

Abstract
Little is known about the association between autosomal-dominant polycystic kidney disease (ADPKD) and coronary artery dissection (CAD). We suggest that the genetic disorder in ADPKD is the main cause of instable artery vasculature. Our case also shows that CAD can be missed in the acute phase. Therefore, we recommend additional investigation in patients with ADPKD presenting with acute chest pain. We report a case of a patient who developed a myocardial infarction due to a spontaneous dissection of the left anterior descending coronary artery. ADPKD was diagnosed during the additional investigation. The patient received medical management.
Keywords: autosomal-dominant polycystic kidney disease, coronary artery angiography, coronary artery dissection, myocardial infarction
Background
Autosomal-dominant polycystic kidney disease (ADPKD) is characterized by cyst formation and occurs primarily in the kidneys. Due to the replacement of the normal renal parenchyma, ADPKD results in end-stage renal failure in 45% of patients [1]. Approximately 7–11% of patients receiving renal replacement therapy in the Western world are doing so due to ADPKD [1]. The underlying cause of ADPKD is a mutation in the polycystin-1 and -2 plasma proteins, located in the primary cilia. This mutation leads to abnormal function of renal tubular epithelia and inadequate calcium influx followed by cyst formation [2].
Cardiovascular disease is the most frequent cause (36%) of mortality in patients with ADPKD [3]. Patients with ADPKD often develop hypertension at an earlier age than the general population before any impairment of kidney function. It is hypothesized that the renin-aldosterone system and endothelial dysfunction caused by impaired nitric oxide release are the important factors in the development of hypertension in ADPKD patients [4]. Arterial dissection is one of the cardiovascular manifestations of the disease [4]. We describe an uncommon case of a middle-aged man with a spontaneous coronary artery dissection (CAD) and ADPKD.
Case report
A 41-year-old Caucasian man presented at an emergency department with acute chest pain. The chest pain began 2 h before presentation. There was no history of trauma or excessive physical exercise. He had no other complaints and used no medication. Medical history recorded a subdural haematoma, as a result of trauma. According to the family history, his mother and aunt were both diagnosed with ADPKD. Our patient was a non-smoker, with no history of hypertension, diabetes mellitus or hypercholesterolaemia.
On physical examination, the blood pressure was 155/102 mmHg and the pulse 72 beats/minute; other vital parameters were normal. Electrocardiography showed a sinus rhythm of 65 beats/minute and ST elevation in the precordial leads (Figure 1). Creatine kinase was 249 U/L, creatine kinase MB was 17.4 U/L and troponin T was 0.21 ng/mL. The other laboratory test results were normal. Twelve hours later, the cardiac enzymes increased to higher levels. Echocardiography revealed a hypokinetic septum and a slightly impaired left ventricular function with an ejection fraction of 45–60%.
Fig. 1.
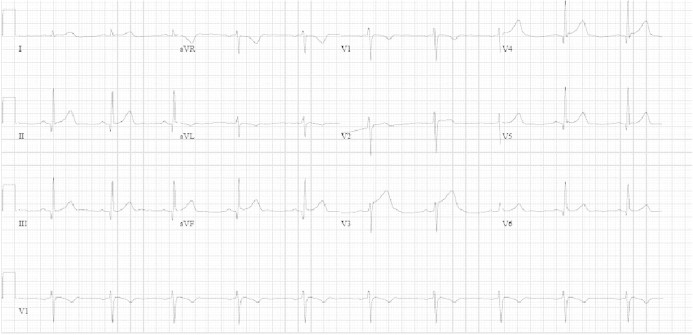
Electrocardiography on presentation shows a sinus rhythm of 65 beats/minute and ST elevation in V3, II and III.
Based on these results, a presumptive diagnosis of acute septal myocardial infarction was made. The patient was immediately transported to the University Medical Centre for cardiac intervention. Coronary artery angiography (CAG) revealed a transient occlusion of the left anterior descending (LAD) coronary artery, most probably as a result of myocardial bridging (Figure 2A). Other coronary arteries appeared normal. Percutaneous coronary intervention was not performed due to small vessel size. The myocardial infarction was treated with anti-platelet therapy, statin and metoprolol.
Fig. 2.

The CAG (A) shows the compression of the LAD coronary artery during the systole resulting in narrowing and (B) performed after the second chest pain attack demonstrating a dissection in the distal left anterior descending coronary artery.
However, the chest pain returned 3 days after the presentation. A second electrocardiography showed persistent inverted T waves in the precordial leads without ST elevation. The cardiac enzyme levels were again increasing. A second CAG was performed because of suspicion of recurrent myocardial infarction. It disclosed an open LAD with a dissection in the distal part and a double lumen, which was not observed during the first angiography (Figure 2B). Myocardial scintigraphy showed a restricted infarction of the septum.
The definitive diagnosis of non-Q-wave anterior infarct as a result of a spontaneous LAD dissection was made. The suspicion of myocardial bridging was rejected.
Unexpectedly, the echocardiography revealed multiple liver cysts. The following abdominal echography also showed multiple renal cysts. Based on the cysts in both kidneys combined with a family history of ADPKD, the diagnosis of ADPKD was made (Ravine’s criteria) [5].
Since cerebral aneurysms are one of the extrarenal manifestations of ADPKD [1], a computerized tomography angiography of the brain was performed, with a negative result for vascular anomalies. An exercise stress test, performed 16 days after the onset of chest pain, was normal. An elective CAG was performed at 6-month follow-up. The LAD was patent without a significant infarction, but still with a double lumen appearance. At 2-year follow-up by a nephrologist, his kidney function is still in the normal range. His blood pressure is also normal with use of perindopril and amlodipine.
Discussion
Our patient, a relatively young man with a negative cardiovascular profile and a history of subdural haematoma, developed a myocardial infarction secondary to dissection of the LAD. ADPKD was diagnosed at presentation. The CAD was not recognized during the first CAG because the relevant coronary artery can obturate the dissection in the acute phase. Myocardial bridging was the initial assumption. This condition is found when the segmental coronary artery has an intramyocardial course, being compressed during systole and restored during diastole [6]. Therefore, one would require a difference in coronary artery contraction in the systolic and diastolic phase which was not observed on the first CAG.
The association between a spontaneous CAD and ADPKD is poorly understood. Four cases were reported on the occurrence of a spontaneous CAD in middle-aged ADPKD patients [7]. Predisposing factors for CAD in ADPKD patients are still undetermined, especially in the absence of traditional cardiovascular risk factors. Furthermore, it is not known if arterial dissection is an extrarenal manifestation of ADPKD or if is just secondary to hypertension.
The estimated prevalence of spontaneous CAD is 0.7%, and it is in 2% of cases, the cause of acute coronary syndrome [8]. The LAD is affected in 80% of the patients with CAD [8, 9]. The majority of patients with a CAD often lack classical risk factors for cardiovascular disease and are female. De Maio et al. [9] identified three groups of patients with CAD: (i) patients with atherosclerotic cardiovascular disease, (ii) women in the postpartum period and (iii) an idiopathic group. Several underlying conditions in the idiopathic group are suggested, such as polyarteritis nodosa, lupus erythematosus, Marfan’s syndrome, Ehlers–Danlos syndrome, intense physical exercise, the use of cocaine, cyclosporin and oral contraceptives. None of these conditions were present in our patient.
Polycystins are also detected in smooth muscle cells of elastic arteries [10]. The tunica media, the middle layer of elastic arteries, include smooth muscles with interposing layers of elastic lamellae. The connection between the intracellular contractile filaments and extracellular elastic fibres is provided by dense plaque sites. Localization in the dense plaques assigns a significant function to polycystins in maintaining vascular integrity [11]. Furthermore, Qian et al. [12] suggested that abnormal intracellular calcium concentration in the vascular smooth muscle cells is linked to vascular phenotype in the case of inactivation of polycystin-2 protein. Literature also confirms the observation of intracranial aneurysms and myocardial infarction secondary to coronary aneurysms in certain families and the occurrence of vascular rupture and haemorrhage in homozygous polycystic kidney disease (PKD)-1 knockout mice [4, 11]. Considering these results, and the high plasma renin activity and impaired nitric oxide release in ADPKD patients [4], we speculate that the vascular abnormalities are most likely a direct result of PKD mutations rather than a secondary cause of hypertension.
In conclusion, polycystins seem to play a main role in the stability of the arterial vasculature. Therefore, a spontaneous CAD should be considered as an extrarenal manifestation of ADPKD. The clinician should be aware of CAD if ADPKD patients present with chest pain or discomfort. CAG can be inconclusive in respect to the mechanism of coronary occlusion in the acute phase and should be repeated, especially when the complaints persist.
Acknowledgments
Conflict of interest statement. None declared.
References
- 1.Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- 2.Patel A, Honoré E. Polycystins and renovascular mechanosensory transduction. Nat Rev Nephrol. 2010;6:530–538. doi: 10.1038/nrneph.2010.97. [DOI] [PubMed] [Google Scholar]
- 3.Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis. 2001;38:777–784. doi: 10.1053/ajkd.2001.27720. [DOI] [PubMed] [Google Scholar]
- 4.Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5:221–228. doi: 10.1038/nrneph.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravine D, Gibson RN, Walker RG, et al. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824–827. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- 6.Tio RA, Ebels T. Ventricular septal rupture caused by myocardial bridging. Ann Thorac Surg. 2001;72:1369–1370. doi: 10.1016/s0003-4975(01)02562-0. [DOI] [PubMed] [Google Scholar]
- 7.Lee CC, Fang CY, Huang CC, et al. Computed tomography angiographic demonstration of an unexpected left main coronary artery dissection in a patient with polycystic kidney disease. J Thorac Imaging. 2011;26:W4–W6. doi: 10.1097/RTI.0b013e3181dc2a53. [DOI] [PubMed] [Google Scholar]
- 8.Mortensen KH, Thuesen L, Kristensen IB, et al. Spontaneous coronary artery dissection: a Western Denmark Heart. Registry study. Catheter Cardiovasc Interv. 2009;74:710–717. doi: 10.1002/ccd.22115. [DOI] [PubMed] [Google Scholar]
- 9.DeMaio SJ, Kinsella SH, Silverman ME. Clinical course and long-term prognosis of spontaneous coronary artery dissection. Am J Cardiol. 1989;64:471–474. doi: 10.1016/0002-9149(89)90423-2. [DOI] [PubMed] [Google Scholar]
- 10.Griffin MD, Torres VE, Grande JP, et al. Vascular expression of polycystin. J Am Soc Nephrol. 1997;8:616–626. doi: 10.1681/ASN.V84616. [DOI] [PubMed] [Google Scholar]
- 11.Qian Q, Li M, Cai Y, et al. Analysis of the polycytins in aortic vascular smooth muscle cells. J Am Soc Nephrol. 2003;14:2280–2287. doi: 10.1097/01.asn.0000080185.38113.a3. [DOI] [PubMed] [Google Scholar]
- 12.Qian Q, Hunter LW, Li M, et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Gen. 2003;12:1875–1880. doi: 10.1093/hmg/ddg190. [DOI] [PubMed] [Google Scholar]