

Dear Editor,
Diastolic heart failure (DHF) and systolic heart failure (SHF) are two main subsets of chronic heart failure that are commonly encountered in clinical practice (Chatterjee and Massie, 2007). Although there have been considerable advances in the treatment of SHF, the molecular and biochemical mechanisms mediating the structural remodeling and principal functional derangement in both SHF and DHF remain unclear. In particular, effective therapeutic regimens are still lacking for DHF; thus, it is imperative to better understand the molecular and biochemical mechanisms of DHF in an effort to seek novel therapeutic avenues for patients suffering from DHF. DHF is manifested by stiffer ventricular walls, while SHF has overly compliant ventricular walls. Therefore, altering ventricular wall stiffness will be a potential therapeutic strategy for both DHF and SHF.
Recently, increasing attention has been paid by researchers and cardiologists to titin in regard to developing an alternative therapy for both DHF and SHF. Titin, a giant sarcomeric protein, is a key determinant of myocardial passive tension and is largely responsible for the diastolic properties of the heart (LeWinter and Granzier, 2014). Two major classes of titin are co-expressed in mammalian cardiac muscles, namely the smaller N2B isoform (3.0 MDa) and the larger N2BA isoform (3.2–3.7 MDa). The larger N2BA isoform is more compliant and develops lower passive tension, while the N2B isoform is stiffer and develops higher passive tension. At varying ratios of N2BA to N2B isoforms, the sarcomeres develop an intermediate level of passive tension affecting myofibrillar extensibility and passive force generation and altering the stiffness of the cardiac walls (Cazorla et al., 2000; Freiburg et al., 2000). Therefore, the manipulation of their ratios has been considered a therapy for reducing pathological diastolic stiffness or increasing pathological systolic stiffness. However, the mechanisms regulating titin isoform transition remain elusive. Recently, RNA binding motif 20 (RBM20), a splicing factor, has been identified as a major regulator of titin isoform transition (Guo et al., 2012), while the thyroid hormone-triiodothyronine (T3) has also been reported to regulate titin isoform transition (Wu et al., 2007; Kruger et al., 2008). Nevertheless, prior to this report, the relationship between RBM20 and T3 in the regulation of titin isoform transition remains unknown. The present study was designed to evaluate the role of RBM20 in T3-regulated titin isoform transition, and the molecular signaling mechanisms linking T3 and RBM20.
There are indications that thyroid gland maturation and titin isoform transition occur in the fetus at approximately the same time (Polk, 1995; Kruger et al., 2006). Indeed, in the presence of RBM20, T3 treatment in primary cultures of neonatal rat cardiomyocytes (CMs) increased the smaller N2B isoform expression (Kruger et al., 2008). However, it is unknown whether T3 can still regulate titin isoform transition in the absence of RBM20. Hence, we examined the influence of T3 on titin isoform transition in the absence of RBM20. In homozygous RBM20 knockout rats (Rbm20−/−), only the largest N2BA isoform (∼3.9 MDa) is present (Guo et al., 2010), and in heterozygous RBM20 knockout rats (Rbm20+/−), the intermediate sized titin N2BA isoforms (3.2–3.7 MDa) are co-expressed with the smaller N2B isoform, while, in adult wild type rats (Rbm20+/+), the small, stiffer N2B isoform predominates (Guo et al., 2012). This makes the RBM20 deficient rat model a unique tool for this study. We isolated and cultured neonatal ventricular cardiomyocytes (NVCMs) of Rbm20+/+, Rbm20+/−, and Rbm20−/− rats. With the addition of T3 in a serum-starved medium, the ratios of N2B to N2BA isoforms increased significantly, from ∼23% to ∼59% of total titin isoforms (N2B+N2BA) in Rbm20+/+ rats (Figure 1A) and from ∼6% to ∼23% of total titin isoforms in Rbm20+/− group (Figure 1B). However, increased N2B expression did not occur in Rbm20−/− NVCMs with T3 supplementation. Virtually no N2B was expressed in either treated or untreated Rbm20−/− NVCMs (Figure 1C). These results suggest that RBM20 plays an indispensable role in the regulation of titin isoform transition triggered by T3.
Figure 1.
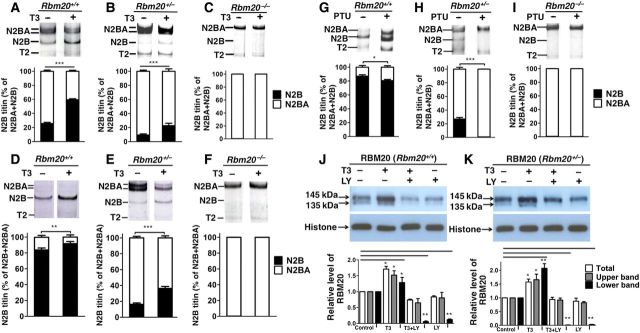
Effect of T3 on titin isoform transition in RBM20 deficient NVCMs and rats. (A, B, D, and E) N2B-titin isoform increased with T3 treatment in primary cultures of Rbm20+/+ and Rbm20+/− NVCMs and rats when compared with control without T3 treatment. (C and F) Virtually no N2B titin was expressed in Rbm20−/− NVCMs and rats, and N2B-titin isoform was not increased by T3 treatment in primary cultures of Rbm20−/− NVCMs and rats when compared with control without T3 treatment. (G and H) N2B-titin isoform decreased in Rbm20+/+ and Rbm20+/− rats with PTU treatment when compared with control groups without PTU supplementation, and N2B-titin isoform was nearly undetectable in Rbm20+/− rats with PTU treatment. (I) The ratios of N2B to N2BA isoforms were not altered by PTU in Rbm20−/− rats. (J and K) RBM20 bands shift on western blot with antibody against RBM20 in both Rbm20+/+ and Rbm20+/− NVCMs; lower band was estimated to be 135 kDa and upper band 145 kDa. The density of the upper band (in gray) and lower band (in black) increased with T3 supplementation, but decreased with LY supplementation when compared with control without T3 and LY in Rbm20+/+ and Rbm20+/− NVCMs. Total RBM20 (in white) increased with T3 supplementation and decreased with LY supplementation when compared with control in Rbm20+/+ and Rbm20+/− NVCMs. T2, degraded titin bands; histone, loading control for nuclear protein RBM20. Bars show means ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
In order to further confirm whether T3-regulated titin isoform transition is RBM20-dependent, we performed in vivo assays. We treated the Rbm20+/+, Rbm20+/−, and Rbm20−/− rats independently with T3 and propylthiouracil (PTU, a drug that inhibits the secretion of thyroid hormone). After a 90-day treatment with subcutaneously implanted T3 pellets in Rbm20+/+, Rbm20+/−, and Rbm20−/− rats, we harvested the hearts, and observed titin isoform transitions by resolving titin bands with a 1% SDS-agarose gel. The N2B isoform was significantly increased in Rbm20+/+ and Rbm20+/− rat hearts (Figure 1D), with increases of N2B isoform from ∼84% to 92% of total titin in Rbm20+/+ rats, and from ∼16% to 36% of total titin in Rbm20+/− rats (Figure 1E). No N2B isoform was expressed in Rbm20−/− rat hearts under T3 treatment when compared with control groups that were implanted with placebo (Figure 1F). An equal number of age-matched rats were used for the PTU treatment by feeding a diet containing 0.15% PTU. After 3 months of treatment, with low thyroid status, the N2BA isoform was increased and the N2B isoform was decreased from ∼87% to 80% of total titin in Rbm20+/+ rats (Figure 1G). The N2B isoform was nearly undetectable with PTU treatment in Rbm20+/− rats (Figure 1H). However, no changes were observed in Rbm20−/− rats treated with PTU when compared with control groups without PTU treatment (Figure 1I). These data further confirm that RBM20 is an essential factor for thyroid hormone-regulated titin isoform transition.
Next, we examined the mechanisms linking RBM20 and T3 in the regulation of titin isoform transition. Western blot analysis using anti-RBM20 antibody indicated two bands (∼135 and 145 kDa) in control primary cultures of Rbm20+/+ and Rbm20+/− NVCMs (Figure 1J and K). With supplementation of T3, the intensity of the 145 kDa band increased in primary cultures of Rbm20+/+ and Rbm20+/− NVCMs when compared with the control without T3, while supplementation of LY 294002 (LY, a PI3K inhibitor) with or without T3 produced slightly decreased intensity of the 145 kDa band, and a significant reduction in the intensity of the 135 kDa band (Figure 1J and K). These results suggest that RBM20 is likely posttranslationally modified. If this is the case, the posttranslational modification of RBM20 could be phosphorylation, given that the phosphorylation of serine–arginine (SR) proteins (Guo et al., 2012) is essential for pre-mRNA splicing (Prasad et al., 1999). A previous study reported that T3 regulates titin isoform transition via the PI3K/Akt pathway (Kruger et al., 2008). Our results also demonstrated that T3 supplementation increased AKT activation and the N2B isoform, while LY supplementation decreased AKT activation and the N2B isoform (Supplementary Figures S1A–C and S2A–C) in primary cultures. In in vivo assays with rats treated with T3 and PTU, we found that the activation and phosphorylation of Akt were increased with T3 treatment, and deceased with PTU treatment (Supplementary Figures S3A–C and S4A–C). These results suggest that the phosphorylation of RBM20 is crucial for titin isoform transition triggered by T3. The mechanism could be that first T3 activates Akt and then Akt phosphorylates RBM20, and finally the phosphorylated RBM20 regulates titin isoform transition. Band density analysis also demonstrated that total RBM20 expression was increased with T3 supplementation when compared with control without T3 supplementation in the medium. Additionally, LY supplementation reduced the total amount of RBM20 when compared with control without T3 or LY supplementation (Figure 1J and K). These results imply that T3-regulated titin isoform transition could also activate the mTOR pathway and increase gene expression of RBM20. The relative expression of Rbm20 mRNA was also validated by real-time PCR, and the results revealed that the mRNA level increased with T3 supplementation while it decreased with LY supplementation (Supplementary Figure S5A and B). Therefore, we conclude that titin isoform transition triggered by T3 is linked to RBM20 via the PI3K/Akt/mTOR signaling pathway, phosphorylating RBM20 and/or increasing gene expression of RBM20 (Supplementary Figure S6).
Taken together, these findings present significant clinical implications for patients with SHF or DHF. Our data suggest that T3 treatment in SHF may increase the stiffness of ventricular walls through upregulating the stiffer N2B isoform. In particular, patients with DHF can be benefited from the treatment to decrease the stiffer N2B isoform. Overall, our data should strengthen the understanding of the regulatory mechanism of titin isoform transition, which might help to decipher the mechanisms of developmental or diseased titin isoform transition, and further, they provide new insights for improving diastolic and systolic dysfunction.
[Supplementary material is available at Journal of Molecular Cell Biology online. We thank Mark D. Hanna for expert technical assistance and laboratory management. We are grateful to the support of Agricultural Experiment Station at University of Wyoming and the Regional Hatch Project (NC1184). This work was supported by a pilot grant from NIH-P20-GM-103432 and Startup Funds from the University of Wyoming.]
Supplementary Material
References
- Cazorla O., Freiburg A., Helmes M., et al. Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ. Res. 2000;86:59–67. doi: 10.1161/01.res.86.1.59. [DOI] [PubMed] [Google Scholar]
- Chatterjee K., Massie B. Systolic and diastolic heart failure: differences and similarities. J. Card. Fail. 2007;13:569–576. doi: 10.1016/j.cardfail.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Freiburg A., Trombitas K., Hell W., et al. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ. Res. 2000;86:1114–1121. doi: 10.1161/01.res.86.11.1114. [DOI] [PubMed] [Google Scholar]
- Guo W., Bharmal S.J., Esbona K., et al. Titin diversity—alternative splicing gone wild. J. Biomed. Biotechnol. 2010;2010:753675. doi: 10.1155/2010/753675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Schafer S., Greaser M.L., et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012;18:766–773. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger M., Kohl T., Linke W.A. Developmental changes in passive stiffness and myofilament Ca2+ sensitivity due to titin and troponin-I isoform switching are not critically triggered by birth. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H496–H506. doi: 10.1152/ajpheart.00114.2006. [DOI] [PubMed] [Google Scholar]
- Kruger M., Sachse C., Zimmermann W.H., et al. Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-kinase/AKT pathway. Circ. Res. 2008;42:186–197. doi: 10.1161/CIRCRESAHA.107.162719. [DOI] [PubMed] [Google Scholar]
- LeWinter M.M., Granzier H.L. Cardiac titin and heart disease. J. Cardiovasc. Pharmacol. 2014;63:207–212. doi: 10.1097/FJC.0000000000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk D.H. Thyroid hormone metabolism during development. Reprod. Fertil. Dev. 1995;7:469–477. doi: 10.1071/rd9950469. [DOI] [PubMed] [Google Scholar]
- Prasad J., Colwill K., Pawson T., et al. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypo-phosphorylation inhibit splicing. Mol. Cell. Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Peng J., Campbell K.B., et al. Hypothyroidism leads to increased collagen-based stiffness and re-expression of large cardiac titin isoforms with high compliance. J. Mol. Cell. Cardiol. 2007;42:186–195. doi: 10.1016/j.yjmcc.2006.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.