). TGF-β1, in particular, has been shown to have functions related to bone repair. TGF-β1 is known to be chemotactic toward MSC, and it induces proliferation of osteoblasts and chondrocytes (15). TGF-β1 regulates bone remodeling (16), and also has been shown to affect angiogenesis through inducing expression of vascular endothelial growth factor during membranous bone fracture healing (17).
Recent work in our laboratory has shown that OPN induces expression of TGF-β1 in MSC (18). This mechanism is dependent on activation of the transcription factor myeloid zinc finger 1 (MZF1), which strongly activates the TGF-β1 promoter. Because of the importance of MSC, OPN, and TGF-β1 in normal fracture healing, we sought to investigate if alcohol (EtOH) exposure interrupted OPN-mediated TGF-β1 expression in MSC. In this study, we demonstrated that EtOH exposure inhibits OPN-induced TGF-β1 mRNA and protein expression. We further demonstrated that EtOH inhibits RNA polymerase II and MZF1 binding to the TGF-β1 promoter, pointing to an explanation for decreased TGF-β1 protein levels. Correlative in vivo studies showed that alcohol-treated mice had decreased levels of TGF-β1 protein within the fracture callus 3 days following an induced tibial fracture. Moreover, we found that EtOH inhibits MZF1 mRNA expression, blocks RNA polymerase II and MZF1 from binding to the MZF1 promoter in a positive feedback mechanism, and abolishes OPN-induced activation of a MZF1-luciferase construct. Next we showed that alteration of the protein kinase A (PKA) signaling pathway influences TGF-β1 protein expression and MZF1 promoter activity in the presence of OPN. Last, we found that OPN and PKA activity were sufficient for stimulation of MZF1 protein phosphorylation.
EXPERIMENTAL PROCEDURES
Materials
The OPN-R3 aptamer and OPN-R3 mutant aptamer were synthesized by Dharmaco (Lafayette, CO). The aptamer was developed in our laboratory to specifically bind to and functionally block extracellular OPN (in vitro Kd = 18 ± 0.2 nm) (19). Mutant aptamer, which lacks OPN binding or IgG served as negative controls. All cysteine and uracil bases are 2′-O-methylated, and the molecules have 5′ cholesterol and 3′ IDT modification. The sequences for the OPN-R3 aptamer and mutant aptamer are as follows: APT, 5′-GCCACAGAAUGAAAAACCUCAUCGAUGUUGCA-3′ and MuAPT, 5′-CGGCCACAGAAUGAAUCAUCGAUGUUGCAUAGUUG-3′.
Recombinant human OPN protein was obtained from R&D Systems (Minneapolis, MN). The αvβ3 integrin antibody, CD44 antibody, and Sp-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt hydrate (SP8) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). H-89 was purchased from Sigma.
Cell Culture
Human MSC, CD105/CD73/CD44 positive and CD34/CD45/CD14/HLA-DR negative, were obtained from the Texas A&M Institute for Regenerative Medicine Health Science Center College of Medicine (Temple, TX). MSC were cultured in MEM-α supplemented with 20% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 mg/ml), and were maintained at 37 °C in a humidified atmosphere of 5% CO2. MSC differentiation experiments were conducted using special media from the StemPro Osteogenesis Differentiation Kit and StemPro Chondrogenesis Differentiation Kit, which were purchased from Life Technologies. Cells were treated with OPN (180 ng/ml), 100% EtOH (25 mm), OPN aptamer (APT) (100 nm), mutant aptamer (MuAPT) (100 nm), SP8 (10 μm), H-89 (50 μm), and TGF-β1 (6 ng/ml). Most experiments were conducted with a 24-h exposure of MSC to the aforementioned treatments. Osteogenesis and chondrogenesis differentiation experiments were performed for a 2-week duration with medium changed and cells re-treated every 3 days.
Quantitative Real-time PCR
RNA were isolated from MSC using TRIzol reagent (Invitrogen) and cDNA was synthesized from 5.0 μg of total RNA using the iScript Select cDNA synthesis kit (Bio-Rad Laboratories), according to the manufacturer's protocol. Quantitative real-time PCR was performed with iQ SYBR Green super mix, using the iCycler iQ Real time PCR Detection System (Bio-Rad), according to the manufacturer's instructions. GAPDH was used as an endogenous control, and ΔΔCT values were calculated after GAPDH normalization.
Quantitative Real-time PCR Primer Sequences
The sequences were as follows: TGF-β1, PCR product length 405 bp: 5′-TGGCGATACCTCAGCAACC-3′ and 5′-CTCGTGGATCCACTTCCAG-3′; MZF1, PCR product length 137 bp: 5′-AGTGTAAGCCCTCACCTCC-3′ and 5′-GGGTCCTGTTCACTCCTCAG-3′; SOX-9, PCR product length 174 bp: 5′-TAAAGGCAACTCGTACCCAA-3′ and 5′-ATTCTCCATCATCCTCCACG-3′; Collagen-1 α1, PCR product length 372 bp: 5′-CTCGAGGTGGACACCACCCT-3′ and 5′-CAGCTGGATGGCCACATCGG-3′; GATA4, PCR product length 255 bp: 5′-CTGTCATCTCACTATGGGCA-3′ and 5′-CAAGTCCGAGCAGGAATTT-3′; GAPDH, PCR product length 146 bp: 5′-CTCTCTGCTCCTCCTGTTCG-3′ and 5′-TTAAAAGCAGCCCTGGTGAC-3′.
Enzyme-linked Immunosorbent Assay (ELISA)
TGF-β1 protein quantification was performed using a Human TGF-β1 Quantikine ELISA kit, following the manufacturer's guidelines (R&D Systems). MSC were grown in serum-free MEM-α, as fetal bovine serum contains TGF-β1. Cells were subjected to appropriate treatments for 24 h, and the resulting supernatant was isolated for TGF-β1 protein analysis. Measurement of the active form did not require any additional steps, whereas measurement of the total protein required acidification using 1 n HCl followed by neutralization with 1.2 n NaOH, 0.5 m HEPES prior to assay.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were performed following treatment of MSC with recombinant human OPN and/or 25 mm EtOH and cultured for 24 h prior to DNA cross-linking and lysis. Chromatin was fixed and immunoprecipitated using the EZ-ChIP assay kit (Millipore, Billerica, MA), following the manufacturer's protocol. The DNA was sonicated to 500-bp fragments using standard settings. Purified chromatin was immunoprecipitated using anti-RNA polymerase II antibody (1 μg), normal goat IgG (5 μg), and anti-MZF1 antibody (5 μg, Santa Cruz Biotechnology). After DNA purification, Real Time-PCR assay was performed to determine the presence of RNA polymerase II bound to the TGF-β1 TATA box, the final TGF-β1 exon, and the MZF1 TATA box. In addition, we used Real Time-PCR to determine the presence of MZF1 bound to the putative binding site for MZF1 on the TGF-β1 promoter. All CT values were normalized to either input CT values to calculate a percent input, or normalized to IgG CT values to calculate fold-increase. PCR DNA products were visualized on a 1% agarose gel. Primers used for Real Time-PCR for ChIP assays are as follows: TGF-β1 promoter/gene: TATA box: PCR product length 145 bp: 5′-CCCAGCCTGACTCTCCTTCCGTTC-3′ 5′-GGTGATCCAGATGCGCTGTGGCTT-3′; final exon: PCR product length 147 bp: 5′-GGTGGAGCAGCTGTCCAACATGAT-3′ and 5′-GCACGGGTGTCCTTAAATACAGCC-3′; MZF1 promoter: TATA box PCR product length 515 bp: 5′-GTGTCTCTTTCATCTGCATCC-3′; 5′-TCCGGCAGATGTAGTCAGCAG-3′; and GATA4 promoter: TATA box: PCR product length 103 bp; 5′-CCCTTCAGCCTTAAGTTCC-3′ and 5′-GCGACACACTTAGGGTTTCTTC-3′.
Construction of Promoter Plasmids
Human genomic DNA was isolated from a normal human lung fibroblast cell line (ATCC, Manassas, VA), and the human MZF1 promoter was cloned using PCR. The promoter region was defined using the NCBI Reference 8 Sequence NM_000660.4. The full-length promoter (−2180/+192) and four deletion constructs (−1578/+192, −599/+192, −490/+192, and −382/+192) were generated, setting the ATG start site as 0. All promoter fragments were then cloned into the pGL4.10(luc2) vector (Promega, Madison, WI) to generate luciferase reporter constructs.
Construction of Promoter Mutations
Point mutations of the putative binding site of MZF1 on the MZF1 promoter were made using the QuikChange II Site-directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA), according to the manufacturer's guidelines. The mutated promoter fragment was also cloned into pGL4.10(luc2) vector, and its sequence was verified with direct sequencing analysis.
Dual Luciferase Reporter Assay
To measure promoter activity, 4 μg of each of the luciferase reporter constructs were co-transfected with 0.4 μg of Renilla into MSC. After 24 h following transfection, cells were exposed to OPN and/or EtOH for an additional 24 h. Using a Dual Luciferase assay system (Promega, Madison, WI), the cells were passively lysed and the Firefly luciferase and Renilla luciferase activity was measured in each lysate using the Modulus Luminometer (Turner Biosystems Inc., Sunnyvale, CA). The reporter activity of each construct is shown as a ratio of Firefly:Renilla luciferase activity.
Dot Blot
Treated cells were lysed with CelLytic M (Sigma) and centrifuged for 20 min at 12,000 × g at 4 °C. 50-μg protein samples were applied to PVDF membrane by following Bio-Dot microfiltration manufactures protocol (Bio-Rad). The membrane was blocked in 5% BSA in TBS (20 mm Tris-HCl, 150 mm NaCl, pH 7.5, and 0.05% Tween 20) and shaken for 1 h at 4 °C. A 1:1000 dilution of phospho-(Ser/Thr) PKA substrate antibody (Cell Signaling) (5% BSA, TBS) was added to the membrane and incubated overnight at 4 °C. Next the membrane was rinsed with TBS, and incubated with anti-rabbit secondary antibody at room temperature for 1 h. Protein was detected using standard horseradish peroxidase system.
Immunoprecipitation of MZF1
Treated cells were lysed with CelLytic M (purchased from Sigma; C2978) and centrifuged for 20 min at 12,000 × g at 4 °C. Then 500 μg of protein/treatment or control group was incubated with 2 μg of MZF1 antibody or IgG control (Abcam) for 4 h at 4 °C. After binding the MZF1 protein with antibody, 20 μl of Protein A-agarose beads (Santa Cruz Biotechnology) were added to the protein-antibody conjugate and incubated overnight at 4 °C. Supernatant was discarded after tubes were spun at 1000 rpm for 30 s. Pellets of agarose bead-MZF1 antibody-MZF1 protein were washed 3 times with 500 μl of RIPA buffer (50 mm Tris-HCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 150 mm NaCl, 2 mm EDTA, 50 mm NaF), with centrifugation and supernatant removal after each wash. After the final supernatant removal, 40 μl of 2× sample buffer was added, samples were boiled for 10 min, and conjugates were centrifuged for 5 min at 1000 rpm to remove the beads from the MZF1 protein. Western blots were performed with anti-phosphoserine/threonine Ab (BD Transduction Laboratories).
ImageStream Flow Cytometry
The recombinant human OPN (R&D Systems) was labeled using the Alexa Fluor 647 microscale protein labeling kit (Life Technologies), according to the manufacturer's instructions. Briefly, 50 μg of human OPN protein (1 mg/ml) was incubated with 7.94 nmol/μl of Alexa Fluor 647 succinimidyl ester for 15 min at room temperature. The Bio-Gel P-6 spin column was used to purify the OPN-Alexa Fluor 647 conjugate from the unreacted dye. The protein concentration and degree of labeling were measured by Nano Drop ND-1000 spectrophotometer, according to the manufacturer's protocol. Human MSC were detached using calcium sequestration under physiological conditions. The cell suspension was blocked with BSA and stained with OPN-Alexa Fluor 647 or Ab αvβ3-FITC and/or Ab CD44-APC conjugates or isotype controls. The cells were illuminated using Aminis ImageStream Mark II (Amnis Corp., Seattle, WA) by a bright-field lamp at a magnification of ×60 and a 488-nm excitation laser (100 milliwatts) for FITC, 642-nm excitation laser (50 milliwatt) for APC or Alexa Fluor 647. A minimum of 500 cells for every population was collected. Data were analyzed by IDEAS software (Amnis Corp., Seattle, WA). Positive cutoff values were calculated on the basis of the background “bright detail similarity” feature of IDEAS software. Staining specificity was determined with the isotype-matched control antibody.
Mouse Alcohol Fracture Model
Male C57Bl/6 mice 6 to 7 weeks of age were obtained from Harlan Laboratories (Indianapolis, IN) and housed in a facility approved by the Institutional Animal Care and Use Committee at Loyola University Medical Center. The mice were allowed to acclimate to the environment for 1 week prior to the initiation of the experimental procedures. Animals were randomly assigned to either the saline + fracture group or the alcohol + fracture group. The acute alcohol exposure used in all experiments consisted of a daily intraperitoneal injection of a 20% (v/v) EtOH/saline solution made from 100% molecular grade absolute EtOH (Sigma) and sterile isotonic saline. Mice were administered the EtOH/saline solution at a dose of 2 g/kg once per day for 3 consecutive days and were weighed daily prior to injection to ensure correct dosage. Mice in the saline control groups were administered sterile isotonic saline only. One hour after the third and final injection, all mice were subjected to the stabilized tibial fracture surgery. Blood alcohol levels averaged ∼200 mg/dl at the time of fracture (1 h post-injection).
Stabilized Tibial Fracture Creation
One hour after administration of the final alcohol or saline injection, mice were given an induction dose of anesthesia (0.5 to 0.75 mg/kg of ketamine and 0.06 to 0.08 mg/kg of xylazine) to facilitate hair removal from the left hind limb of the animal. Mice were given 5 mg/kg of prophylactic gentamicin subcutaneously and anesthetized completely with isofluorane for the duration of the procedure. Under sterile conditions, the surgery site was swabbed with povidone-iodine solution followed by 70% EtOH. A small incision was made to expose the patellar tendon and a 27-gauge needle was used to ream a hole into the medullary cavity at the proximal aspect of the tibia. An insect pin, 0.25 mm in diameter (Fine Science Tools, Inc., Foster City, CA), was inserted into the reamed hole to stabilize the tibia. A pair of angled bone scissors (Fine Science Tools) was used to surgically create a mid-diaphyseal tibial fracture. The insect pin was cut flush with the bone, and the wound was sutured closed. Mice were then placed in clean cages on heating pads with free access to food and water. All animals received postoperative buprenex subcutaneously (0.05 mg/kg) for pain control.
Fracture Callus Histology
Injured tibias were harvested from the mice 7 days post-fracture and placed in 10% neutral buffered formalin for 48 h. The tibias were decalcified in 10% EDTA with agitation for 5 days, processed through a graded series of alcohol solutions and xylene, and infiltrated overnight with melted paraffin at 56 to 58 °C. The tibias were oriented identically during paraffin embedding to identify mid-callus sections. Five-micrometer sections were placed onto Superfrost® Plus slides (Fisher Scientific, Pittsburgh, PA) and baked on a 60 °C slide warmer overnight. Sections from each group were stained with hematoxylin and eosin, using standard protocols. Immunohistochemistry was also performed in all sections, staining for OPN, TGF-β1, and α-smooth muscle actin (α-SMA).
Mouse Fracture Callus TGF-β1 Protein Quantification
Injured tibias were harvested from mice 3 and 7 days post-fracture and snap-frozen in liquid nitrogen. Fracture callus tissue was isolated using a Dremel tool (Dremel Inc., Racine, WI) while frozen and pulverized in lysis buffer using a freezer mill (SPEX CertiPrep Inc., Metuchen, NJ). An equivalent fragment of bone was isolated from contralateral tibias, which served as baseline levels in uninjured control tissue. Total protein was measured using a bicinchoninic acid assay (Thermo Fisher Scientific Inc.). Fifteen micrograms of total protein from each sample was used for analysis of active TGF-β1 using a Mouse TGF-β1 Quantikine ELISA, following manufacturer's guidelines (R&D Systems).
Statistical Analysis
All experiments were done in triplicate. Data are presented as mean ± S.E. Student's t tests were used for analysis. Values of p < 0.05 were considered significant.
RESULTS
EtOH Inhibits OPN-mediated TGF-β1 Protein and mRNA Expression, and OPN-mediated MZF1 mRNA Expression
The effects of EtOH on TGF-β1 expression were assessed by analyzing both protein and mRNA levels. MSC were exposed to varying combinations of OPN (180 ng/ml), EtOH (25 mm), OPN-R3 aptamer (100 nm), and mutant aptamer (100 nm). The concentration of secreted active and total TGF-β1 protein was measured after 24 h of exposure. The mean concentration of active TGF-β1 was 991.7 pg/ml following OPN treatment, whereas exposure to EtOH significantly reduced active TGF-β1 to 33.0 pg/ml, exposure of MSC to R3 aptamer significantly reduced OPN-mediated active TGF-β1 expression to 48.0 pg/ml, whereas exposure to mutant R3 aptamer had no significant effect on TGF-β1 expression (Fig. 1A). Similar results were found when measuring the total concentration of TGF-β1 protein, which includes the inactive latent form. OPN-treated MSC displayed mean total TGF-β1 concentration of 4,938.0 pg/ml, whereas exposure to EtOH significantly reduced the total TGF-β1 to 295.4 pg/ml. R3 aptamer also markedly reduced OPN-mediated TGF-β1 expression to 347.3 pg/ml, whereas mutant R3 aptamer had no significant effect on total TGF-β1 expression (Fig. 1B).
FIGURE 1.

EtOH inhibits OPN-mediated TGF-β1 protein and mRNA expression and OPN-mediated MZF1 mRNA expression. A, active TGF-β1 protein in MSC medium. MSC were exposed to OPN (180 ng/ml), EtOH (25 mm), APT (100 nm), and MuAPT (100 nm) for 24 h and secreted protein was measured with ELISA (*, p < 0.0001). B, total TGF-β1 protein in MSC medium. MSC were exposed to identical aforementioned treatments and secreted protein was measured with ELISA (*, p < 0.0001). C, expression of TGF-β1 mRNA. MSCs were exposed to OPN (180 ng/ml), EtOH (25 mm), APT (100 nm), and MuAPT (100 nm) for 24 h and TGF-β1 mRNA levels were measured using qRT-PCR (*, p < 0.0001). D, expression of MZF1 mRNA. MSCs were exposed to OPN, EtOH, APT, and MuAPT for 24 h and MZF1 mRNA levels were measured using qRT-PCR (*, p < 0.0001). Data are presented as mean ± S.E. of three experiments.
Next we sought to determine the effects of OPN, EtOH, and R3 aptamer on TGF-β1 mRNA levels in MSC using qRT-PCR. Exposure of MSC to OPN significantly increased TGF-β1 mRNA compared with MSC alone (ΔΔCT, 14.4 versus 2.3). OPN-mediated TGF-β1 mRNA expression was decreased in EtOH and R3 aptamer-treated cells, as compared with MSC treated with OPN alone (ΔΔCT, 14.4 versus 2.3 and 2.0, respectively) (Fig. 1C). These results demonstrate the ability of 25 mm EtOH exposure to inhibit OPN-mediated TGF-β1 mRNA and protein expression in MSC.
MZF1 has been shown to play a key role in the transcriptional regulation of TGF-β1 expression. Thus, we next evaluated the effects of OPN, EtOH, and R3 aptamer on MZF1 mRNA levels in MSC using qRT-PCR. OPN-treated MSC showed significantly increased MZF1 mRNA compared with untreated MSC (ΔΔCT, 9.5 versus 1.5). MSC exposure to EtOH and R3 aptamer significantly reduced OPN-mediated MZF1 mRNA expression (ΔΔCT, 9.5 versus 3.1 and 4.5, respectively), whereas R3 mutant aptamer had no significant effect on MZF1 mRNA levels (Fig. 1D). EtOH appears to have a significant inhibitory effect on MZF1 mRNA expression, which is induced by OPN.
EtOH Blocks RNA Polymerase II and MZF1-mediated TGF-β1 Promoter Activation and RNA Polymerase II-mediated MZF1 Promoter Activation
To further understand the mechanism by which EtOH alters TGF-β1 expression, chromatin immunoprecipitation (ChIP) Real Time-PCR was used to determine the role of OPN and EtOH in transcriptional regulation of the TGF-β1 promoter in MSC. Two primer sets were designed to include either one of the consensus TATA boxes in the TGF-β1 promoter region, or the third and final exon of the TGF-β1 coding region. DNA amplification utilized by Real Time-PCR following ChIP determined recruitment of RNA polymerase II to the TATA box and the last exon of the TGF-β1 gene. We have previously demonstrated that MZF1 is required for TGF-β1 promoter activation in MSC (18). Thus, we also investigated the recruitment of MZF1 to the TGF-β1 promoter region via PCR following ChIP assay. Quantification of RNA polymerase II and MZF1 binding activity was determined by calculating the percent input following Real Time-PCR DNA amplification. OPN was found to significantly induce binding of RNA pol II to the TGF-β1 promoter (0.73% input) and to the last exon of TGF-β1 (0.75% input). EtOH exposure diminished this OPN-mediated binding of RNA polymerase II (Fig. 2, A and B). Using the ChIP assay, we further demonstrated that OPN induces binding of MZF1 to the TGF-β1 promoter following Real Time-PCR amplification of the TGF-β1 TATA box and the last exon (0.45 and 0.55% input, respectively). EtOH exposure was found to completely ablate OPN induced binding of MZF1 to the TGF-β1 promoter (Fig. 2, A–D). These results suggest that EtOH exposure is sufficient to completely block TGF-β1 promoter induction following treatment with OPN.
FIGURE 2.

EtOH blocks RNA polymerase II and MZF1-mediated TGF-β1 promoter activation and RNA polymerase II-mediated MZF1 promoter activation. A, binding of RNA pol II and MZF1 to the TGF-β1 promoter. ChIP Real Time-PCR was performed using primers that included the TGF-β1 promoter TATA box to determine the role of OPN and EtOH in transcriptional regulation of TGF-β1 expression. B, graphical representation of RNA pol II and MZF1 binding to the TGF-β1 promoter. The percent input was calculated from ChIP Real Time-PCR CT values using input samples as internal controls for the amount of chromatin. C, binding of RNA pol II and MZF1 to the TGF-β1 third exon. ChIP Real Time-PCR was performed using primers that included the TGF-β1 third and final exon to determine the role of OPN and EtOH in transcriptional regulation of TGF-β1 expression. D, graphical representation of RNA pol II and MZF1 binding to the TGF-β1 third exon. The percent input was calculated from ChIP Real Time-PCR CT values using input samples as internal controls for the chromatin amount. E, binding of RNA pol II to the MZF1 promoter. ChIP Real Time-PCR was performed using primers that included the MZF1 TATA box. F, graphical representation of RNA pol II binding to the MZF1 promoter. The percent input was calculated from ChIP Real Time-PCR CT values using input samples as internal controls for the amount of chromatin. Data are presented as mean ± S.E. of three experiments. Gels are representative of three experiments.
To further assess the role of OPN and EtOH in modulating MZF1 activity, ChIP Real Time-PCR was used to examine transcriptional regulation of the MZF1 gene in MSC. A primer set was designed to include the MZF1 promoter TATA box. OPN was found to significantly induce binding of RNA polymerase II to the MZF1 promoter (1.2% input), whereas EtOH blocked OPN-mediated RNA polymerase II binding (Fig. 2, E and F). This supports the role of EtOH as a transcriptional repressor of MZF1.
EtOH Inhibits OPN-mediated MZF1-luciferase Construct Activation
Additional information regarding the effects of OPN and EtOH on MZF1 promoter activation was gathered by utilizing a luciferase reporter plasmid. The human MZF1 promoter sequence was amplified, and the PCR products (2.2, 1.8, 0.7, 0.6, 0.5, and 0.4 kb) were sequenced and subcloned into the pGL3 luciferase reporter (Fig. 3A). Luciferase induction was quantified as relative luciferase activity (RLA) using Renilla signal to control for transfection efficiency. Stimulation with OPN significantly induced luciferase activation of the 2.3 (21,409 RLA), 1.8 (29,664 RLA), and 0.7 kb (20,007 RLA) MZF1 promoter sequences, but was not sufficient for induction of the shorter MZF1 promoter sequences. MSC exposure to EtOH completely ablated OPN-mediated luciferase activation of the 2.3-, 1.8-, and 0.7-kb MZF1 promoter sequences (Fig. 3B). Next the MZF1 promoter DNA sequence was analyzed for known transcription factor binding sequences. A MZF1 binding sequence within the MZF1 gene promoter region was identified and then mutated (Fig. 3C). The 2.2-kb fragment of the MZF1 promoter containing the mutated MZF1 DNA binding site was subcloned into the pGL3 luciferase reporter. Luciferase assay demonstrated that mutation of the MZF1 binding site was sufficient to nearly completely ablate the luciferase activity of the 2.2-kb MZF1 promoter fragment when stimulated with OPN (Fig. 3D). These results show that EtOH interferes with MZF1 promoter activation in the presence of stimulating OPN. The results also further suggest that EtOH exerts its effects by interfering with a positive regulation or feed-forward type of mechanism, which we showed is important for MZF1 gene activation.
FIGURE 3.

EtOH inhibits OPN-mediated MZF1-luciferase construct activation. A, schematic depiction of MZF1 promoter-luciferase reporter constructs. B, luciferase activity of MZF1 promoter constructs in MSC (*, p < 0.0001). C, schematic representation of the wild type and mutated MZF1 binding site within the MZF1 promoter sequence. D, luciferase activity of the wild type and mutant MZF1 promoter constructs in MSC (*, p < 0.0001). Data are presented as mean ± S.E. of at least three experiments.
EtOH Blocks MZF1 Binding to the MZF1 Promoter
DNA sequence analysis revealed a putative MZF1 binding site within the MZF1 promoter region. ChIP Real Time-PCR was used to determine the effect of OPN and EtOH on the interactions of MZF1 with its binding site located within the MZF1 promoter. ChIP and subsequent pulldown with an anti-MZF1 antibody showed that OPN exposure induced binding of MZF1 to its own promoter at the expected MZF1 binding site. EtOH exposure blocked OPN-mediated binding of MZF1 to its promoter sequence (fold-enrichment, MSC + OPN, 31305.0 versus MSC + OPN + EtOH, 2.6). Binding of RNA polymerase to the MZF1 promoter was similarly induced by OPN, but this was significantly reduced when exposed to EtOH (fold-enrichment: MSC + OPN, 82614.6 versus MSC + OPN + EtOH, 3.0) (Fig. 4, A and B). These results support a positive feedback mechanism in which the MZF1 protein induces its own transcription via binding to the MZF1 promoter, and exposure to EtOH interferes with this regulatory process.
FIGURE 4.

EtOH blocks MZF1 binding to the MZF1 promoter. A, binding of MZF1 to the MZF1 promoter. ChIP Real Time-PCR was done using primers that amplified the promoter region at −545 to −537 base pairs upstream of the MZF1 start codon that contains an MZF1 binding sequence. B, graphical representation of RNA pol II and MZF1 binding to the MZF1 promoter. Fold-enrichment was calculated using nonspecific IgG binding as an internal control (*, p < 0.0001). Data are presented as mean ± S.E. of three experiments. The gel is representative of three experiments.
Manipulation of the PKA Signaling Pathway Alters OPN-mediated TGF-β1 Protein and mRNA Expression, and MZF1 mRNA Expression and Promoter Activation
A three-dimensional structural analysis of the MZF1 protein sequence by PhosphoSitePlus revealed a phosphorylation site on MZF1 with a high likelihood of being targeted by the PKA signaling pathway. We assessed the effect of PKA modulation in the presence of varying concentrations of OPN and EtOH by measuring TGF-β1 mRNA and active and total TGF-β1 protein. SP8 is a small molecule known to increase PKA signaling activity (20, 21). SP8 treatment of MSC increased active and total TGF-β1 protein expression in the presence of OPN, and partially rescued EtOH inhibition of OPN-mediated TGF-β1 protein expression at 24 and 72 h post-treatment (Fig. 5, A and B). Inhibition of the PKA signaling pathway was achieved by treating MSC with a known PKA inhibitor, H-89. PKA inhibition with H-89 resulted in decreased active and total TGF-β1 protein expression at 24 and 72 h post-treatment in MSC treated with OPN (Fig. 5, C and D). Similar results were found with TGF-β1 mRNA expression following PKA signaling modulation. SP8 enhanced TGF-β1 mRNA levels in the presence of OPN and partially rescued EtOH-mediated inhibition of TGF-β1 mRNA expression. H-89 treatment significantly reduced TGF-β1 mRNA expression in the presence of OPN (Fig. 5E). Treatment of MSC with SP8 in the presence of OPN enhanced MZF1 mRNA expression, and SP8 exposure partially reversed EtOH-mediated inhibition of MZF1 mRNA expression. Exposure of OPN-treated MSC to the PKA inhibitor H-89 resulted in decreased MZF1 mRNA expression when compared with OPN treatment alone (Fig. 5F). These findings suggest that PKA activation may function to enhance TGF-β1 protein expression via the OPN-MZF1-dependent signaling pathway.
FIGURE 5.

Manipulation of the PKA signaling pathway alters OPN-mediated TGF-β1 protein and mRNA expression, and MZF1 mRNA expression and promoter activation. A–D, active and total secreted TGF-β1 protein following varying treatments of MSC with OPN, EtOH, the PKA activator, SP8, and the PKA inhibitor, H-89. SP8 partially rescues EtOH inhibition of OPN-mediated TGF-β1 expression. H-89 reduced the level of TGF-β1 protein expression following treatment with OPN. E, expression of TGF-β1 mRNA following varying treatments of OPN, EtOH, SP8, and H-89. F, MZF1 mRNA expression following varying treatments of OPN, EtOH, SP8, and H-89 (*, p < 0.0001). G, binding of MZF1 to the MZF1 promoter in the presence of OPN, EtOH, and PKA modulators SP8 and H-89. ChIP Real Time-PCR was performed using primers that amplified the MZF1 binding site within the MZF1 promoter. H, graphical representation of MZF1 binding the MZF1 promoter. Fold-enrichment values were calculated using IgG pulldown CT values as internal control (*, p < 0.0001). Data are presented as mean ± S.E. of three experiments. The gel is representative of three experiments.
ChIP was then performed to investigate the role of PKA signaling in influencing activation of the MZF1 promoter. ChIP Real Time-PCR was used to assess the binding of MZF1 protein to the MZF1 promoter region following MSC treatment with SP8, H-89, and varying combinations of OPN and EtOH. As previously demonstrated, OPN treatment of MSC induced binding of MZF1 to its own promoter, and SP8 increased this binding (fold-enrichment: MSC + OPN, 47341.8 versus MSC + OPN + SP8, 1326120.0), and also partially rescued EtOH inhibition of MZF1 binding (fold-enrichment: MSC + OPN + EtOH, 1.0 versus MSC + OPN + EtOH + SP8, 5940.5), thus enhancing MZF1 transcriptional activation. Treatment with H-89 was found to significantly reduce OPN induced binding of MZF1 to its promoter (fold-enrichment: MSC + OPN, 47341.8 versus MSC + OPN + H-89, 6998.5) (Fig. 5, G and H).
OPN and EtOH Influence Phosphorylation of PKA Downstream Targets and MZF1
With experimental evidence showing that PKA signaling can enhance MZF1 activity and TGF-β1 expression, we further explored the effects of OPN and EtOH on PKA activity by examining phosphorylation of PKA substrates. An antibody specific for phosphorylated PKA target proteins was used to identify the effects of OPN and EtOH on downstream phosphorylation by PKA. OPN treatment of MSC was shown to strongly induce phosphorylation of downstream PKA targets, whereas EtOH exposure reversed the effect of OPN on phosphorylation. As expected, the PKA activator SP8 was sufficient for inducing phosphorylation of PKA substrates, as well as reversing the inhibitory effect of EtOH on OPN-induced phosphorylation (Fig. 6A). Next we sought to determine whether MZF1 exists in a phosphorylated state, and if so, how phosphorylation of MZF1 might be affected by OPN, EtOH, and PKA. This experiment consisted of co-immunoprecipitation of the MZF1 protein with anti-MZF1 antibody, followed by precipitation with anti-phosphoserine/threonine antibody. OPN treatment resulted in the presence of phosphorylated MZF1 (p-MZF1), which was detectable by Western blot. MSC alone yielded no detectable p-MZF1. EtOH treatment inhibited any detection of p-MZF1 from MSC stimulated with OPN. Next we showed that PKA stimulation with SP8 was sufficient for detection of p-MZF1. SP8 was also able to partially reverse the effect of EtOH on blocking OPN-mediated phosphorylation of MZF1 (Fig. 6B). Last, a similar co-immunoprecipitation study was performed using an anti-MZF1 antibody followed by pulldown with a phospho-(Ser/Thr) PKA substrate antibody. This experiment failed to detect phosphorylated MZF1 on Western blots, indicating that MZF1 is not likely a direct substrate of PKA (data not shown).
FIGURE 6.
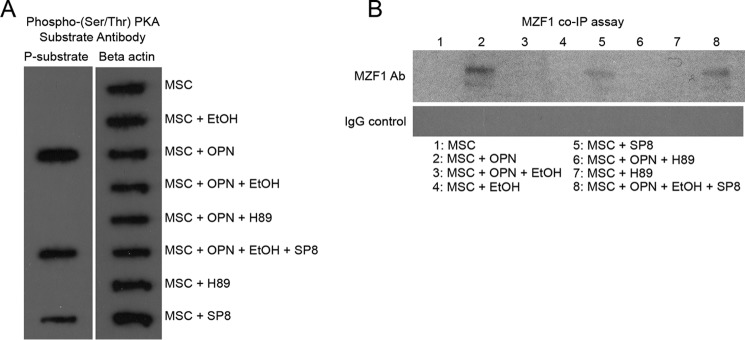
OPN and EtOH influence phosphorylation of PKA downstream targets and MZF1. A, dot blot showing the effect of OPN, EtOH, SP8, and H-89 on the phosphorylation state of downstream PKA substrate proteins. PKA substrate-specific anti-phosphoserine/threonine Ab was utilized to show that OPN stimulates PKA substrate phosphorylation, an effect reversed by EtOH. B, Western blot of phosphorylated MZF1 protein following co-immunoprecipitation with anti-MZF1 Ab and anti-phosphoserine/threonine Ab. OPN and PKA activator SP8 stimulated phosphorylation of MZF1 protein, and the effect of OPN was blocked by both EtOH exposure as well as PKA inhibition with H-89 the gel is representative of three experiments.
Global Transcription in MSC Is Not Affected by EtOH Exposure
Although the literature supports the use of EtOH in the 25–100 mm range as a reasonable concentration for in vitro models, with no reported increase in cell death or apoptotic rate in human monocytes or dendritic cells, we conducted additional control experiments to screen for the effects of EtOH on global transcription. We performed control experiments with the GATA4 gene, which encodes a transcription factor containing a zinc finger motif, similar to MZF1. MSC were exposed to standard combinations of OPN and EtOH for 24 h. ChIP Real Time-PCR was used to examine the effect of EtOH on GATA4 transcriptional activation via binding of RNA polymerase II to the GATA4 promoter. RNA polymerase II was consistently activated and bound to the GATA4 promoter. EtOH exposure did not perturb this interaction (Fig. 7A). Fold-enrichment was calculated to graphically represent the effect of OPN and EtOH on RNA polymerase II binding to the GATA4 promoter (Fig. 7B). qRT-PCR was then used to measure GATA4 mRNA levels. There was no significant difference in GATA4 mRNA among the four different treatment groups. EtOH exhibited no discernible effect on GATA4 mRNA expression (Fig. 7C).
FIGURE 7.

Global transcription in MSC is not affected by EtOH exposure. A, ChIP showing binding of RNA pol II to the GATA4 promoter in MSC following treatment with OPN and EtOH. EtOH did not alter RNA pol II binding to GATA4 promoter and subsequent transcriptional activation. B, graphical representation of MZF1 binding to the MZF1 promoter. Fold-enrichment was calculated using nonspecific IgG binding as an internal control. C, expression of GATA4 mRNA in OPN- and EtOH-treated MSC. GATA4 mRNA levels were unaffected by exposure to EtOH. Data are presented as mean ± S.E. of three experiments. Teh gel is representative of three experiments.
EtOH Does Not Interfere with OPN-Receptor Interactions or OPN Receptor Cell Surface Density
The effect of EtOH exposure on OPN cell surface receptor density was measured by fluorescently labeling antibodies specific for two confirmed OPN receptors, specifically CD44 and αvβ3. Fluorescence intensity measured by AMNIS demonstrated that both receptors have identical cell surface density across all treatment groups, suggesting that EtOH plays no role in OPN-receptor down-regulation (Fig. 8A). To investigate the kinetic interactions of OPN with its receptors in the presence of EtOH, OPN was fluorescently labeled with FITC and then added to MSC in the presence of EtOH. This showed equivalent cell surface OPN levels with or without EtOH present, thus supporting the lack of effect of EtOH on OPN-receptor binding interactions (Fig. 8B).
FIGURE 8.
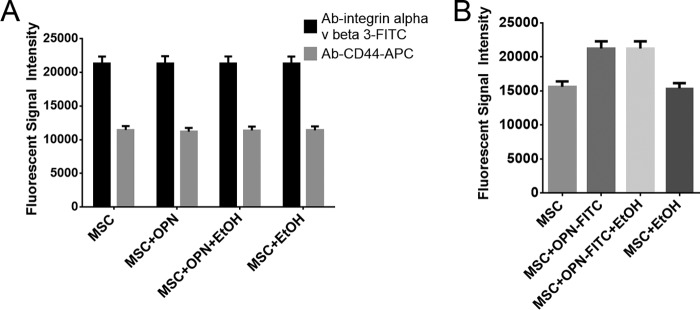
EtOH does not interfere with OPN-receptor interactions or OPN receptor cell surface density. A, integrin CD44 and αvβ3 receptor density on MSC in the absence or presence of EtOH. A fluorescently tagged antibody binding to OPN receptors CD44 and αvβ3 was unaffected by OPN and EtOH exposure, indicating a constant receptor population among treatment groups. B, fluorescent signal of FITC-labeled OPN in the absence and presence of EtOH in MSC. OPN receptor interactions in the presence of EtOH were analyzed using OPN conjugated to FITC. EtOH did not alter OPN-receptor interactions. Data are presented as mean ± S.E. of three experiments.
OPN, EtOH, and TGF-β1 Regulate SOX-9 and Collagen-1 mRNA Expression
Data were obtained regarding the influence of OPN and EtOH on MSC expression profiles indicative of differentiation into the osteoblast and chondrocyte lineages. SOX-9 and collagen-1 were chosen as markers for chondrocyte and osteoblast differentiation, respectively, and mRNA levels were measured using qRT-PCR. MSC treated with OPN showed a significant increase in SOX-9 (ΔΔCT, 4.1 versus 1.3) and collagen-1 mRNA (ΔΔCT, 4.8 versus 1.5) compared with untreated MSC. MSC exposure to EtOH significantly reduced OPN-mediated SOX-9 (ΔΔCT,4.1 versus 1.1) and collagen-1 (ΔΔCT, 4.8 versus 1.1) mRNA expression (Fig. 9A).
FIGURE 9.

OPN, EtOH, and TGF-β1 regulate SOX-9 and collagen-1 mRNA expression. A, expression of SOX-9 and collagen-1 mRNA. MSC were exposed to OPN and EtOH for 24 h, and SOX-9 and collagen-1 mRNA levels were measured using qRT-PCR (*, p < 0.0001). B, effect of TGF-β1 on collagen-1 mRNA expression in MSC grown in osteogenic inducing cell culture medium. Application of exogenous TGF-β1 to MSC partially reversed EtOH inhibition of OPN-induced collagen-1 expression (*, p < 0.0001). C, effect of TGF-β1 on SOX-9 mRNA expression in MSC grown in chondrogenic inducing cell culture medium. Exogenous TGF-β1 treatment also partially reversed EtOH inhibition of OPN-induced SOX-9 mRNA expression (*, p < 0.0001). Data are presented as mean ± S.E. of three experiments.
Next, differentiation studies were carried out by treating MSC with specific cell medium (Life Technologies) designed to induce differentiation into either the osteogenic or chondrogenic lineages. Standard treatments with OPN and EtOH were applied under both culture conditions, and exogenous TGF-β1 was administered to determine whether TGF-β1 could counteract the inhibitory effect of EtOH on OPN-induced collagen-1 and SOX-9 mRNA expression. Under the osteogenic differentiation conditions, treatment of MSC with TGF-β1 alone significantly increased collagen-1 mRNA expression compared with untreated MSC (ΔΔCT, 6.4 versus 4.4). TGF-β1 was also able to increase collagen-1 in the presence of OPN and EtOH (ΔΔCT, 6.3 versus 4.5), thus partially reversing the inhibitory effect of EtOH on OPN-induced collagen-1 expression (Fig. 9B). Under the chondrogenic differentiation conditions, similar results were found regarding TGF-β1 effects on SOX-9 expression. Exogenous TGF-β1 treatment was able to partially reverse inhibition of EtOH of the OPN-mediated SOX-9 expression (ΔΔCT, 6.2 versus 5.4), however, TGF-β1 application to untreated MSC did not significantly increase SOX-9 mRNA levels (ΔΔCT, 4.2 versus 4.1) (Fig. 9C). These results suggest that OPN stimulates expression of known protein markers associated with osteoblast and chondrocyte differentiation, and EtOH exposure partially inhibits expression of these protein markers. Exogenous TGF-β1 application to MSC appears to enhance expression of collagen-1 and SOX-9 in the presence of EtOH.
EtOH Exposure Affects Mouse Fracture Callus Protein Composition
In vivo studies were conducted using a mouse model system that allowed for histological analysis of the bone fracture environment.
Mice were treated with either saline or EtOH and then subjected to a tibial fracture. Tibial fracture callus sections from saline and EtOH-treated mice were isolated 7 days post-fracture and sliced for histological analysis of the fracture environment. Hematoxylin and esosin staining of the healing callus 7 days post-fracture showed normal post-fracture healing with well formed new bone trabeculae (Fig. 10A). The fracture from EtOH-treated mice, however, showed a marked decrease in callus formation and new bone deposition (Fig. 10B), supporting previous work by Lauing et al. (4). OPN immunohistochemistry revealed considerable OPN expression in the fracture environment from both saline- and EtOH-treated mice (Fig. 10, C and D). Immunohistochemistry for TGF-β1 demonstrated visible focal staining in the normal fracture callus environment. Focal staining of TGF-β1 in the EtOH-treated fracture callus appeared to be reduced or nearly absent (Fig. 10, E and F). Staining for the MSC marker, α-SMA, showed strong expression in the normal healing fracture environment. Fracture callus from EtOH-treated mice demonstrated decreased α-SMA staining, correlating to a decreased activated fibroblast phenotype in the fracture callus (Fig. 10, G and H). Tibial fracture callus sections from saline- and EtOH-treated mice also underwent a protein isolation procedure 3 and 7 days post-fracture, and then subsequent quantification of TGF-β1 protein using ELISA. EtOH-treated mice showed significantly decreased active TGF-β1 protein in the fracture callus environment on day 3 post-fracture compared with saline-treated control mice (883.6 versus 547.1 pg/ml) (Fig. 9I).
FIGURE 10.

EtOH exposure affects mouse fracture callus protein composition. A–H, histological analysis of binge EtOH treatment on mouse fracture callus composition. Fracture callus sections were stained with hematoxylin and esosin, and antibodies against OPN, TGF-β1, and α-SMA. The black bar indicates the length of 60 μm. I, active TGF-β1 protein in mouse fracture callus. The concentration of active TGF-β1 protein in the tibial fracture callus from saline- and EtOH-treated mice 3 and 7 days post-fracture was measured using ELISA (*, p < 0.01).
DISCUSSION
EtOH exposure has been shown to interfere with numerous aspects of fracture healing across a broad scope of experimental models. However, the signaling mechanisms responsible for the effects of EtOH are still not completely understood. In this study, we have demonstrated that EtOH inhibits MSC production of the OPN-dependent TGF-β1 protein, and that EtOH mediates its activity by interfering with the phosphorylation and transcriptional activation of MZF1 gene. OPN was found to induce MZF1 to stimulate its own transcription via a positive feedback mechanism. We also provided evidence supporting that MZF1 can exist in a phosphorylated state, which can be induced by OPN. In addition, we demonstrated that PKA signaling enhances TGF-β1 protein expression and MZF1 promoter activation and phosphorylation in the presence of OPN. Last, we found that EtOH-treated mice demonstrate a altered fracture callus environment and protein composition. Our laboratory was the first to describe the OPN-MZF1-TGF-β1 linked pathway, and we have shown here that EtOH exposure impedes this pathway in the production of TGF-β1 in MSC.
Although EtOH consumption in alcoholic patients has been shown to have a range of negative effects on bone physiology, including decreased bone mineral density and increased risk for osteoporosis, the effect of EtOH on fracture healing is of particular clinical relevance. Acute alcohol intoxication is a common presentation to emergency departments, and it is associated with traumatic injuries requiring longer hospital stays (1, 22). Intoxication increases risks for both sustaining fracture injuries as well as adversely affecting the healing process. Human studies have shown that healing and recovery from fractures is adversely affected and is prolonged with higher rates of complications in patients with significant EtOH exposure (3, 23, 24).
OPN and TGF-β1 have been widely implicated in normal bone formation and remodeling. Both signaling molecules promote MSC migration from bone marrow, and TGF-β1 stimulates MSC differentiation vital for proper fracture healing (25). A particular spatial-temporal pattern of TGF-β1 expression in the bone and stromal environment is necessary for efficient bone remodeling. Genetic mutations in TGF-β1 signaling are implicated in the pathogenesis of a number of skeletal defects and bone diseases. TGF-β1 has been shown to influence fracture healing (26), stimulate angiogenesis within the fracture site (17), and recruit MSC from bone marrow (27). OPN is believed to be involved in numerous aspects of bone function, including migration, attachment, and osteoclastogenesis during bone remodeling (28, 29). OPN deficiency also has adverse effects on fracture healing, as OPN null mice exhibit decreased size and reduced biomechanical properties of the healing fracture callus (12).
MSC play an essential role in fracture healing, both by their strong expression of TGF-β1 and by their ability to differentiate into cell types that constitute new bone, specifically osteoblasts and chondrocytes. Many studies have outlined the ability of MSC to migrate and attach to a fracture environment, thereby contributing to callus formation and healing (30, 31). There is much clinical interest in using MSC as an exogenous source to enhance fracture healing, and this has been already shown to partially reverse EtOH-induced deficits in fracture healing in animal models (9). It has been shown that TGF-β1 contributes to induction of MSC differentiation into several cell lineages (25). It has also been previously shown that EtOH exposure specifically can inhibit MSC differentiation into the osteogenic lineage (32), and instead induces adipogenic differentiation (33), which is an important finding related to EtOH-induced deficient bone formation. We examined the role of EtOH in affecting MSC differentiation and showed that in the low TGF-β1 environment following EtOH administration, MSC exhibited reduced levels of osteoblast and chondrocyte markers collagen-1 and SOX-9, respectively. In addition, treatment of MSC with exogenous TGF-β1 was able to partially reverse the inhibitory effect of EtOH on OPN-induced collagen-1 and SOX-9 mRNA expression. These findings suggest that downstream signaling of OPN leading to TGF-β1 expression plays an important role in stimulating collagen-1 and SOX-9 expression. TGF-β1 may be acting in a feedback mechanism on MSC, which may ultimately play an important role in MSC differentiation into the osteogenic and chondrogenic lineages.
Although the link between OPN and TGF-β1 has been described extensively in numerous settings including inflammation, fibrosis, and cancer, our laboratory was the first to identify MZF1 as the primary transcription factor activated by OPN, which then induces TGF-β1 expression (18). MZF1 belongs to the Kruppel family of zinc finger proteins, and has been shown to be expressed in totipotent hematopoietic cells as well as in myeloid progenitors (34). In this study, MZF1 function was found to be an important target of EtOH. Exposure of OPN-treated MSC to EtOH leads to decreased TGF-β1 protein expression and negligible binding of MZF1 to the TGF-β1 promoter. Further investigation showed that EtOH interferes with MZF1 expression and promoter activation. In addition to decreasing MZF1 mRNA levels, EtOH blocked binding of RNA polymerase II and MZF1 to its own promoter, thus shutting down transcriptional activation. Luciferase studies demonstrated that the MZF1 binding site between −545 and −537 bp upstream of the MZF1 start codon is necessary for full promoter activation, as directed mutations at this site blocked promoter activity. ChIP revealed that EtOH exposure in OPN-treated MSC ablated MZF1 binding to this promoter site, thus inhibiting the autoinduction mechanism responsible for MZF1 expression.
To date there have not been any studies linking OPN signaling and MZF1 activation. OPN interacts with receptors CD44 and integrin αvβ3 on MSC (35), and it was shown in this study that EtOH exposure neither interferes with OPN-receptor interactions or alters the cell surface receptor density. Previous work in our laboratory demonstrated that antibody blockade of the αvβ3 integrin receptor specifically prevented MSC production of TGF-β1 in response to OPN treatment. It is possible that downstream signaling from the αvβ3 receptor in response to OPN leads to MZF1 activation, and thus is targeted by EtOH. However, with little known about the intracellular signaling cascade leading to MZF1 activation, we used predictive modeling to determine potential target sites on MZF1 for phosphorylation. The PKA signaling pathway was chosen as a candidate based off the computational analysis, and its activity was experimentally modulated using the small activator and inhibitor molecules SP8 and H-89, respectively. Both molecules were found to significantly influence MZF1 promoter activation as well as subsequent expression of TGF-β1 mRNA and active and total protein. SP8, a cAMP activator, was shown to partially rescue EtOH inhibition of OPN-dependent TGF-β1 expression. To the contrary, exposure of MSC to the PKA inhibitor H-89 resulted in reduced MZF1 activation and decreased TGF-β1 expression in the presence of OPN.
This study further demonstrated the relationship between OPN, PKA, and MZF1 in showing that OPN alone can stimulate phosphorylation of known PKA downstream substrate proteins, a phenomenon that can be blocked by EtOH. We also demonstrated that MZF1 can exist in a phosphorylated state by utilizing co-immunoprecipitation techniques. Both OPN treatment and PKA activation by SP8 induces phosphorylation of MZF1, and EtOH exposure blocks the observed effect of OPN and reverses MZF1 phosphorylation. Taken together, these findings link OPN to PKA activity and subsequent phosphorylation of downstream PKA targets, as well as to phosphorylation of MZF1. Co-immunoprecipitation studies using a phosphoantibody specific for phosphorylated PKA substrates (data not shown) failed to bind MZF1, thus indicating that MZF1 is not a direct substrate of PKA. Phosphorylation of MZF1 is more likely performed by one of downstream target kinases of the PKA. The ability of PKA signaling to augment and enhance MZF1 activity and subsequent TGF-β1 expression of MSC in the presence of EtOH opens up exciting possibilities, as modulation of this pathway can be achieved using well known small molecules that are amenable to future animal models and potential clinical scenarios.
EtOH exposure to rodents prior to bone fracture has been shown to alter the protein composition and biomechanical properties of the healing fracture callus (4, 6). Here we showed that intraperitoneal injection of EtOH in mice prior to fracture injury leads to altered fracture callus composition. EtOH exposure decreased deposition of new bone and aberrant signaling molecule expression in the fracture callus environment. Focal TGF-β1 expression was suggestively decreased, and α-SMA, a marker of activated fibroblasts in response to TGF-β1, was markedly decreased in the callus of EtOH-treated mice. Interestingly, OPN staining appeared to be similar across treatment groups.
In summary, we described a novel mechanism by which EtOH interferes with OPN-dependent MZF1 and TGF-β1 function in MSC. EtOH appears to exert its inhibitory effects in the signaling pathway downstream of OPN-receptor binding but upstream of MZF1 phosphorylation and promoter activation. Further characterization of the OPN-MZF1-TGF-β1 signaling pathway will be required to better understand the exact mechanism by which EtOH acts. This study also found that PKA signaling potentiates OPN stimulation and is linked to MZF1 function. This finding may serve as an avenue for continued experimental investigation and potential clinical intervention.
Footnotes
- MSC
- mesenchymal stem cell
- OPN
- osteopontin
- APT
- aptamer
- MuAPT
- mutant aptamer
- MZF1
- myeloid zinc finger 1
- SP8
- Sp-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt hydrate
- α-SMA
- α-smooth muscle actin
- RLA
- relative luciferase activity
- Ab
- antibody
- qRT
- quantitative RT.
REFERENCES
- 1. Rivara F. P., Jurkovich G. J., Gurney J. G., Seguin D., Fligner C. L., Ries R., Raisys V. A., Copass M. (1993) The magnitude of acute and chronic alcohol abuse in trauma patients. Arch. Surg. 128, 907–912; discussion 912–903 [DOI] [PubMed] [Google Scholar]
- 2. Demetriades D., Gkiokas G., Velmahos G. C., Brown C., Murray J., Noguchi T. (2004) Alcohol and illicit drugs in traumatic deaths: prevalence and association with type and severity of injuries. J. Am. Coll. Surg. 199, 687–692 [DOI] [PubMed] [Google Scholar]
- 3. Nyquist F., Berglund M., Nilsson B. E., Obrant K. J. (1997) Nature and healing of tibial shaft fractures in alcohol abusers. Alcohol Alcohol. 32, 91–95 [DOI] [PubMed] [Google Scholar]
- 4. Lauing K. L., Roper P. M., Nauer R. K., Callaci J. J. (2012) Acute alcohol exposure impairs fracture healing and deregulates β-catenin signaling in the fracture callus. Alcohol. Clin. Exp. Res. 36, 2095–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Volkmer D. L., Sears B., Lauing K. L., Nauer R. K., Roper P. M., Yong S., Stover M., Callaci J. J. (2011) Antioxidant therapy attenuates deficient bone fracture repair associated with binge alcohol exposure. J. Orthop. Trauma 25, 516–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chakkalakal D. A., Novak J. R., Fritz E. D., Mollner T. J., McVicker D. L., Garvin K. L., McGuire M. H., Donohue T. M. (2005) Inhibition of bone repair in a rat model for chronic and excessive alcohol consumption. Alcohol 36, 201–214 [DOI] [PubMed] [Google Scholar]
- 7. Chakkalakal D. A., Strates B. S., Mashoof A. A., Garvin K. L., Novak J. R., Fritz E. D., Mollner T. J., McGuire M. H. (1999) Repair of segmental bone defects in the rat: an experimental model of human fracture healing. Bone 25, 321–332 [DOI] [PubMed] [Google Scholar]
- 8. Yoo J. U., Johnstone B. (1998) The role of osteochondral progenitor cells in fracture repair. Clin. Orthop. Relat. Res. 355, S73–S81 [DOI] [PubMed] [Google Scholar]
- 9. Obermeyer T. S., Yonick D., Lauing K., Stock S. R., Nauer R., Strotman P., Shankar R., Gamelli R., Stover M., Callaci J. J. (2012) Mesenchymal stem cells facilitate fracture repair in an alcohol-induced impaired healing model. J. Orthop. Trauma 26, 712–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirakawa K., Hirota S., Ikeda T., Yamaguchi A., Takemura T., Nagoshi J., Yoshiki S., Suda T., Kitamura Y., Nomura S. (1994) Localization of the mRNA for bone matrix proteins during fracture healing as determined by in situ hybridization. J. Bone Miner. Res. 9, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 11. Shin H., Zygourakis K., Farach-Carson M. C., Yaszemski M. J., Mikos A. G. (2004) Attachment, proliferation, and migration of marrow stromal osteoblasts cultured on biomimetic hydrogels modified with an osteopontin-derived peptide. Biomaterials 25, 895–906 [DOI] [PubMed] [Google Scholar]
- 12. Duvall C. L., Taylor W. R., Weiss D., Wojtowicz A. M., Guldberg R. E. (2007) Impaired angiogenesis, early callus formation, and late stage remodeling in fracture healing of osteopontin-deficient mice. J. Bone Miner. Res. 22, 286–297 [DOI] [PubMed] [Google Scholar]
- 13. Blum A., Zarqh O., Peleg A., Sirchan R., Blum N., Salameh Y., Ganaem M. (2012) Vascular inflammation and endothelial dysfunction in fracture healing. Am. J. Orthop. 41, 87–91 [PubMed] [Google Scholar]
- 14. Cho T. J., Gerstenfeld L. C., Einhorn T. A. (2002) Differential temporal expression of members of the transforming growth factor β superfamily during murine fracture healing. J. Bone Miner. Res. 17, 513–520 [DOI] [PubMed] [Google Scholar]
- 15. Lieberman J. R., Daluiski A., Einhorn T. A. (2002) The role of growth factors in the repair of bone. Biology and clinical applications. J. Bone Joint Surg. 84, 1032–1044 [DOI] [PubMed] [Google Scholar]
- 16. Filvaroff E., Erlebacher A., Ye J., Gitelman S. E., Lotz J., Heillman M., Derynck R. (1999) Inhibition of TGF-β receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 126, 4267–4279 [DOI] [PubMed] [Google Scholar]
- 17. Saadeh P. B., Mehrara B. J., Steinbrech D. S., Dudziak M. E., Greenwald J. A., Luchs J. S., Spector J. A., Ueno H., Gittes G. K., Longaker M. T. (1999) Transforming growth factor-β1 modulates the expression of vascular endothelial growth factor by osteoblasts. Am. J. Physiol. 277, C628–637 [DOI] [PubMed] [Google Scholar]
- 18. Weber C. E., Kothari A. N., Wai P. Y., Li N. Y., Driver J., Zapf M. A., Franzen C. A., Gupta G. N., Osipo C., Zlobin A., Syn W. K., Zhang J., Kuo P. C., Mi Z. (2014) Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 10.1038/onc.2014.410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mi Z., Guo H., Russell M. B., Liu Y., Sullenger B. A., Kuo P. C. (2009) RNA aptamer blockade of osteopontin inhibits growth and metastasis of MDA-MB231 breast cancer cells. Mol. Ther. 17, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dostmann W. R., Taylor S. S., Genieser H. G., Jastorff B., Døskeland S. O., Ogreid D. (1990) Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3′,5′-cyclic phosphorothioates. J. Biol. Chem. 265, 10484–10491 [PubMed] [Google Scholar]
- 21. Yang D. C., Tsay H. J., Lin S. Y., Chiou S. H., Li M. J., Chang T. J., Hung S. C. (2008) cAMP/PKA regulates osteogenesis, adipogenesis and ratio of RANKL/OPG mRNA expression in mesenchymal stem cells by suppressing leptin. PloS One 3, e1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levy R. S., Hebert C. K., Munn B. G., Barrack R. L. (1996) Drug and alcohol use in orthopedic trauma patients: a prospective study. J. Orthop. Trauma 10, 21–27 [DOI] [PubMed] [Google Scholar]
- 23. Askew A., Chakkalakal D., Fang X., McGuire M. (2011) Delayed fracture healing in alcohol abusers: a preliminary retrospective study. Open Bone J. 10.2174/1876525401103010001 [DOI] [Google Scholar]
- 24. Tønnesen H., Pedersen A., Jensen M. R., Møller A., Madsen J. C. (1991) Ankle fractures and alcoholism: the influence of alcoholism on morbidity after malleolar fractures. J. Bone Joint Surg. Br. 73, 511–513 [DOI] [PubMed] [Google Scholar]
- 25. Park J. S., Chu J. S., Tsou A. D., Diop R., Tang Z., Wang A., Li S. (2011) The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-β. Biomaterials 32, 3921–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lind M., Schumacker B., Søballe K., Keller J., Melsen F., Bünger C. (1993) Transforming growth factor-beta enhances fracture healing in rabbit tibiae. Acta Orthop. Scand. 64, 553–556 [DOI] [PubMed] [Google Scholar]
- 27. Tang Y. (2009) TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 15, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giachelli C. M., Steitz S. (2000) Osteopontin: a versatile regulator of inflammation and biomineralization. Matrix Biol. 19, 615–622 [DOI] [PubMed] [Google Scholar]
- 29. Standal T., Borset M., Sundan A. (2004) Role of osteopontin in adhesion, migration, cell survival and bone remodeling. Exp. Oncol. 26, 179–184 [PubMed] [Google Scholar]
- 30. Granero-Moltó F., Weis J. A., Miga M. I., Landis B., Myers T. J., O'Rear L., Longobardi L., Jansen E. D., Mortlock D. P., Spagnoli A. (2009) Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells 27, 1887–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruder S. P., Jaiswal N., Ricalton N. S., Mosca J. D., Kraus K. H., Kadiyala S. (1998) Mesenchymal stem cells in osteobiology and applied bone regeneration. Clin. Orthop. Relat. Res. 355, S247–S256 [DOI] [PubMed] [Google Scholar]
- 32. Gong Z., Wezeman F. H. (2004) Inhibitory effect of alcohol on osteogenic differentiation in human bone marrow-derived mesenchymal stem cells. Alcohol. Clin. Exp. Res. 28, 468–479 [DOI] [PubMed] [Google Scholar]
- 33. Wezeman F. H., Gong Z. (2004) Adipogenic effect of alcohol on human bone marrow-derived mesenchymal stem cells. Alcohol. Clin. Exp. Res. 28, 1091–1101 [DOI] [PubMed] [Google Scholar]
- 34. Gaboli M., Kotsi P. A., Gurrieri C., Cattoretti G., Ronchetti S., Cordon-Cardo C., Broxmeyer H. E., Hromas R., Pandolfi P. P. (2001) Mzf1 controls cell proliferation and tumorigenesis. Genes Dev. 15, 1625–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Denhardt D. T., Guo X. (1993) Osteopontin: a protein with diverse functions. FASEB J. 7, 1475–1482 [PubMed] [Google Scholar]