

Abstract
In the past decade, research has advanced our understanding how endothelin contributes to proteinuria and glomerulosclerosis. Data from pre-clinical and clinical studies now provide evidence that proteinuric diseases such as focal segmental glomerulosclerosis and diabetic nephropathy as well as hypertension nephropathy are sensitive to treatment with endothelin receptor antagonists (ERAs). Like blockade of the renin–angiotensin system, ERA treatment—under certain conditions—may even cause disease regression, effects that could be achieved on top of renin–angiotensin–aldosterone system blockade, suggesting independent therapeutic mechanisms by which ERAs convey nephroprotection. Beneficial effects of ERAs on podocyte function, which is essential to maintain the glomerular filtration barrier, have been identified as one of the key mechanisms by which inhibition of the endothelin ETA receptor ameliorates renal structure and function. In this article, we will review pre-clinical studies demonstrating a causal role for endothelin in proteinuric chronic kidney disease (with a particular focus on functional and structural integrity of podocytes in vitro and in vivo). We will also review the evidence suggesting a therapeutic benefit of ERA treatment on the functional integrity of podocytes in humans.
Keywords: diabetes, epithelial cell, focal segmental glomerulosclerosis, glomerular, hypertension
Endothelins are ubiquitously expressed stress-responsive regulators acting in both a paracrine and autocrine fashion and are in involved in the pathogenesis of chronic kidney disease (CKD).
Glomerulosclerosis as the main underlying cause of CKD is associated with activation of endothelin, and its detrimental effects on renal structure and function are predominantly mediated by the endothelin ETA receptor.
Podocytes are gatekeepers of the glomerular filtration barrier and express a fully functional endothelin system. CKD leads to podocyte effacement and disruption of the podocyte actin cytoskeleton. Blockade of endothelin ETA receptors can prevent and reverse podocyte injury and actin cytoskeleton disruption.
Endothelin receptor antagonists interfere with glomerulosclerosis and proteinuria and may even cause reversal of renal disease under certain conditions by mechanisms that are only in part pressure-dependent.
Endothelin receptor antagonists are a new class of drugs undergoing clinical testing for the treatment of proteinuric renal disease. First clinical studies have shown reversal of proteinuria on top of standard RAAS inhibitor therapy in patients with diabetic nephropathy and those with non-diabetic CKD that were largely independent of systemic arterial blood pressure.
Introduction
The prevalence and incidence of chronic kidney disease (CKD), one of the most serious clinical consequences of patients with diabetes or hypertension, have been steadily rising over the past decades [1]. CKD is characterized by progressive loss of functional glomerular tissue, defects in the glomerular filter function, and subsequent proteinuria [2]. CKD also aggravates pre-existing cardiovascular risk factors such as hypertension and dyslipidaemia, and as a result, accelerates atherosclerosis progression; in fact most CKD patients die from cardiovascular complications [3]. In view of an ageing population and the predicted increase of the world's population from currently 7 to 9 billion people by the year 2050 [4], health care providers and political decision makers need to emphasize the importance of primary and secondary prevention of CKD even more [4].
In the past decade, inhibitors of the renin–angiotensin–aldosterone system (RAAS) such as angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) have been firmly established as anti-proteinuric therapeutics [5]. Because inhibitors of the renin–angiotensin system only partially interfere with progression of CKD of diabetic or hypertensive aetiology [5], further preventive measures and additional therapeutic approaches are urgently needed [6]—particularly in view of the ongoing obesity/diabetes pandemic. Inhibiting the cellular activity of endothelin-1 in renal disease through endothelin receptor antagonists (ERAs) may be a new and promising approach [7]. In several recent articles [8–14], on which the present article is based, we have addressed molecular and physiological mechanisms by which endothelin contributes to proteinuria and podocyte injury and reviewed the clinical trials on ERAs in patients with CKD ongoing at the time [8, 9, 12].
Podocyte dysfunction as a determinant of CKD
Podocyte injury and chronic proteinuric nephropathies
Podocyte loss and podocyturia have been proposed as potential diagnostic markers of glomerular disease severity [15, 16]. Indeed, as disease progresses, viable—in addition to senescent—podocytes are shed in the urine [17] and thus provide a sensitive indicator of the degree of glomerular injury [16]. CKD is characterized by mesangial cell injury as well as by defects of the glomerular filtration barrier [15]. This includes injury of the glomerular endothelial cells, the glomerular basement membrane (GBM), glomerular epithelial cells, and the slit diaphragm (Figure 1) [11, 15].
Fig. 1.
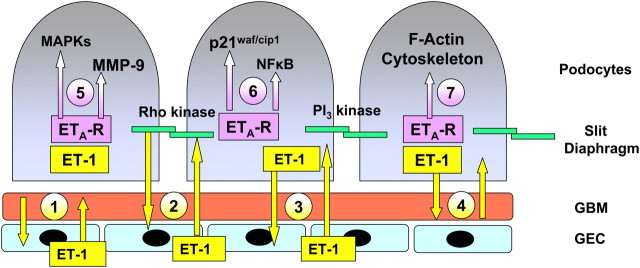
Proposed interactions of endothelin (ET-1) with the glomerular basement membrane (GBM) of the glomerular capillary, its endothelial cells (GEC) and podocytes/slit diaphragm. ET-1 is produced by cells on both sides of the GBM, namely GEC and podocytes. ET-1 is released from GEC and may interact either directly with GBM (1) the slit diaphragm (2) or the podocyte, also in the reverse direction (3). Podocyte-derived ET-1 (4) may also affect the GBM and vice versa. ET-1 activates podocyte endothelin ETA receptors (ETA R) activating mitogen-activated protein kinases (MAPKs) p38 and p44/p42 (5), growth promoters (p21waf/cip1), or inflammation (NF-kappaB) (6). ET-1 also causes disruption of the podocyte F-actin cytoskeleton (7) and slit diaphragm dysfunction via activation of rho kinase and PI3-kinase. Figure reproduced from reference [11] with permission of the publisher.
Glomerular epithelial cells can be divided into parietal and visceral (glomerular) epithelial cells [15]. Visceral epithelial cells have been termed ‘podocytes’ due the appearance of ‘interdigiting’ foot processes adhering to the glomerular capillary [13]. Podocyte injury is a hallmark of proteinuric renal disease such as diabetic nephropathy and focal segmental glomerulosclerosis (FSGS) [18, 19]. Although the term ‘podocytopathy’ has recently been used to describe proteinuria in association with podocyte injury [20], proteinuria in some cases may also occur in the absence of podocyte effacement [13, 21]. Since podocyte injury precedes the development of glomerulosclerosis, it not only represents a therapeutic target but can also be used for diagnostic purposes [22–26].
Previous dogma stated that because podocytes—once injured—cannot be replaced, glomerulosclerosis is progressive and irreversible [27, 28]. However, podocyte proliferation and migration have been observed under certain conditions, including inflammatory activation during renal injury [29, 30]; in the past decade numerous studies have clearly shown that glomerulosclerosis is a reversible disease condition [10, 31–40]. Podocytes have a defined lifetime since they are terminally differentiated, however, they may be continuously replaced by parietal epithelial cells (PECs) residing in the inner portion of Bowman's capsule, which can migrate onto the glomerulus [41, 42]. Thus far, whether migration of PECs is beneficial or detrimental for glomerular structure and function remains a matter of debate. These novel and unexpected findings have provided one explanation for many studies reporting reversal/regression of established glomerular disease using different pharmacological approaches [10, 31–40].
Podocyte function and the actin cytoskeleton
The podocyte actin cytoskeleton is being increasingly recognized as a key modulator of the functional integrity of the glomerulus [43]. Disruption of the actin cytoskeleton is a feature of podocyte injury in proteinuric nephropathies. The actin cytoskeleton of podocytes not only provides structural support for the cells but also contributes to podocyte signalling [43, 44]. Reorganization of the podocyte actin cytoskeleton appears to be a response to cell stress [45], and loss of podocytes results in uncoupling of podocyte-specific proteins in the remaining cells from the actin cytoskeleton [46, 47].
Podocytes are linked to the GBM through their foot processes and form the structural basis of the glomerular filtration barrier responsible for filtering proteins (Figure 1). Podocyte injury results in a reduction in podocyte number, which is characteristic of many forms of proteinuric renal disease [21, 48, 49] and subsequent increased accumulation of GBM matrix, which is derived from podocytes [30]. Accordingly, in cases of regression of proteinuria induced by pharmacological intervention, a reduction in the degree of podocyte injury and of GBM thickening has been observed [39].
In addition to podocyte injury, proliferation of glomerular mesangial cells and hypertrophy of the GBMs contribute to the progression of proteinuric nephropathies [10]. Accordingly, mutations of the actinin-4 (actn4) gene, which encodes the podocyte-localized actin-binding protein α-actinin, have been linked to autosomal-dominant proteinuria associated with familial FSGS [50, 51]. Similarly, variants of the apolipoprotein L-1 (apol1) gene have been identified as a risk factor for FSGS and hypertension nephropathy in blacks; it is present in African chromosomes and absent in European chromosomes [52]. Black subjects are at higher risk to develop hypertension, diabetes and renal and vascular disease and hypertensive blacks show higher circulating endothelin-1 (ET-1) levels than Caucasians [53].
Integrin-mediated adhesion of podocytes to the basement membrane of the glomerular capillary has recently been reported to require the tretraspanin cd151, which protects from podocyte effacement and renal disease development in a podocyte-dependent fashion [27, 54]. It is well possible, if not likely, that anti-proteinuric effects of drugs such as ARBs, ACEIs, or ERAs involving drug-induced glomerular stabilization of preventing podocyte loss in end-stage kidney disease [55] involve mechanisms requiring integrins and/or cd151 [56]. Research is ongoing to identify the mechanisms involved in podocyte injury, many of which converge at the level of the actin cytoskeleton [43]. One mechanism that was recently identified is podocyte-induced secondary injury of ‘remnant’ intact podocytes [57], which leads to podocyte depletion in CKD.
Role for endothelin in the development of CKD
Endothelin-1: vasoconstrictor and promoter of inflammation and growth
Endothelins are ubiquitously expressed stress-responsive regulators acting in both a paracrine and autocrine fashion [58]. Within a year after Furchgott and Zawadzki's had discovered an endothelium-derived vasorelaxing factor (later identified as nitric oxide) [59–61], endothelium-derived vasoconstrictor activity was reported by de Mey and Vanhoutte [59, 60]. A potent peptidergic vasoconstrictor activity isolated from endothelial cell supernatants was reported in 1985 [62], and the gene and peptide sequence of this vasoconstrictor, named endothelin-1, were published by Yanagisawa et al. in 1988 [63]. Endothelin-1 is the biologically most relevant isoform of three endothelin isopeptides, which bind to endothelin receptors (designated ETA and ETB) [59], that were cloned in the early 1990s [58]. ETA receptors have primarily vasoconstrictor and growth-promoting functions, whereas ETB receptors mainly mediate vasodilation and inhibition of growth and inflammation, via release of nitric oxide and prostacyclin [58]. Identification of these receptors allowed the development of orally active ERAs, which are now firmly established in pulmonary medicine [9] and currently in clinical trials for CKD [9, 12].
Numerous endothelin-dependent mechanisms contribute to proteinuria and CKD [9, 64]. Endothelin promotes collagen production and stimulates glomerular fibronectin synthesis. Endothelin becomes activated under conditions associated with renal disease progression, such as diabetes, insulin resistance, obesity, dyslipidaemia, reactive oxygen species formation and inflammation [10]. In fact, inflammation may be a unifying detrimental mechanism by which endothelin causes kidney injury. Indeed, inflammation is crucial for glomerulosclerosis progression and can be attenuated by ETA receptor antagonist treatment, which reduces circulating cytokines in a model of acute allograft rejection after solid organ transplantation, even in the absence of immunosuppression [65]; ERA treatment also limits inflammation in experimental proliferative nephritis [66]. Consistent with these effects, chronic infusion of endothelin at non-pressor doses increases pro-inflammatory mediators such as intercellular adhesion molecule-1 (ICAM-1) and monocyte chemotactic protein-1 (MCP-1) and the number of macrophages in the renal cortex, effects that are largely abrogated by pre-treatment with an ETA receptor antagonist [67], and similar effects were obtained in a model of diabetes-associated renal inflammation [68, 69]. Interestingly, only selective ETA, but not non-selective ET, receptor antagonists inhibited the renal inflammatory response [70].
Endothelin also increases formation of other vasoactive and growth factors such as angiotensin II by increasing the activity of ACE [71]. On the other hand, angiotensin II activates renal endothelin formation [72], compatible with a vicious cycle between the renin–angiotensin–aldosterone and the endothelin systems [73]. Mesangial cell proliferation and GBM hypertrophy (Figure 1) are indirectly mediated via podocyte injury [44] and represent an important indicator of glomerular stability [18]. Wiggins et al. recently reported that combined ARB/ACEI treatment reduces podocyte loss and thereby contributes to glomerular stablization in experimental end-stage renal disease [55].
Endothelins and endothelin receptors in the kidney
In the normal kidney, endothelin regulates blood pressure, vascular tone and natriuresis, the latter of which is mediated via the ETB receptor [74], and is influenced by sex [75]. In the systemic and renal vasculature, endothelin exerts basal (‘tonic’) ETA receptor-mediated vasoconstriction [74]. Under physiological conditions, endogenous renal endothelin controls water and sodium excretion and acid–base balance and maintains normal renal cell proliferation and tonic vasoconstriction [74]. Endothelin also stimulates proliferation of vascular smooth muscle cells, a cellular function facilitating the development of hypertension and renal disease [58]. Endothelial cell-derived endothelin controls blood pressure: mice with endothelial cell-specific deficiency of endothelin-1 are hypotensive [76], while those with endothelial cell-specific overexpression are hypertensive [77, 78].
With ageing, renal expression of endothelins increases at the messenger RNA (mRNA) and peptide level [79]. Radiolabelled ET-1 binds within 10 min after injection onto glomerular podocytes [80], cells that also produce and secrete endothelin [81, 82]. Endothelin binding to podocytes results in changes in intracellular calcium [83], and ETB-specific binding on podocyte foot processes [84] and expression of mRNA for ETA and ETB receptors, preproendothelin-1, and endothelin-converting enzymes in podocytes have been reported [10]. The exact role of the podocyte endothelin system in podocytes warrants further investigation.
Endothelin contributes to glomerulosclerosis progression
For almost 20 years, different experimental models of renal disease (renal mass reduction, hypertension, salt-sensitive hypertension, RAAS-dependent hypertension, nitric oxide (NO)-deficient hypertension, ageing, FSGS) have been studied to test whether endothelin inhibition can interfere with renal disease progression [85]. In 1993, Benigni et al. [86] published the first pre-clinical study to suggest a causal role for endothelin in chronic proteinuric renal disease. In a prevention-type study, remarkable reductions in proteinuria and glomerulosclerosis were observed after selective blockade of ETA receptors in a rat renal mass reduction model of hypertensive glomerular disease [86]. Hocher et al. [87] subsequently demonstrated glomerulosclerosis in normotensive mice systemically overexpressing the human preproendothelin gene. Data from pre-clinical studies indicate that endothelin—via activation of the ETA receptor—importantly contributes to renal disease progression under hypertensive [24, 34, 88, 89] as well as under normotensive conditions [39, 90].
Involvement of endothelin in podocyte dysfunction and injury
Podocyte growth factor receptors and glomerular disease
Injury to podocytes results in glomerular filtration barrier dysfunction and subsequent proteinuria, a hallmark of all glomerular diseases [15]. Podocytes express receptors for vasconstrictors and growth factors such as angiotensin II, thromboxane A2, endothelin-1 and prostaglandins [13]. Angiotensin II increases renal endothelin synthesis [10] and glomerular permeability for albumin, and causes podocyte actin cytoskeleton disruption and podocyte apoptosis [10]. A role for growth factor receptors is also suggested by the observation that either ETA receptor [39] or angiotensin AT1 receptor antagonists [49] prevent podocyte injury.
The activity of endothelin or angiotensin may also be inhibited indirectly. Nephroprotective peptides such as bone morphogenetic protein-7 (BMP-7) [91, 92] reduce expression of the growth-promoting ETA receptor, both at the RNA and the protein level, alleviate hyperglycaemia-mediated podocyte injury and improve podocyte survival (reviewed in [10]). BMP-7 also stimulates glomerular capillary formation that has been implicated in ARB- or ACEI-induced regression of glomerulosclerosis [37, 93]. Similar to BMP-7, nephroprotective effects inhibition of epidermal growth factor (EGF) receptor transactivation in podocytes [94] (EGF stimulates endothelin-mediated cell growth and constriction [95, 96]) might also reduce renal injury—at least in part—via interfering with the endothelin-1-ETA receptor axis.
Podocyte injury involves endothelin signalling
We have previously proposed that the neighbouring cells in close proximity of podocytes—which also produce endothelin in vitro [81, 82]—affect podocyte function and structure through endothelin-mediated interactions [10, 13] (Figure 1). Disruption of the podocyte actin cytoskeleton has been observed following in vitro exposure to endothelin [39, 97, 98]. Interestingly, exposure of podocytes to protein in vitro induces synthesis of endothelin that results in increases in glomerular permselectivity, an effect antagonized by ETA receptor blockade [97, 98]. These findings are reinforced by recent work from Pollock et al. demonstrating that exogenous endothelin—both acutely as well as chronically—increases glomerular permeability via ETA receptor-mediated mechanisms [67]. This effect appears to be independent of whether the ETB receptor is blocked or not [70]. Thus, ETA receptor-dependent effects mediate proteinuria.
Similarly, ETA receptor antagonists prevent disruption of the podocyte actin cytoskeleton following exposure to puromycin aminonucleoside [39] (Figures 2 and 3), an effect that can also be obtained with angiotensin AT1 receptor antagonists [46, 99, 100]. Puromycin aminonucleoside (which causes FSGS in vivo) [101, 102] results in podocyte apoptosis and up-regulation of matrix metalloproteinase-9 (MMP-9) as a marker of podocyte injury [103]; up-regulation of MMP-9 and disruption of the actin cytoskeleton can be completely prevented by ETA receptor (but not ETB) antagonists or ETA receptor gene silencing (Figure 2 and 3) [39]. These observations were confirmed by Benigni's group [39, 97] showing similar cytoskeleton-protective effects of ETA receptor blockade [98]. Little is known about the mechanisms that would explain how endothelin might regulate podocyte proliferation or motility [44]. We found that ETA receptor gene silencing stimulates podocyte growth (Figure 2E), a finding compatible with the notion that endogenous endothelin produced by the podocyte itself [81, 82] acts as a negative regulator of podocyte cell proliferation.
Fig. 2.

Effect of ETA receptor inhibition in laser-dissected glomeruli of rats with FSGS in vivo (a) and in podocytes in vitro (b-f). (a) Darusentan reduced MMP-9 gene expression (a marker of podocyte injury [103]) compared with placebo-treated rats. (b) In vitro treatment with puromycin aminonucleoside (PAN) caused podocyte apoptosis and up-regulation of MMP-9; (c) peptide (BQ) or non-peptide (LU, darusentan) ETA receptor antagonists prevented this effect, as did ETA receptor gene silencing (siRNA) (d). (e) Gene silencing of ETA receptors increased podocyte growth as determined by de novo DNA synthesis. (f) Actin cytoskeleton disruption was largely prevented by ETA antagonists; representative examples of these experiments are shown in Figure 3. ncRNA, non-coding RNA control; O, old; AU, arbitrary units; OLU, old, darusentan. *P < 0.05 versus control (CTL); †P < 0.05 versus PAN alone/old. Figure reproduced from reference [39] with permission of the publisher.
Fig. 3.
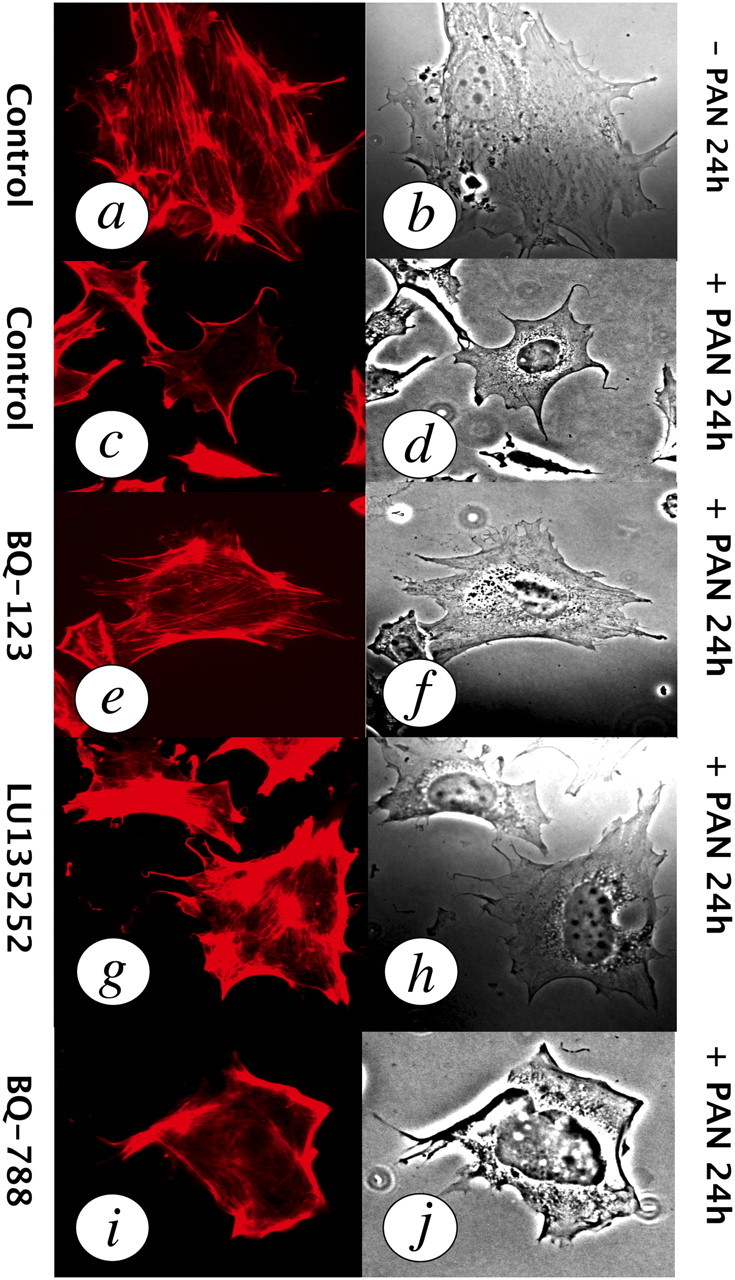
Endothelin mediates podocyte injury in vitro. (a, b) Actin-phalloidin immunofluorescence (left) and phase-contrast images of actin cytoskeleton fibres and cell morphology (right panels) in a normal podocyte. (c, d) Puromycin aminonucleoside (PAN)-induced foot process effacement, cytoskeleton disruption (disappearance of actin fibres) and cell body shrinkage. (e, f) Prevention of PAN-induced injury by the ETA antagonist BQ-123. (g, h) Prevention of PAN-induced injury by the ETA antagonist darusentan/ LU135252. (i, j) The ETB receptor antagonist BQ-788 had no effect on PAN-induced injury. Figure reproduced from reference [39] and reproduced with permission of the publisher.
CKDs involving podocyte injury and endothelin
Assessing podocyte injury in patients is difficult and so far can be done only indirectly through measurements of proteinuria or albuminuria. Since most of the recent renal trials with ERAs have been extensively discussed in our previous articles, we will only briefly address clinical outcomes.
Hypertension nephropathy
Hypertensive renal disease was one of the first targets in pre-clinical studies to explore the therapeutic potential of ERAs. Renal preproET-1 mRNA expression and urinary ET-1 excretion increase in hypertension due to renal mass reduction, a condition associated with proteinuria and impairment of renal function (reviewed in [13]). Using the same experimental model, Benigni et al. [86] demonstrated that selective ETA receptors antagonists reduce the degree of glomerulosclerosis and proteinuria, albeit with treatment also having anti-hypertensive effects. Clozel et al. [104], using combined blockade of ETA and ETB receptors, found only protective effects on renal function, but no effect on proteinuria, suggesting possible therapeutic advantages of selective ETA selective antagonists. Other prevention-type studies found nephroprotective effects in a variety of forms of hypertension (angiotensin-II-dependent, renin-dependent, salt-loaded renin-dependent, aldosterone-induced, genetically salt-sensitive and deoxycorticosterone–salt-induced) as well as chronic NO deficiency, with effects usually being associated with a partial reduction in blood pressure [105].
The first study to demonstrate reversal of indices of hypertensive nephropathy using ERAs was performed by Boffa et al. [34] in a rat model of NO-deficient hypertension. The same group also demonstrated that established glomerulosclerosis caused by chronic hypertension due to NO deficiency was also reversed by non-selective ERA in a manner that was independent of blood pressure [40]. In this model, increased ET-1 synthesis in the kidney was found primarily in pre-glomerular arterioles and a downstream sclerotic action on glomerular cells was unraveled with non-selective ERAs [106]. Vaneckova et al. have provided evidence that selective ETA receptor blockade maintains its profound anti-proteinuric effects even when initiated in the presence of established malignant hypertension and that an attenuation of podocyte injury (which precedes proteinuria) can be observed even in the absence of glomerulosclerosis, i.e. at early stages of renal disease [24, 89]. In contrast, non-selective ERA treatment did not prevent podocyte injury nor did it reduce proteinuria. Importantly, the protective effects were observed in the presence of sustained hypertension [24, 89].
Podocyte injury before the development of glomerulosclerosis has also been observed in aldosterone-infused rats [26] and in hypertensive Dahl salt-sensitive hypertensive rats [23, 107]. Podocyte protection in Dahl rats was achieved by either inhibition of the mineralocorticoid receptor [23] or the endothelin ETA receptor [107]. In contrast, simple anti-hypertensive treatment of these animals with hydralazine had no effect on podocyte injury [23], indicating that the nephroprotective effects of aldosterone blockade and endothelin blockade are specific for these vasoactive and growth-promoting factors and in part blood pressure independent. The concept of a blood pressure-independent mode of action of ERAs is further supported by the glomerulosclerosis despite normotension in endothelin-overexpressing mice [87] and blood pressure-independent nephroprotective effects of ERAs in diabetes [58]. A reduction in proteinuria has also been observed in patients with resistant hypertension on triple anti-hypertensive therapy additionally treated with the ETA antagonist darusentan [108].
Focal segmental glomerulosclerosis
FSGS is a widely varying, clinicopathological entity characterized by injury of the glomerular filtration barrier [19]. Urinary excretion of ET-1 increases in primary FSGS patients and glomerular ET-1 expression is enhanced in experimental FSGS (reviewed in [13]). Podocyte-specific mechanisms have been proposed as mechanisms underlying FSGS development [22, 109].
Ageing in rodents and humans is associated with spontaneous development of FSGS [22], which is characterized by podocyte injury and hypertrophy, glomerular enlargement and glomerulosclerosis [110, 111]. Pharmacologically induced FSGS results in podocyte injury leading to cytoplasmic accumulation of functional synaptic-like vesicles, which occurs even before detachment of the cells from the GBM [112]. These vesicles or vacuoles are also present in spontaneous age-dependent FSGS [39] (Figure 4, untreated animals, left panels). Interestingly, susceptibility to develop age-dependent FSGS has recently been linked to autophagy-related mechanisms controlling podocyte vacuolation [113].
Fig. 4.
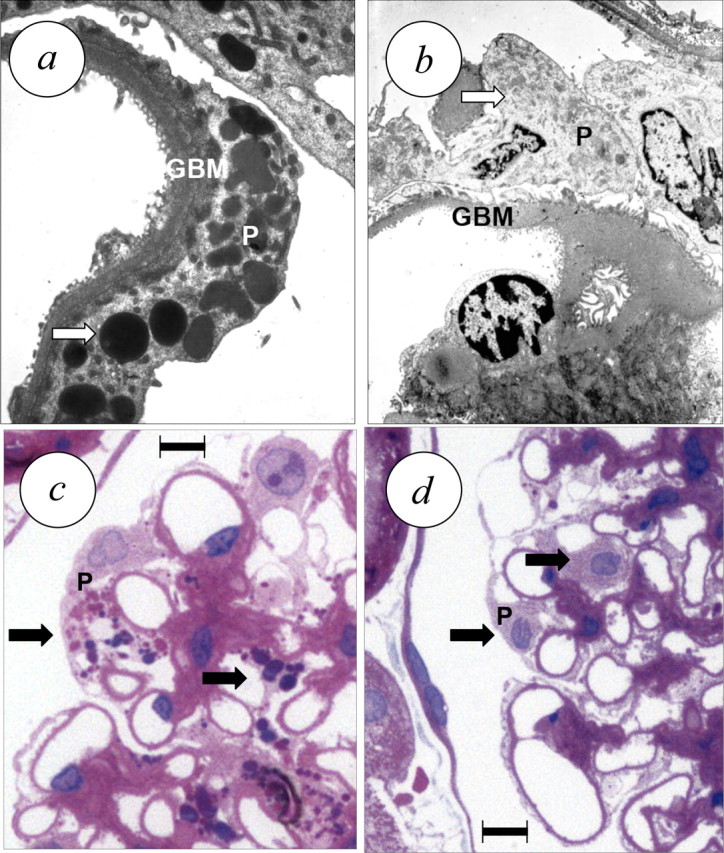
Effect of 4 weeks of treatment with the ERA darusentan on renal structure in established FSGS [39]. (a) Untreated animal, transmission electron microscopy demonstrates GBM hypertrophy and podocyte injury with diffuse foot process effacement and vacuolar degeneration involving autophagy [113]. (b) ERA-treated animal, showing regression of GBM hypertrophy and disappearance of podocyte vacuoles. (c) Untreated animal, light microscopy image (haematoxilin/eosin) demonstrating hypertrophy of podocytes with enlarged nuclei, prominent nucleoles and vacuolar degeneration due to autophagy. (d) Treated animal, showing normal sized podocyte nuclei and virtually complete disappearance of vacuolar degeneration (arrows). Scale bar, 10 μm (c, d). Panels adapted from reference [39] and reproduced with permission of the publisher.
Pre-clinical studies in aged rats suggest that treatment with ETA receptor antagonists can in part restore podocyte morphology (Figure 4, treated animals, right panels), reverse FSGS and proteinuria independently of renal or systemic haemodynamics or renal function [39]. Disease regression was associated with reduced glomerular expression of MMP-9, a podocyte injury marker [39] and of cortical p21waf/cip1 [39], a cdk-inhibitor and promoter of glomerulosclerosis [114]. Most notably, the regression of FSGS achieved by ERA treatment was associated with normalization of the thickness of the hypertrophied GBM and almost complete disappearance of podocyte vacuolization [39] (Figure 4).
We have recently proposed mechanisms that could contribute to the partial restoration of podocyte and glomerular structure and protein loss after ERA treatment [8, 10]. Though terminally differentiated, podocytes may be replaced from PECs [41, 115, 116]. This concept, initially put forward by Fogo et al. using podocyte-specific Lac-Z expression and demonstrating that PECs replace glomerular epithelial cells [42], was independently confirmed by Appel et al. [41]. However, they are still lacking evidence that former PECs migrating to the glomerular capillaries can become functional podocytes. In fact, studies from Bart Smeets and Marcus Moeller suggest that PECs' invasion into the glomerular capillary tuft promotes scarring and FSGS. After these recent studies, we can wonder whether manipulation of the endothelin system would affect migration of PECs and their subsequent fate towards a desirable podocyte phenotype or a scarring phenotype. In addition, improved glomerular capillary architecture may result from ERA therapy and thereby accelerate disease regression [69].
Clinical data regarding effects of ERAs in FSGS patients are scarce. In patients with non-diabetic renal disease on standard anti-proteinuric RAAS inhibitor therapy, ETA antagonism reduces proteinuria [117] independent of changes in glomerular filtration rate (GFR). The same authors reported that the effect depends on the degree of RAAS inhibition in this particular patient population with renal disease of different aetiologies [118], which complicates reaching a conclusion regarding the mechanisms involved.
Diabetic Nephropathy
Diabetes remains the main cause of CKD and is likely to increase even further in prevalence due to the ongoing obesity pandemic [119]. Hyperglycaemia—like endothelin [58]—results in disassembly of the podocyte actin cytoskeleton [120] as well as podocyte depletion and apoptosis involving reactive oxygen species (ROS)-dependent mechanisms [121]. In animals with diabetic nephropathy, even viable podocytes are shed in the urine [17, 122]. Nephrin, a podocyte-specific protein contributing to functional stability of the slit diaphragm (Figure 1), is involved in podocyte shedding as recombinant endothelin triggers nephrin loss from human podocytes, which is prevented by ETA receptor antagonists [123]. A large number of experimental prevention studies demonstrated beneficial effects of ERA therapy on diabetic nephropathy and proteinuria [90, 124]. Whether these effects are pressure dependent is not clear: in some studies, partial or even complete normalization of blood pressure was observed [125, 126], while in other studies, little or no effect on blood pressure or GFR was observed, suggesting possible non-haemodynamic but rather structural effects. Indeed, Gross et al. [126] reported that darusentan was as effective as ACE inhibition in reducing glomerulosclerosis in insulin-treated streptozotocin-diabetic rats, but that ACE inhibition produced greater reductions in blood pressure, proteinuria, and glomerular volume. However, only ETA receptor blockade completely restored the number of glomeruli per kidney to the normal expected number and increased the number of podocytes per glomerulus, suggesting that endothelin blockade has specific glomeruloprotective effects at the level of the pocoyte [126]. Indeed, Gagliardini et al. [69] found that ERA treatment in part prevents glomerular podocyte loss in experimental diabetes and reduced podocyte injury despite having only negligible effects on arterial blood pressure [127]. Thus, anti-proteinuric effects are likely to be due to structural or anti-inflammatory [124] properties of ERAs. A pressure-independent mode of action would also be compatible with findings from recent clinical trials in patients with diabetic nephropathy in which impressive reversal of proteinuria was observed [74, 119, 128].
Obesity nephropathy
Obesity has become a major health issue in all parts of the world and is increasingly affecting children and juveniles [129, 130]. Other diseases such as arterial hypertension and diabetes, which are independent risk factors for CKD [131, 132] are aggravated by obesity. Secondary FSGS has been linked to obesity nephropathy [133, 134]. Accordingly, podocyte loss has been described in patients with obesity nephropathy [135] as well as in obese rodents [121]. In patients with obesity nephropathy, albuminuria is inversely correlated with plasma levels of the adipocytokine adiponectin [136]. On the other hand, albuminuria in adiponectin-deficient mice can be normalized by administering recombinant adiponectin [137]. Endothelin, which promotes proteinuria [67] and increases locally in the kidney [73, 138], inhibits adiponectin production [139], suggesting a possible mechanistic link. Obesity-associated diabetes often is associated with high circulating levels of insulin, a potent stimulus of endothelin production [58]. Furthermore, endothelin-1 has insulin-antagonizing properties [140]; thus, high local tissue or circulating levels of endothelin-1 could promote the development of insulin resistance thus promoting hyperglycaemia and subsequent podocyte injury. Another common concern of obesity-associated FSGS as well as with diabetic nephropathy is the initial chronic hyperfiltration leading to increased glomerular capillary pressure and likely podocyte stretch. ERA therapy may alleviate such local change in the glomerular haemodynamics. Finally, renal ACE becomes activated in obesity, an effect that can be completely prevented by ETA receptor antagonists [73]. This suggests that ERAs function as ACEIs under certain pathological conditions and reinforcing the use of ACEI in patients with obesity nephropathy. Importantly, in most of the clinical ERA studies in patients with diabetic nephropathy showing beneficial effects on proteinuria, patients were either overweight or obese [74, 119, 128]. Thus, future studies in renal patients should also assess and compare effects of ERA therapy in individuals with normal and pathological body-mass indices. It should also be noted that in experimental studies, calorie restriction can limit or even largely prevent FSGS-associated podocyte injury [110, 111] suggesting that calorie restriction and/or weight loss are likely to be of great importance for the primary and possibly secondary prevention of CKD in obese patients.
Immune-mediated and minimal-change nephropathies
The mitogenic and co-mitogenic effects of ET-1 on mesangial cells were reported early [10]. In the model of glomerulonephritis with mesangial proliferation induced by the injection of anti-thymocyte antibodies, an increase is observed in the glomerular expression of ET-1 and ETB receptors [141]. ETA antagonists also prevent proteinuria and histological lesions due to mesangial proliferation following the repeated systemic injection of ovalbumin in rats [66]. Work by Benigni's group suggests that albumin causes podocyte cytoskeleton disruption and up-regulation of podocyte ET-1 expression [97].
Glomerulonephritis due to streptococcal infection is characterized by intense endocapillary mesangial proliferation [13]. In children with streptococcal infection, investigators found positive correlations between plasma ET-1 concentration and arterial blood pressure, consistent with a role for endothelial activation and endothelin participating in the disease process of acute post-streptococcal glomerulonephritis [142]. It is likely that plasma concentrations only in part reflect local tissue concentrations also in this disease, given the paracrine actions of this peptide [76]. Increased mesangial expression of ET-1 in humans with lupus-associated nephropathy with moderate mesangial proliferation and after kidney transplantation with cyclosporine treatment has been reported (reviewed in [13]). Remarkably, even in the absence of immunosuppressant, ETA receptor blockade can alleviate much of the transplant-associated mesangial proliferation and glomerulosclerosis [143].
Minimal-change disease represents a podocyte disease without an inflammatory infiltrate [13, 19]. Semi-quantitative morphological and immunohistochemical studies assessed renal ET-1 expression patients with minimal-change disease in the absence or presence of acute renal failure [144]. Renal failure patients showed higher ET-1 levels in glomerular and tubular endothelial cells. Thus, local increase in renal ET-1 production, which has been found in experimental studies of acute renal failure [145], appears to be involved in acute renal failure also in this setting.
Sickle cell nephropathy
FSGS is becoming epidemic in subjects affected by sickle cell disease (SCD) as life expectancy increases. In a case-control study comparing patients with chronic renal insufficiency and healthy subjects, urinary ET-1 excretion levels were significantly higher in the patients than the controls [146]. High urinary endothelin concentrations have also been reported in subjects with SCD and microalbuminuria with glomerular hyperfiltration. In these subjects, the elevated urinary ET-1 excretion, which was four times higher than that in carefully matched healthy controls, was correlated with microalbuminuria, suggesting a pathological link between the loss of glomerular permeability/selectivity and the renal synthesis of ET-1 in this context preceding progression to FSGS [147]. No studies have yet addressed possible interactions between the ET system and polymorphisms of the apoL1 gene, which is associated with an increased risk of FSGS in carriers of African chromosomes [52]. Notably, SCD condition with initial hyperfiltration and glomerulomegaly may be related in part to the pathophysiology of incipient diabetic nephropathy and obesity-related FSGS as discussed above. ETA or combined ETA/ETB blockade displayed contrasted effectiveness in blunting albuminuria and injury in experimental diabetic models with initial hyperfiltration [124, 148]. The increase in renal (mostly endothelial) ET-1 synthesis has been demonstrated in a mouse model of SCD [149] characterized by glomerulomegaly and glomerular hyperfiltration complicated by FSGS may be due to mechanically transduced stimulation. To date, clinical studies testing the potential anti-proteinuric and nephroprotective efficacy of ERAs in SCD patients have not been performed.
Conclusion and perspective
Endothelin is essential in the physiological regulation of renal blood flow and cell growth. There is solid pre-clinical and beginning clinical evidence that ERAs bear the potential as anti-proteinuric drugs and which also have beneficial effects at the level of the podocyte and the glomerular capillary in diseases such as diabetic nephropathy, hypertension nephropathy, FSGS and sickle cell nephropathy [150]. One of the current unsolved limitations of ERA therapy in patients is oedema formation. This appears to involve ETA and ETB receptor-dependent mechanisms [150, 151], which, however, can be circumvented by carefully adjusting diuretic therapy [12, 119]. Provided that clinical drug development continues at a careful level, the addition of ERAs to the therapeutic armamentarium of nephrologists might ultimately allow the reversal—at least in part—proteinuria and possible glomerulosclerosis.
Acknowledgments
This work is support by Swiss National Science Foundation grants 108 258 and 122 504 to M.B., and by a grant from INSERM and grant ANR-06-PHYSIO, l'Agence Nationale de la Recherche, France, both to P.-L.T.
Conflict of interest statement. None declared.
References
- 1.Bakris GL, Ritz E. The message for World Kidney Day 2009: hypertension and kidney disease–a marriage that should be prevented. J Hum Hypertens. 2009;23:222–225. doi: 10.1038/jhh.2008.169. [DOI] [PubMed] [Google Scholar]
- 2.Reiser J, Gupta V, Kistler AD. Toward the development of podocyte-specific drugs. Kidney Int. 2010;77:662–668. doi: 10.1038/ki.2009.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiner DE, Tighiouart H, Amin MG, et al. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: a pooled analysis of community-based studies. J Am Soc Nephrol. 2004;15:1307–1315. doi: 10.1097/01.asn.0000123691.46138.e2. [DOI] [PubMed] [Google Scholar]
- 4.Barton M. Ageing as a determinant of renal and vascular disease: role of endothelial factors. Nephrol Dial Transplant. 2005;20:485–490. doi: 10.1093/ndt/gfh689. [DOI] [PubMed] [Google Scholar]
- 5.Reiser J, Mundel P. Dual effects of RAS blockade on blood pressure and podocyte function. Curr Hypertens Rep. 2007;9:403–408. doi: 10.1007/s11906-007-0074-7. [DOI] [PubMed] [Google Scholar]
- 6.Turner JM, Bauer C, Abramowitz MK, et al. Treatment of chronic kidney disease. Kidney Int. 2011 doi: 10.1038/ki.2011.380. doi: 10.1038/ki.2011. [DOI] [PubMed] [Google Scholar]
- 7.Kohan DE, Pritchett Y, Molitch M, et al. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy. J Am Soc Nephrol. 2011;22:763–772. doi: 10.1681/ASN.2010080869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barton M. Endothelin antagonism and reversal of proteinuric renal disease in humans. Contrib Nephrol. 2011;172:210–222. doi: 10.1159/000328702. [DOI] [PubMed] [Google Scholar]
- 9.Barton M. Endothelin receptor antagonists in cardiovascular medicine: challenges and opportunities. In: Abraham D, Handler C, Dashwood MR, et al., editors. Translational Vascular Medicine. London: Springer; 2012. pp. 231–259. [Google Scholar]
- 10.Barton M. Reversal of proteinuric renal disease and the emerging role of endothelin. Nature Clin Pract Nephrol. 2008;4:490–501. doi: 10.1038/ncpneph0891. [DOI] [PubMed] [Google Scholar]
- 11.Barton M. Therapeutic potential of endothelin receptor antagonists for chronic proteinuric renal disease in humans. Biochim Biophys Acta. 2010;1802:1203–1213. doi: 10.1016/j.bbadis.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Barton M, Kohan DE. Endothelin antagonists in clinical trials: lessons learned. Contrib Nephrol. 2011;172:255–260. doi: 10.1159/000328859. [DOI] [PubMed] [Google Scholar]
- 13.Fligny C, Barton M, Tharaux PL. Endothelin and podocyte injury in chronic kidney disease. Contrib Nephrol. 2011;172:120–138. doi: 10.1159/000328692. [DOI] [PubMed] [Google Scholar]
- 14.Tharaux PL. Endothelin in renal injury due to sickle cell disease. Contrib Nephrol. 2011;172:185–199. doi: 10.1159/000328699. [DOI] [PubMed] [Google Scholar]
- 15.Kriz W. Podocyte is the major culprit accounting for the progression of chronic renal disease. Microsc Res Tech. 2002;57:189–195. doi: 10.1002/jemt.10072. [DOI] [PubMed] [Google Scholar]
- 16.Petermann A, Floege J. Podocyte damage resulting in podocyturia: a potential diagnostic marker to assess glomerular disease activity. Nephron Clin Pract. 2007;106:c61–c66. doi: 10.1159/000101799. [DOI] [PubMed] [Google Scholar]
- 17.Mundel P. Urinary podocytes: lost and found alive. Kidney Int. 2003;64:1529–1530. doi: 10.1046/j.1523-1755.2003.00339.x. [DOI] [PubMed] [Google Scholar]
- 18.Asanuma K, Mundel P. The role of podocytes in glomerular pathobiology. Clin Exp Nephrol. 2003;7:255–259. doi: 10.1007/s10157-003-0259-6. [DOI] [PubMed] [Google Scholar]
- 19.Meyrier A. Mechanisms of disease: focal segmental glomerulosclerosis. Nat Clin Pract Nephrol. 2005;1:44–54. doi: 10.1038/ncpneph0025. [DOI] [PubMed] [Google Scholar]
- 20.Ziyadeh FN, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4:39–45. doi: 10.2174/157339908783502370. [DOI] [PubMed] [Google Scholar]
- 21.Kalluri R. Proteinuria with and without renal glomerular podocyte effacement. J Am Soc Nephrol. 2006;17:2383–2389. doi: 10.1681/ASN.2006060628. [DOI] [PubMed] [Google Scholar]
- 22.Floege J, Hackmann B, Kliem V, et al. Age-related glomerulosclerosis and interstitial fibrosis in Milan normotensive rats: a podocyte disease. Kidney Int. 1997;51:230–243. doi: 10.1038/ki.1997.28. [DOI] [PubMed] [Google Scholar]
- 23.Nagase M, Shibata S, Yoshida S, et al. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;47:1084–1093. doi: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- 24.Opocensky M, Kramer HJ, Backer A, et al. Late-onset endothelin-A receptor blockade reduces podocyte injury in homozygous Ren-2 rats despite severe hypertension. Hypertension. 2006;48:965–971. doi: 10.1161/01.HYP.0000245117.57524.d6. [DOI] [PubMed] [Google Scholar]
- 25.Shibata S, Nagase M, Fujita T. Fluvastatin ameliorates podocyte injury in proteinuric rats via modulation of excessive Rho signaling. J Am Soc Nephrol. 2006;17:754–764. doi: 10.1681/ASN.2005050571. [DOI] [PubMed] [Google Scholar]
- 26.Shibata S, Nagase M, Yoshida S, et al. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–364. doi: 10.1161/01.HYP.0000255636.11931.a2. [DOI] [PubMed] [Google Scholar]
- 27.Pozzi A, Zent R. Hold tight or you’ll fall off: CD151 helps podocytes stick in high-pressure situations. J Clin Invest. 2012;122:13–16. doi: 10.1172/JCI61858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riede UN, Wehner H. Allgemeine und spezielle Pathologie (Textbook) Stuttgart: Thieme; 1986. [Google Scholar]
- 29.Reiser J, Oh J, Shirato I, et al. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J Biol Chem. 2004;279:34827–34832. doi: 10.1074/jbc.M401973200. [DOI] [PubMed] [Google Scholar]
- 30.Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 31.Adamczak M, Gross ML, Amann K, et al. Reversal of glomerular lesions involves coordinated restructuring of glomerular microvasculature. J Am Soc Nephrol. 2004;15:3063–3072. doi: 10.1097/01.ASN.0000146121.72699.86. [DOI] [PubMed] [Google Scholar]
- 32.Adamczak M, Gross ML, Krtil J, et al. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol. 2003;14:2833–2842. doi: 10.1097/01.asn.0000095248.91994.d3. [DOI] [PubMed] [Google Scholar]
- 33.Boffa JJ, Lu Y, Placier S, et al. Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol. 2003;14:1132–1144. doi: 10.1097/01.asn.0000060574.38107.3b. [DOI] [PubMed] [Google Scholar]
- 34.Boffa JJ, Tharaux PL, Dussaule JC, et al. Regression of renal vascular fibrosis by endothelin receptor antagonism. Hypertension. 2001;37:490–496. doi: 10.1161/01.hyp.37.2.490. [DOI] [PubMed] [Google Scholar]
- 35.Cruzado JM, Lloberas N, Torras J, et al. Regression of advanced diabetic nephropathy by hepatocyte growth factor gene therapy in rats. Diabetes. 2004;53:1119–1127. doi: 10.2337/diabetes.53.4.1119. [DOI] [PubMed] [Google Scholar]
- 36.Francois H, Placier S, Flamant M, et al. Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. Faseb J. 2004;18:926–928. doi: 10.1096/fj.03-0702fje. [DOI] [PubMed] [Google Scholar]
- 37.Ma LJ, Nakamura S, Aldigier JC, et al. Regression of glomerulosclerosis with high-dose angiotensin inhibition is linked to decreased plasminogen activator inhibitor-1. J Am Soc Nephrol. 2005;16:966–976. doi: 10.1681/ASN.2004060492. [DOI] [PubMed] [Google Scholar]
- 38.Remuzzi G, Benigni A, Remuzzi A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J Clin Invest. 2006;116:288–296. doi: 10.1172/JCI27699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ortmann J, Amann K, Brandes RP, et al. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension. 2004;44:974–981. doi: 10.1161/01.HYP.0000149249.09147.b4. [DOI] [PubMed] [Google Scholar]
- 40.Placier S, Boffa JJ, Dussaule JC, et al. Reversal of renal lesions following interruption of nitric oxide synthesis inhibition in transgenic mice. Nephrol Dial Transplant. 2006;21:881–888. doi: 10.1093/ndt/gfk004. [DOI] [PubMed] [Google Scholar]
- 41.Appel D, Kershaw DB, Smeets B, et al. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol. 2009;20:333–343. doi: 10.1681/ASN.2008070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asano T, Niimura F, Pastan I, et al. Permanent genetic tagging of podocytes: fate of injured podocytes in a mouse model of glomerular sclerosis. J Am Soc Nephrol. 2005;16:2257–2262. doi: 10.1681/ASN.2004121134. [DOI] [PubMed] [Google Scholar]
- 43.Welsh GI, Saleem MA. The podocyte cytoskeleton-key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2012;8:14–21. doi: 10.1038/nrneph.2011.151. [DOI] [PubMed] [Google Scholar]
- 44.Shankland SJ, Pippin JW, Reiser J, et al. Podocytes in culture: past, present, and future. Kidney Int. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 45.Endlich N, Kress KR, Reiser J, et al. Podocytes respond to mechanical stress in vitro. J Am Soc Nephrol. 2001;12:413–422. doi: 10.1681/ASN.V123413. [DOI] [PubMed] [Google Scholar]
- 46.Benigni A, Gagliardini E, Remuzzi G. Changes in glomerular perm-selectivity induced by angiotensin II imply podocyte dysfunction and slit diaphragm protein rearrangement. Semin Nephrol. 2004;24:131–140. doi: 10.1016/j.semnephrol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 47.Takeda T, McQuistan T, Orlando RA, et al. Loss of glomerular foot processes is associated with uncoupling of podocalyxin from the actin cytoskeleton. J Clin Invest. 2001;108:289–301. doi: 10.1172/JCI12539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YH, Goyal M, Kurnit D, et al. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001;60:957–968. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]
- 49.Macconi D, Abbate M, Morigi M, et al. Permselective dysfunction of podocyte-podocyte contact upon angiotensin II unravels the molecular target for renoprotective intervention. Am J Pathol. 2006;168:1073–1085. doi: 10.2353/ajpath.2006.050701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gubler MC. Podocyte differentiation and hereditary proteinuria/nephrotic syndromes. J Am Soc Nephrol. 2003;14(Suppl 1):S22–S26. doi: 10.1097/01.asn.0000067648.75923.68. [DOI] [PubMed] [Google Scholar]
- 51.Meyrier A. An update on the treatment options for focal segmental glomerulosclerosis. Expert Opin Pharmacother. 2009;10:615–628. doi: 10.1517/14656560902754029. [DOI] [PubMed] [Google Scholar]
- 52.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ergul S, Parish DC, Puett D, et al. Racial differences in plasma endothelin-1 concentrations in individuals with essential hypertension. Hypertension. 1996;28:652–655. doi: 10.1161/01.hyp.28.4.652. [DOI] [PubMed] [Google Scholar]
- 54.Sachs N, Kreft M, van den Bergh Weerman MA, et al. Kidney failure in mice lacking the tetraspanin CD151. J Cell Biol. 2006;175:33–39. doi: 10.1083/jcb.200603073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fukuda A, Wickman LT, Venkatareddy MP, et al. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int. 2011;81:40–55. doi: 10.1038/ki.2011.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sachs N, Claessen N, Aten J, et al. Blood pressure influences end-stage renal disease of Cd151 knockout mice. J Clin Invest. 2012;122:348–358. doi: 10.1172/JCI58878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matsusaka T, Sandgren E, Shintani A, et al. Podocyte injury damages other podocytes. J Am Soc Nephrol. 2011;22:1275–1285. doi: 10.1681/ASN.2010090963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can J Physiol Pharmacol. 2008;86:485–498. doi: 10.1139/Y08-059. [DOI] [PubMed] [Google Scholar]
- 59.Barton M. The discovery of endothelium-dependent contraction: the legacy of Paul M. Vanhoutte Pharmacol Res. 2011;63:455–462. doi: 10.1016/j.phrs.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 60.Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. Faseb J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 61.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;299:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 62.Hickey KA, Rubanyi GM, Paul RJ, et al. Characterization of a coronary vasoconstrictor produced by cultured endothelial cells. Am J Physiol. 1985;248:C550–C5C6. doi: 10.1152/ajpcell.1985.248.5.C550. [DOI] [PubMed] [Google Scholar]
- 63.Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 64.Benigni A, Remuzzi G. How renal cytokines and growth factors contribute to renal disease progression. Am J Kidney Dis. 2001;37:S21–S24. doi: 10.1053/ajkd.2001.20734. [DOI] [PubMed] [Google Scholar]
- 65.Lattmann T, Hein M, Horber S, et al. Activation of pro-inflammatory and anti-inflammatory cytokines in host organs during chronic allograft rejection: role of endothelin receptor signaling. Am J Transplant. 2005;5:1042–1049. doi: 10.1111/j.1600-6143.2005.00807.x. [DOI] [PubMed] [Google Scholar]
- 66.Gomez-Garre D, Largo R, Liu XH, et al. An orally active ETA/ETB receptor antagonist ameliorates proteinuria and glomerular lesions in rats with proliferative nephritis. Kidney Int. 1996;50:962–972. doi: 10.1038/ki.1996.397. [DOI] [PubMed] [Google Scholar]
- 67.Saleh MA, Boesen EI, Pollock JS, et al. Endothelin-1 increases glomerular permeability and inflammation independent of blood pressure in the rat. Hypertension. 2010;56:942–949. doi: 10.1161/HYPERTENSIONAHA.110.156570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saleh MA, Boesen EI, Pollock JS, et al. Endothelin receptor A-specific stimulation of glomerular inflammation and injury in a streptozotocin-induced rat model of diabetes. Diabetologia. 2011;54:979–988. doi: 10.1007/s00125-010-2021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gagliardini E, Corna D, Zoja C, et al. Unlike each drug alone, lisinopril if combined with avosentan promotes regression of renal lesions in experimental diabetes. Am J Physiol Renal Physiol. 2009;297:F1448–F1456. doi: 10.1152/ajprenal.00340.2009. [DOI] [PubMed] [Google Scholar]
- 70.Saleh MA, Pollock JS, Pollock DM. Distinct actions of endothelin A-selective versus combined endothelin A/B receptor antagonists in early diabetic kidney disease. J Pharmacol Exp Ther. 2011;338:263–270. doi: 10.1124/jpet.111.178988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawaguchi H, Sawa H, Yasuda H. Endothelin stimulates angiotensin I to angiotensin II conversion in cultured pulmonary artery endothelial cells. J Mol Cell Cardiol. 1990;22:839–842. doi: 10.1016/0022-2828(90)90115-i. [DOI] [PubMed] [Google Scholar]
- 72.Barton M, Shaw S, d'Uscio LV, et al. Angiotensin II increases vascular and renal endothelin-1 and functional endothelin converting enzyme activity in vivo: role of ETA receptors for endothelin regulation. Biochem Biophys Res Commun. 1997;238:861–865. doi: 10.1006/bbrc.1997.7394. [DOI] [PubMed] [Google Scholar]
- 73.Barton M, Carmona R, Morawietz H, et al. Obesity is associated with tissue-specific activation of renal angiotensin-converting enzyme in vivo: evidence for a regulatory role of endothelin. Hypertension. 2000;35:329–336. doi: 10.1161/01.hyp.35.1.329. [DOI] [PubMed] [Google Scholar]
- 74.Kohan DE, Rossi NF, Inscho EW, et al. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakano D, Pollock DM. Contribution of endothelin A receptors in endothelin 1-dependent natriuresis in female rats. Hypertension. 2009;53:324–330. doi: 10.1161/HYPERTENSIONAHA.108.123687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kisanuki YY, Emoto N, Ohuchi T, et al. Low blood pressure in endothelial cell-specific endothelin 1 knockout mice. Hypertension. 2010;56:121–128. doi: 10.1161/HYPERTENSIONAHA.109.138701. [DOI] [PubMed] [Google Scholar]
- 77.Amiri F, Virdis A, Neves MF, et al. Endothelium-restricted overexpression of human endothelin-1 causes vascular remodeling and endothelial dysfunction. Circulation. 2004;110:2233–2240. doi: 10.1161/01.CIR.0000144462.08345.B9. [DOI] [PubMed] [Google Scholar]
- 78.Leung JW, Wong WT, Koon HW, et al. Transgenic mice over-expressing ET-1 in the endothelial cells develop systemic hypertension with altered vascular reactivity. PLoS One. 2011;6:e26994. doi: 10.1371/journal.pone.0026994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lattmann T, Shaw S, Munter K, et al. Anatomically distinct activation of endothelin-3 and the L-arginine/nitric oxide pathway in the kidney with advanced aging. Biochem Biophys Res Commun. 2005;327:234–241. doi: 10.1016/j.bbrc.2004.11.160. [DOI] [PubMed] [Google Scholar]
- 80.Furuya S, Naruse S, Nakayama T, et al. Effect and distribution of intravenously injected 125I-endothelin-1 in rat kidney and lung examined by electron microscopic radioautography. Anat Embryol (Berl) 1992;185:87–96. doi: 10.1007/BF00213604. [DOI] [PubMed] [Google Scholar]
- 81.Cybulsky AV, Stewart DJ, Cybulsky MI. Glomerular epithelial cells produce endothelin-1. J Am Soc Nephrol. 1993;3:1398–1404. doi: 10.1681/ASN.V371398. [DOI] [PubMed] [Google Scholar]
- 82.Kasinath BS, Fried TA, Davalath S, et al. Glomerular epithelial cells synthesize endothelin peptides. Am J Pathol. 1992;141:279–283. [PMC free article] [PubMed] [Google Scholar]
- 83.Rebibou JM, He CJ, Delarue F, et al. Functional endothelin 1 receptors on human glomerular podocytes and mesangial cells. Nephrol Dial Transplant. 1992;7:288–292. doi: 10.1093/oxfordjournals.ndt.a092130. [DOI] [PubMed] [Google Scholar]
- 84.Yamamoto T, Hirohama T, Uemura H. Endothelin B receptor-like immunoreactivity in podocytes of the rat kidney. Arch Histol Cytol. 2002;65:245–250. doi: 10.1679/aohc.65.245. [DOI] [PubMed] [Google Scholar]
- 85.Schneider MP, Boesen EI, Pollock DM. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu Rev Pharmacol Toxicol. 2007;47:731–759. doi: 10.1146/annurev.pharmtox.47.120505.105134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Benigni A, Zoja C, Corna D, et al. A specific endothelin subtype A receptor antagonist protects against injury in renal disease progression. Kidney Int. 1993;44:440–444. doi: 10.1038/ki.1993.263. [DOI] [PubMed] [Google Scholar]
- 87.Hocher B, Thone-Reineke C, Rohmeiss P, et al. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J Clin Invest. 1997;99:1380–1389. doi: 10.1172/JCI119297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barton M, Vos I, Shaw S, et al. Dysfunctional renal nitric oxide synthase as a determinant of salt-sensitive hypertension: mechanisms of renal artery endothelial dysfunction and role of endothelin for vascular hypertrophy and glomerulosclerosis. J Am Soc Nephrol. 2000;11:835–845. doi: 10.1681/ASN.V115835. [DOI] [PubMed] [Google Scholar]
- 89.Vernerova Z, Kramer HJ, Backer A, et al. Late-onset endothelin receptor blockade in hypertensive heterozygous REN-2 transgenic rats. Vascul Pharmacol. 2008;48:165–173. doi: 10.1016/j.vph.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 90.Benigni A, Colosio V, Brena C, et al. Unselective inhibition of endothelin receptors reduces renal dysfunction in experimental diabetes. Diabetes. 1998;47:450–456. doi: 10.2337/diabetes.47.3.450. [DOI] [PubMed] [Google Scholar]
- 91.Wang S, Chen Q, Simon TC, et al. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003;63:2037–2049. doi: 10.1046/j.1523-1755.2003.00035.x. [DOI] [PubMed] [Google Scholar]
- 92.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 93.Fogo AB. New capillary growth: a contributor to regression of sclerosis? Curr Opin Nephrol Hypertens. 2005;14:201–203. doi: 10.1097/01.mnh.0000165883.08675.ab. [DOI] [PubMed] [Google Scholar]
- 94.Bollee G, Flamant M, Schordan S, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med. 2011;17:1242–1250. doi: 10.1038/nm.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chansel D, Ciroldi M, Vandermeersch S, et al. Heparin binding EGF is necessary for vasospastic response to endothelin. Faseb J. 2006;20:1936–1938. doi: 10.1096/fj.05-5328fje. [DOI] [PubMed] [Google Scholar]
- 96.Portik-Dobos V, Harris AK, Song W, et al. Endothelin antagonism prevents early EGFR transactivation but not increased matrix metalloproteinase activity in diabetes. Am J Physiol Regul Integr Comp Physiol. 2006;290:R435–R441. doi: 10.1152/ajpregu.00300.2005. [DOI] [PubMed] [Google Scholar]
- 97.Morigi M, Buelli S, Angioletti S, et al. In response to protein load podocytes reorganize cytoskeleton and modulate endothelin-1 gene: implication for permselective dysfunction of chronic nephropathies. Am J Pathol. 2005;166:1309–1320. doi: 10.1016/S0002-9440(10)62350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morigi M, Buelli S, Zanchi C, et al. Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol. 2006;169:1965–1975. doi: 10.2353/ajpath.2006.051331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Benigni A, Perico N, Remuzzi G. The potential of endothelin antagonism as a therapeutic approach. Expert Opin Investig Drugs. 2004;13:1419–1435. doi: 10.1517/13543784.13.11.1419. [DOI] [PubMed] [Google Scholar]
- 100.Liang XB, Ma LJ, Naito T, et al. Angiotensin type 1 receptor blocker restores podocyte potential to promote glomerular endothelial cell growth. J Am Soc Nephrol. 2006;17:1886–1895. doi: 10.1681/ASN.2005020205. [DOI] [PubMed] [Google Scholar]
- 101.Diamond JR, Karnovsky MJ. Focal and segmental glomerulosclerosis following a single intravenous dose of puromycin aminonucleoside. Am J Pathol. 1986;122:481–487. [PMC free article] [PubMed] [Google Scholar]
- 102.Fogo AB. Animal models of FSGS: lessons for pathogenesis and treatment. Semin Nephrol. 2003;23:161–171. doi: 10.1053/snep.2003.50015. [DOI] [PubMed] [Google Scholar]
- 103.von Luttichau I, Djafarzadeh R, Henger A, et al. Identification of a signal transduction pathway that regulates MMP-9 mRNA expression in glomerular injury. Biol Chem. 2002;383:1271–1275. doi: 10.1515/BC.2002.142. [DOI] [PubMed] [Google Scholar]
- 104.Clozel M, Qiu C, Osterwalder R, et al. Effects of nonpeptide endothelin receptor antagonists in rats with reduced renal mass. J Cardiovasc Pharmacol. 1999;33:611–618. doi: 10.1097/00005344-199904000-00014. [DOI] [PubMed] [Google Scholar]
- 105.Schiffrin EL. State-of-the-Art lecture. Role of endothelin-1 in hypertension. Hypertension. 1999;34:876–881. doi: 10.1161/01.hyp.34.4.876. [DOI] [PubMed] [Google Scholar]
- 106.Tharaux PL, Chatziantoniou C, Casellas D, et al. Vascular endothelin-1 gene expression and synthesis and effect on renal type I collagen synthesis and nephroangiosclerosis during nitric oxide synthase inhibition in rats. Circulation. 1999;99:2185–2191. doi: 10.1161/01.cir.99.16.2185. [DOI] [PubMed] [Google Scholar]
- 107.Barton M, d'Uscio L, Shaw S, et al. ETA receptor blockade prevents increased tissue endothelin-1, vascular hypertrophy and endothelial dysfunction in salt-sensitive hypertension. Hypertension. 1998;31:499–504. doi: 10.1161/01.hyp.31.1.499. [DOI] [PubMed] [Google Scholar]
- 108.Weber MA, Black H, Bakris G, et al. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1423–1431. doi: 10.1016/S0140-6736(09)61500-2. [DOI] [PubMed] [Google Scholar]
- 109.Zhu L, Jiang R, Aoudjit L, et al. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22:1621–1630. doi: 10.1681/ASN.2010111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wiggins J. Podocytes and glomerular function with aging. Semin Nephrol. 2009;29:587–593. doi: 10.1016/j.semnephrol.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wiggins JE, Goyal M, Sanden SK, et al. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J Am Soc Nephrol. 2005;16:2953–2966. doi: 10.1681/ASN.2005050488. [DOI] [PubMed] [Google Scholar]
- 112.Mahan JD, Sisson-Ross S, Vernier RL. Glomerular basement membrane anionic charge site changes early in aminonucleoside nephrosis. Am J Pathol. 1986;125:393–401. [PMC free article] [PubMed] [Google Scholar]
- 113.Hartleben B, Godel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2011;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Megyesi J, Price PM, Tamayo E, et al. The lack of a functional p21(WAF1/CIP1) gene ameliorates progression to chronic renal failure. Proc Natl Acad Sci USA. 1999;96:10830–10835. doi: 10.1073/pnas.96.19.10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dijkman H, Smeets B, van der Laak J, et al. The parietal epithelial cell is crucially involved in human idiopathic focal segmental glomerulosclerosis. Kidney Int. 2005;68:1562–1572. doi: 10.1111/j.1523-1755.2005.00568.x. [DOI] [PubMed] [Google Scholar]
- 116.Smeets B, Angelotti ML, Rizzo P, et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol. 2009;20:2593–2603. doi: 10.1681/ASN.2009020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dhaun N, Macintyre IM, Melville V, et al. Blood pressure-independent reduction in proteinuria and arterial stiffness after acute endothelin-a receptor antagonism in chronic kidney disease. Hypertension. 2009;54:113–119. doi: 10.1161/HYPERTENSIONAHA.109.132670. [DOI] [PubMed] [Google Scholar]
- 118.Dhaun N, Macintyre IM, Melville V, et al. Effects of endothelin receptor antagonism relate to the degree of renin-angiotensin system blockade in chronic proteinuric kidney disease. Hypertension. 2009;54:e19–e20. doi: 10.1161/HYPERTENSIONAHA.109.138263. [DOI] [PubMed] [Google Scholar]
- 119.Rabelink TJ, Kohan DE. Endothelin receptor blockade in patients with diabetic nephropathy. Contrib Nephrol. 2011;172:235–242. doi: 10.1159/000328703. [DOI] [PubMed] [Google Scholar]
- 120.Zhou X, Hurst RD, Templeton D, et al. High glucose alters actin assembly in glomerular mesangial and epithelial cells. Lab Invest. 1995;73:372–383. [PubMed] [Google Scholar]
- 121.Susztak K, Raff AC, Schiffer M, et al. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 122.Petermann AT, Krofft R, Blonski M, et al. Podocytes that detach in experimental membranous nephropathy are viable. Kidney Int. 2003;64:1222–1231. doi: 10.1046/j.1523-1755.2003.00217.x. [DOI] [PubMed] [Google Scholar]
- 123.Collino F, Bussolati B, Gerbaudo E, et al. Pre-eclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am J Physiol Renal Physiol. 2008 doi: 10.1152/ajprenal.00442.2007. [DOI] [PubMed] [Google Scholar]
- 124.Sasser JM, Sullivan JC, Hobbs JL, et al. Endothelin A receptor blockade reduces diabetic renal injury via an anti-inflammatory mechanism. J Am Soc Nephrol. 2007;18:143–154. doi: 10.1681/ASN.2006030208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ding SS, Qiu C, Hess P, et al. Chronic endothelin receptor blockade prevents both early hyperfiltration and late overt diabetic nephropathy in the rat. J Cardiovasc Pharmacol. 2003;42:48–54. doi: 10.1097/00005344-200307000-00008. [DOI] [PubMed] [Google Scholar]
- 126.Gross ML, El-Shakmak A, Szabo A, et al. ACE-inhibitors but not endothelin receptor blockers prevent podocyte loss in early diabetic nephropathy. Diabetologia. 2003;46:856–868. doi: 10.1007/s00125-003-1106-8. [DOI] [PubMed] [Google Scholar]
- 127.Gross ML, Ritz E, Schoof A, et al. Renal damage in the SHR/N-cp type 2 diabetes model: comparison of an angiotensin-converting enzyme inhibitor and endothelin receptor blocker. Lab Invest. 2003;83:1267–1277. doi: 10.1097/01.lab.0000085188.23709.29. [DOI] [PubMed] [Google Scholar]
- 128.Mann JF, Green D, Jamerson K, et al. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol. 2010;21:527–535. doi: 10.1681/ASN.2009060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barton M. Childhood obesity—a life-long health risk. Acta Pharmacol Sin. 2012;33:189–93. doi: 10.1038/aps.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Barton M. Obesity and aging: determinants of endothelial cell dysfunction and atherosclerosis. Pflugers Arch. 2010;460:825–837. doi: 10.1007/s00424-010-0860-y. [DOI] [PubMed] [Google Scholar]
- 131.Griffin KA, Kramer H, Bidani AK. Adverse renal consequences of obesity. Am J Physiol Renal Physiol. 2008;294:F685–F696. doi: 10.1152/ajprenal.00324.2007. [DOI] [PubMed] [Google Scholar]
- 132.Wolf G. Obesity and the kidney. Contrib Nephrol. 2006;151:1–260. doi: 10.1159/000095317. , Karger, Basel. [DOI] [PubMed] [Google Scholar]
- 133.Kramer H. Obesity and chronic kidney disease. Contrib Nephrol. 2006;151:1–18. doi: 10.1159/000095315. [DOI] [PubMed] [Google Scholar]
- 134.Praga M, Hernandez E, Morales E, et al. Clinical features and long-term outcome of obesity-associated focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2001;16:1790–1798. doi: 10.1093/ndt/16.9.1790. [DOI] [PubMed] [Google Scholar]
- 135.Chen HM, Liu ZH, Zeng CH, et al. Podocyte lesions in patients with obesity-related glomerulopathy. Am J Kidney Dis. 2006;48:772–779. doi: 10.1053/j.ajkd.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 136.Saraheimo M, Forsblom C, LT, et al. Serum adiponectin and prgression of diabetic nephropathy in patients with type 1 diabetes. Diabetes Care. 2008;31:1165–1169. doi: 10.2337/dc07-2306. [DOI] [PubMed] [Google Scholar]
- 137.Sharma K, RamachandraRao S, Qiu G, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;188:1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Barton M, Carmona R, Ortmann J, et al. Obesity-associated activation of angiotensin and endothelin in the cardiovascular system. Int J Biochem Cell Biol. 2003;35:826–837. doi: 10.1016/s1357-2725(02)00307-2. [DOI] [PubMed] [Google Scholar]
- 139.Bedi D, Clarke KJ, Dennis JC, et al. Endothelin-1 inhibits adiponectin secretion through a phosphatidylinositol 4,5-bisphosphate/actin-dependent mechanism. Biochem Biophys Res Commun. 2006;345:332–339. doi: 10.1016/j.bbrc.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 140.Idris I, Patiag D, Gray S, et al. Tissue- and time-dependent effects of endothelin-1 on insulin-stimulated glucose uptake. Biochem Pharmacol. 2001;62:1705–1708. doi: 10.1016/s0006-2952(01)00815-2. [DOI] [PubMed] [Google Scholar]
- 141.Yoshimura A, Iwasaki S, Inui K, et al. Endothelin-1 and endothelin B type receptor are induced in mesangial proliferative nephritis in the rat. Kidney Int. 1995;48:1290–1297. doi: 10.1038/ki.1995.413. [DOI] [PubMed] [Google Scholar]
- 142.Nicolaidou P, Georgouli H, Matsinos Y, et al. Endothelin-1 in children with acute poststreptococcal glomerulonephritis and hypertension. Pediatr Int. 2003;45:35–38. doi: 10.1046/j.1442-200x.2003.01661.x. [DOI] [PubMed] [Google Scholar]
- 143.Orth SR, Odoni G, Amann K, et al. The ET(A) receptor blocker LU 135252 prevents chronic transplant nephropathy in the “Fisher to Lewis” model. J Am Soc Nephrol. 1999;10:387–391. doi: 10.1681/ASN.V102387. [DOI] [PubMed] [Google Scholar]
- 144.Chen CL, Fang HC, Chou KJ, et al. Increased endothelin 1 expression in adult-onset minimal change nephropathy with acute renal failure. Am J Kidney Dis. 2005;45:818–825. doi: 10.1053/j.ajkd.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 145.Ruschitzka F, Shaw S, Gygi D, et al. Endothelial dysfunction in acute renal failure: role of circulating and tissue endothelin-1. J Am Soc Nephrol. 1999;10:953–962. doi: 10.1681/ASN.V105953. [DOI] [PubMed] [Google Scholar]
- 146.Ohta K, Hirata Y, Shichiri M, et al. Urinary excretion of endothelin-1 in normal subjects and patients with renal disease. Kidney Int. 1991;39:307–311. doi: 10.1038/ki.1991.38. [DOI] [PubMed] [Google Scholar]
- 147.Tharaux PL, Hagege I, Placier S, et al. Urinary endothelin-1 as a marker of renal damage in sickle cell disease. Nephrol Dial Transplant. 2005;20:2408–2413. doi: 10.1093/ndt/gfi111. [DOI] [PubMed] [Google Scholar]
- 148.Hocher B, Lun A, Priem F, et al. Renal endothelin system in diabetes: comparison of angiotensin-converting enzyme inhibition and endothelin-A antagonism. J Cardiovasc Pharmacol. 1998;31(Suppl 1):S492–S495. doi: 10.1097/00005344-199800001-00141. [DOI] [PubMed] [Google Scholar]
- 149.Sabaa N, de Franceschi L, Bonnin P, et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. J Clin Invest. 2008;118:1924–1933. doi: 10.1172/JCI33308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Barton M, Kohan DE. Endothelin in renal physiology and disease. Contrib Nephrol. 2011;172:1–266. , Karger, Basel. [Google Scholar]
- 151.Kohan DE, Strait K, Stricklett P, et al. Identification of the site of endothelin A receptor antagonist-induced fluid retention. Proceedings of the British Pharmacological Society 2011: http://www.pA2online.org/abstracts/Vol9Issue1abst015P.pdf. [Google Scholar]