

Summary
Human embryonic stem cell (hESC)-derived pancreatic progenitor cells effectively reverse hyperglycemia in rodent models of type 1 diabetes, but their capacity to treat type 2 diabetes has not been reported. An immunodeficient model of type 2 diabetes was generated by high-fat diet (HFD) feeding in SCID-beige mice. Exposure to HFDs did not impact the maturation of macroencapsulated pancreatic progenitor cells into glucose-responsive insulin-secreting cells following transplantation, and the cell therapy improved glucose tolerance in HFD-fed transplant recipients after 24 weeks. However, since diet-induced hyperglycemia and obesity were not fully ameliorated by transplantation alone, a second cohort of HFD-fed mice was treated with pancreatic progenitor cells combined with one of three antidiabetic drugs. All combination therapies rapidly improved body weight and co-treatment with either sitagliptin or metformin improved hyperglycemia after only 12 weeks. Therefore, a stem cell-based therapy may be effective for treating type 2 diabetes, particularly in combination with antidiabetic drugs.
Highlights
-
•
High-fat diets (HFDs) caused rapid metabolic dysfunction in immunodeficient mice
-
•
HFDs and/or antidiabetic drugs did not affect function of hESC-derived beta cells
-
•
hESC-derived beta cells improved glucose tolerance in HFD-fed mice
-
•
Cell therapy combined with an antidiabetic drug was the most effective treatment
Kieffer and colleagues demonstrate the utility of human embryonic stem cell (hESC)-derived pancreatic progenitor cells for treating high-fat diet (HFD)-induced hyperglycemia and obesity, either alone or combined with antidiabetic drugs. Exposure to HFDs and antidiabetic drugs did not impact the maturation and function of hESC-derived insulin-secreting cells, supporting the potential of a stem cell-based therapy for type 2 diabetes.
Introduction
The International Diabetes Federation estimates that up to 95% of the ∼380 million people worldwide who are affected by diabetes suffer from type 2 diabetes (International Diabetes Federation, 2014). Thus, the potential impact of a novel treatment for type 2 diabetes is enormous. Despite obvious differences in the pathogenesis of type 1 and 2 diabetes, both diseases are characterized by impaired glucose homeostasis resulting from insufficient insulin production by pancreatic beta cells. In type 1 diabetes, beta cell destruction by the immune system is rapid and extensive, causing severe insulin deficiency. In contrast, beta cell failure in type 2 diabetes occurs gradually over time and is associated with peripheral insulin resistance. Clinical studies have shown that patients with type 2 diabetes also have reduced beta cell mass (Butler et al., 2003; Yoon et al., 2003) and declining beta cell function during the progression from pre-diabetes to overt diabetes (Weyer et al., 1999; Ferrannini et al., 2005). Therefore, treatment strategies for type 2 diabetes should be aimed at restoring beta cell mass and/or function, in addition to improving insulin sensitivity (Halban, 2008; Kahn et al., 2014).
Transplantation of cadaveric human islets can restore insulin-independence in patients with type 1 diabetes (Shapiro et al., 2000; Ryan et al., 2001), but this approach has not been actively pursued for type 2 diabetes, likely due to the inadequate supply of donor islets, risk of immunosuppression, and perceived hurdle of insulin resistance. The obstacle of an insufficient cell supply may be overcome with the use of human embryonic stem cells (hESCs). We previously demonstrated that hESC-derived pancreatic progenitor cells reversed hyperglycemia in a mouse model of type 1 diabetes characterized by severe beta cell destruction and insulin deficiency (Rezania et al., 2012, 2013; Bruin et al., 2013). However, the efficacy of this stem cell-based therapy for treating hyperglycemia in an obesogenic and insulin-resistant environment, such as in type 2 diabetes, has not been reported. Based on evidence that intensive insulin therapy improves insulin sensitivity, glycemic control, and beta cell function in patients with type 2 diabetes (Weng et al., 2008; Kramer et al., 2013), we hypothesized that hESC-derived insulin-secreting cells may also be effective for this patient population.
Our first aim was to establish a model of type 2 diabetes in immunodeficient mice that would be compatible with xenotransplantation. Different strains of rodents have widely variable susceptibility to high-fat diet (HFD)-induced obesity and/or hyperglycemia (Srinivasan and Ramarao, 2007; Svenson et al., 2007; Hariri and Thibault, 2010). Moreover, insulin resistance, a hallmark feature of type 2 diabetes (Kahn et al., 2006), is thought to be driven primarily by obesity-associated inflammation (reviewed in Kalupahana et al., 2012; Osborn and Olefsky, 2012), and recruitment of T cells (Feuerer et al., 2009; Nishimura et al., 2009; Winer et al., 2009) and B cells (Winer et al., 2011) to insulin-sensitive tissues. SCID-beige mice are a spontaneous double-mutant model in which the scid mutation results in a lack of both T and B lymphocytes, and the beige mutation causes defects in cytotoxic T cells, macrophages, and NK cells (http://www.taconic.com). To our knowledge, the susceptibility of double-mutant SCID-beige mice to HFDs has not previously been examined as a potential model of type 2 diabetes.
An important consideration in translating a stem cell-derived pancreatic progenitor therapy to clinical practice is the variability that will be encountered within the patient environment during the period of cell engraftment and maturation in vivo. This is particularly relevant given that macroencapsulated hESC-derived pancreatic progenitor cells are now being tested for safety, tolerability, and efficacy in a phase 1/2 clinical trial by Viacyte (ClinicalTrials.gov, Identifier: NCT02239354). We hypothesized that exposure to HFDs may impair the development of hESC-derived insulin-secreting cells, since obesity-associated lipotoxicity and inflammation contribute to beta cell dysfunction in patients with type 2 diabetes (reviewed in Potter et al., 2014). Furthermore, both human and rodent islets displayed beta cell dysfunction following transplant into HFD-fed rodents (Hiramatsu and Grill, 2001; Gargani et al., 2013). Here, we examined the impact of HFDs on hESC-derived progenitor cell development in vivo, and assessed whether a stem cell-based insulin therapy could improve glycemic control in mice with diet-induced obesity, insulin resistance, and hyperglycemia. We also investigated the efficacy of combining the cell therapy with one of three antidiabetic drugs: sitagliptin (a dipeptidyl peptidase-4 [DPP4 inhibitor]), metformin (suppresses hepatic gluconeogenesis and enhances insulin sensitivity), and rosiglitazone (a PPARγ agonist from the thiazolidinedione [TZD] class). Our studies demonstrated that a combination therapy was more effective in HFD-fed mice than either antidiabetic drugs or progenitor cell transplants alone. Moreover, neither HFDs nor antidiabetic drugs impacted the ability of hESC-derived cells to mature in vivo and appropriately secrete insulin in response to glucose.
Results
SCID-Beige Mice Rapidly Developed Diet-Induced Obesity, Insulin Resistance, and Hyperglycemia
All three of the HFDs used in this study (45% fat, 60% fat, and Western) induced rapid increases in fasting body weight (BW; Figure 1A) and blood glucose levels (Figure 1B) compared with low-fat diet (LFD) controls (10% fat). Moreover, after only 5 days, mice in all three HFD groups were severely glucose intolerant relative to LFD controls (Figure S1A), even though no differences in BW were observed at that time (Figure S1B). At 32 days, HFD mice were both glucose intolerant (Figure S1C) and significantly heavier (Figure S1D) than LFD controls. Mice fed 45% and 60% fat diets were overtly insulin resistant at day 42 (higher glucose levels at 10 and 60–120 min post-insulin, and reduced area above the curve relative to LFD controls), whereas mice on the Western diet only showed significant insulin resistance at 10 min after insulin administration (Figure 1E).
Figure 1.
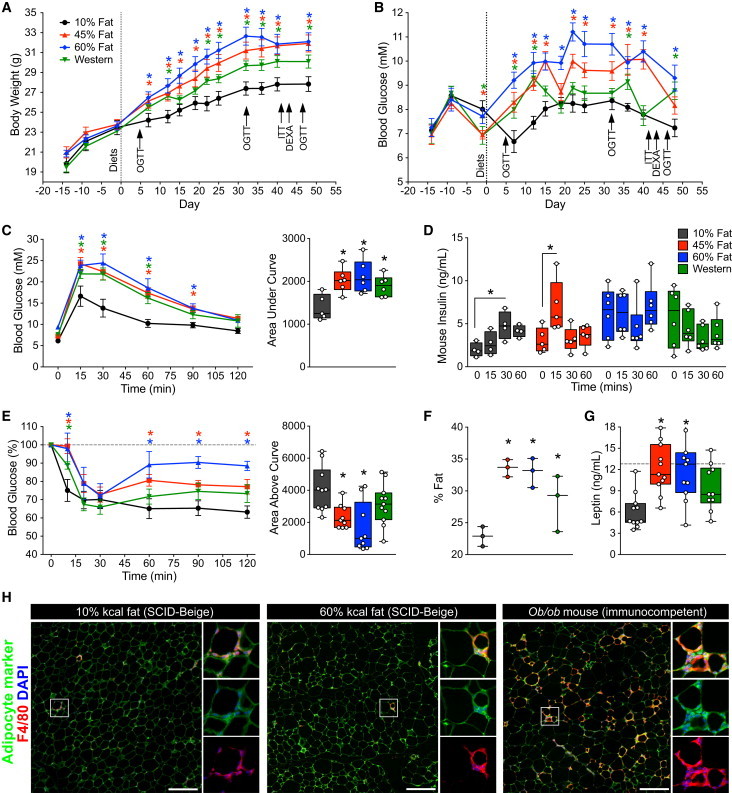
SCID-Beige Mice Rapidly Develop Obesity, Fasting Hyperglycemia, Glucose Intolerance, and Insulin Resistance Following Exposure to HFDs
(A and B) Body weight (A) and fasting blood glucose levels (B) were measured during a 14-day acclimation period on normal chow and for 51 days following administration of one of the following diets: 10% fat (black; n = 11 mice), 45% fat (red; n = 11 mice), 60% fat (blue; n = 11 mice), or Western diet (green; n = 11 mice).
(C and D) Blood glucose (C, raw values and area under the curve) and plasma mouse insulin levels (D) were assessed during an oral glucose tolerance test (OGTT; n = 4–6 mice per group) on day 47. See Figure S1 for OGTTs at days 5 and 32.
(E) An insulin tolerance test (ITT) was performed on day 42 (n = 9–11 mice per group). Glucose levels are presented as a percentage of basal glucose levels (at time 0) and the area above the curve was calculated using 100% as the baseline.
(F) Adiposity (% fat) was assessed by DEXA at day 43 in a subset of mice (n = 3 mice per group).
(G) Mouse leptin levels were measured on days 47–49 following a 4- to 6-hr morning fast (dashed line indicates the highest standard concentration in the leptin assay). See Figures S1E–S1G for circulating lipid levels.
(H) Immunofluorescent staining of epididymal fat from immunocompromised SCID-beige mice fed either a 10% or 60% fat diet for 36 weeks, and from an immunocompetent adult ob/ob mouse. F4/80 and FGF21 are shown as macrophage and adipocyte markers, respectively. Crown-like structures, representing macrophage infiltration, are shown in insets. Scale bars, 200 μm.
(A–C and E–G) ∗p < 0.05 versus 10% controls (two-way ANOVA for comparison of multiple time points in line graphs, and one-way ANOVA for bar graphs); (D) ∗p < 0.05 versus time 0 (one-way ANOVA). Data are represented as mean ± SEM (line graphs) or as box-and-whisker plots with individual mice shown as separate data points.
In the week preceding transplantation (day 51), mice in all HFD groups were glucose intolerant (day 47, Figure 1C) and showed insulin-secretion kinetics that differed from LFD controls (either no glucose-induced insulin secretion or altered timing of peak insulin levels; Figure 1D). All HFD mice were significantly overweight (Figure 1A) and had increased adiposity (Figure 1F) compared with LFD controls, and mice fed 45% or 60% fat diets also had significantly elevated circulating leptin levels (Figure 1G). Exposure to HFDs caused dyslipidemia, including significantly reduced plasma free fatty acid levels in all HFD-fed mice (Figure S1E), reduced triglyceride levels in 45% and 60% fat groups (Figure S1F), and elevated cholesterol levels in the Western-diet group compared with LFD controls (Figure S1G). Interestingly, the HFD-induced metabolic defects in immunodeficient mice were not associated with macrophage infiltration in adipose tissue (marked by F4/80 immunoreactivity; Weisberg et al., 2003), whereas significant accumulation of F4/80-positive crown-like structures was observed in the epididymal fat of ob/ob mice, an immunocompetent model of type 2 diabetes (Figure 1H). Fibroblast growth factor 21 (FGF21) was used as an adipocyte marker because it is highly expressed in white adipose tissue (Markan et al., 2014).
Taken together, these results indicate that 7 weeks of exposure to HFDs generated a type 2-like diabetes model characterized by hyperglycemia, insulin resistance, obesity, and dyslipidemia in immunodeficient mice. Given the phenotypic similarities between mice on the 45% and 60% fat diets, these groups were combined for all further analysis (hereafter referred to as the 45%–60% fat group).
Exposure to HFDs Did Not Affect the Function of hESC-Derived Endocrine Cells In Vivo
Prior to transplantation, differentiated hESC-derived pancreatic progenitor cells were assessed by fluorescence-activated cell sorting (FACS) and immunofluorescent staining (Figure S2). Following in vitro differentiation, ∼95% of cells expressed PDX1 and ∼65% expressed NKX6.1 (Figures S2A, S2C, S2D, and S2G), two key markers of pancreatic endoderm. Approximately 20% of PDX1-positive cells were in the cell cycle, as indicated by Ki67 or PCNA expression (Figures S2A and S2D), and the pluripotency marker OCT3/4 was not detected (Figure S2A). Although ∼14% of progenitor cells expressed endocrine markers (Figures S2A and S2B), only 2.5% of synaptophysin-positive cells co-expressed NKX6.1 (Figure S2A) and most were polyhormonal (Figure S2F), indicative of an immature endocrine population. Indeed, insulin/C-peptide-positive cells only rarely co-expressed PAX6 (Figure S2E) or NKX6.1 (Figure S2C) at this stage of differentiation. These data are consistent with the characteristics of hESC-derived pancreatic progenitor cells described previously by our group (Rezania et al., 2012, 2013; Bruin et al., 2013).
Progenitor cells were encapsulated in Theracyte devices and transplanted subcutaneously into immunodeficient mice from each of the four diet regimens. We used immunodeficient mice because although macroencapsulation devices are predicted to protect human cells from allogeneic immune rejection (Tibell et al., 2001), they are unlikely to protect them from xenograft rejection (Brauker et al., 1996; Mckenzie et al., 2001). The immunoisolation devices allowed us to mimic studies in patients receiving macroencapsulated pancreatic progenitor cells (ClinicalTrials.gov, Identifier: NCT02239354). Following transplantation, hESC-derived cells from all diet groups secreted similar levels of human C-peptide under basal and fed conditions between 8 and 20 weeks (Figure 2A), and produced robust glucose-stimulated human C-peptide secretion at 18 weeks (Figures 2B and 2C). Similarly, human insulin secretion was induced by an arginine challenge in all diet groups at 24 weeks, although due to high variability, the Western-diet group did not reach statistical significance (Figure 2D). We observed a trend toward increased basal glucagon secretion in the HFD groups, but as four out of five mice in the LFD group had undetectable fasting glucagon levels, it was not possible to perform a statistical analysis (Figure 2E). Arginine-stimulated glucagon levels were similar between diet groups (Figures 2E and 2F), and we estimate that approximately half of the circulating glucagon may have originated from hESC-derived cells, as indicated by the difference between glucagon levels in sham-treated mice and transplant recipients (Tx; Figure 2F).
Figure 2.
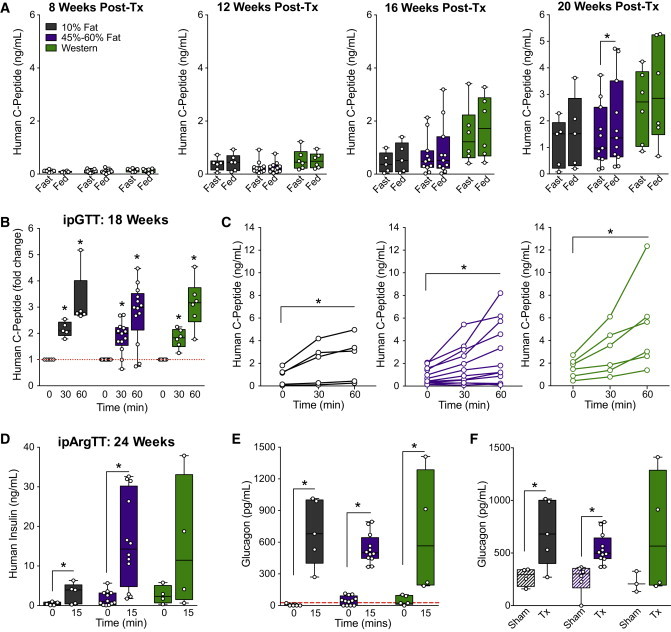
Exposure to HFDs Does Not Affect the Function of hESC-Derived Pancreatic Endocrine Cells In Vivo
The development of hESC-derived progenitor cells into pancreatic endocrine cells was assessed in mice fed a 10% fat (black), 45% or 60% fat (purple), or Western (green) diet. See Figure S2 for characterization of the progenitor cells pre-transplantation.
(A) Human C-peptide levels were measured after an overnight fast and 40 min following an oral mixed-meal challenge (“fed”) at 8, 12, 16, and 20 weeks post-transplantation. ∗p < 0.05, paired t test (fast versus fed).
(B and C) At 18 weeks post-transplantation, human C-peptide levels were measured during an i.p. glucose tolerance test (ipGTT). In (B) data are normalized to baseline levels, and in (C) raw levels (ng/ml) are presented for individual animals, with each diet group shown on a separate plot. ∗p < 0.05, one-way repeated-measures ANOVA (versus time 0).
(D–F) At 24 weeks post-transplantation, an i.p. arginine tolerance test (ipArgTT) was performed. Plasma was collected after a 4-hr fast and 15 min following arginine administration to measure human insulin (D) and glucagon (E and F) levels. (E) shows glucagon levels at 0 and 15 minutes in transplant recipients, and (F) shows glucagon levels in sham-treated mice (Sham, striped bars) and transplant recipients (Tx, solid bars) at 15 minutes only. The red line indicates the lower limit of detection for the glucagon assay. (D and E) ∗p < 0.05, one-tailed paired t test (0 versus 15 min); (F) ∗p < 0.05, two-tailed t test (sham versus Tx). Data points from individual mice are shown as box-and-whisker plots.
See also Table S2.
hESC-Derived Endocrine Cells Were Similar following LFD or HFD Exposure, but Grafts from HFD-Fed Mice Contained More Polyhormonal Cells
At 29 weeks post-transplantation, hESC-derived grafts had similar or significantly higher levels of islet-related genes compared with human islets and there were no significant differences between grafts from mice fed LFDs or HFDs (Figure 3). The majority of cells within the harvested devices were immunoreactive for the endocrine marker synaptophysin, and a small proportion expressed the ductal marker CK19. Trypsin-positive exocrine cells were rarely observed (Figure 4A). The grafts were largely composed of cells expressing insulin, glucagon, or somatostatin (Figures 4B and 4E), and the percentage of mono-hormonal insulin-positive and glucagon-positive cells was similar between diet groups (Figure 4C). However, we did note a minor but significantly higher percentage of cells that were immunoreactive for both insulin and glucagon in the HFD grafts compared with LFD grafts (Figures 4C and 4D). Aside from these rare polyhormonal cells, exposure to HFDs did not appear to generally influence the maturation state of hESC-derived insulin-secreting cells: the majority of insulin-positive cells in all transplant recipients co-expressed PDX1 (Figure 4F), NKX2.2 (Figure 4G), NKX6.1 (Figure 4H), and MAFA (Figure 4I) at 29 weeks post-transplantation.
Figure 3.
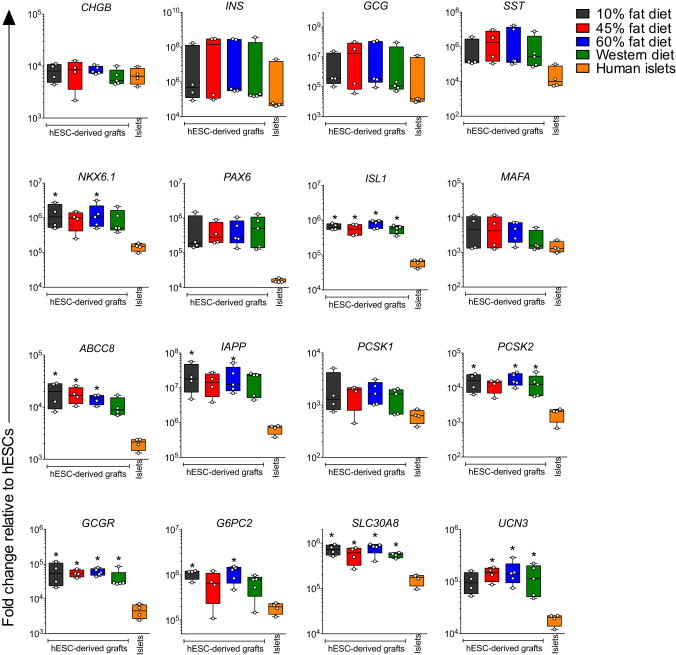
Gene Expression Profiles Are Similar for hESC-Derived Graft Tissues Exposed to Different LFDs or HFDs In Vivo
The gene expression of islet-related genes was assessed in macroencapsulated hESC-derived grafts at 29 weeks post-transplantation from mice fed a LFD (10% fat) or HFD (45% fat, 60% fat, and Western), as well as in adult human islet preparations. All data are presented as the fold change relative to undifferentiated hESCs (H1 cells) using a log scale. Data points from individual mice or different human islet donors are shown as box-and-whisker plots. ∗p < 0.05 for hESC-derived grafts from each diet group versus human islets (one-way ANOVA). See also Tables S2 and S3.
Figure 4.
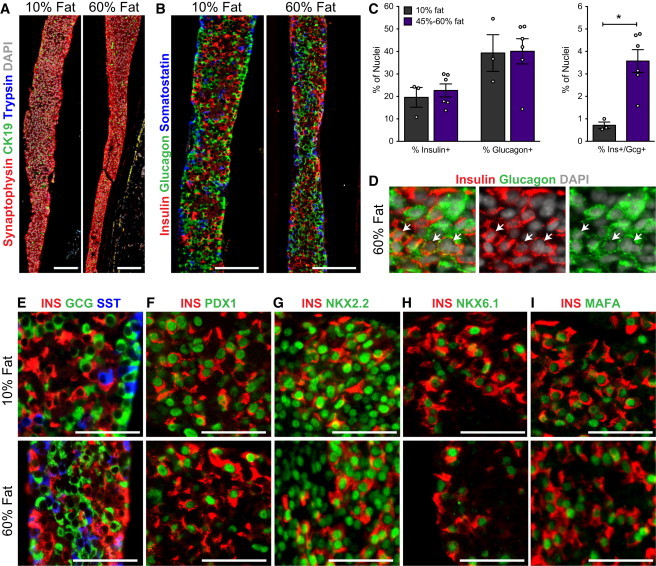
The Morphology of Macroencapsulated hESC-Derived Pancreatic Endocrine Cells Is Similar in Grafts from Mice Fed a LFD or HFD
(A, B, and D–I) Representative immunofluorescent images of Theracyte devices at 29 weeks post-transplantation from mice fed 10% fat or 60% fat diets.
(A) The majority of hESC-derived cells within devices from both diet groups were endocrine cells. The expression of synaptophysin (endocrine marker, red), CK19 (ductal marker, green), trypsin (exocrine marker, blue), and DAPI (nuclear marker, white) is shown. Scale bars, 100 μm.
(B) The endocrine compartment was mainly composed of cells expressing either insulin (red, guinea pig antibody), glucagon (green, rabbit antibody), or somatostatin (blue, Ms antibody); scale bars, 100 μm. Higher-magnification images are shown in (E); scale bars, 50 μm.
(C) The percentage of cells (% of DAPI+ nuclei) within devices that were immunoreactive for insulin (insulin+), glucagon (glucagon+), or both hormones (ins+/gcg+) was quantified in grafts from mice fed 10% fat or 45%–60% fat diets. ∗p < 0.05, two-tailed t test. Data are represented as mean ± SEM and data points from individual mice are also shown.
(D) Example of graft tissue from the 60% fat diet group with a region of endocrine cells that expressed both insulin and glucagon (white arrows); scale bars, 25 μm.
(F–I) The majority of insulin-positive cells (INS, red) show a mature beta cell phenotype, including co-expression of key beta cell transcription factors (green): (F) PDX1, (G) NKX2.2, (H) NKX6.1, and (I) MAFA; scale bars, 50 μm. See also Figure S2 and Tables S2 and S4.
hESC-Derived Insulin-Secreting Cells Improved Diet-Induced Dysglycemia and Insulin Resistance
Long-term tracking revealed that mice in all HFD groups continued to be overweight (Figure S3A) and hyperglycemic under fasting conditions (Figure S3E) compared with LFD controls throughout the duration of the study. Transplantation of hESC-derived cells did not affect either BW (Figures S3B–S3D) or fasting blood glucose levels (Figures S3F–S3H) compared with sham surgery. However, we did observe significant improvements in long-term glycemic control, as measured by HbA1C, following transplantation (Figures 5A and 5B). HbA1C levels were elevated at 12 and 24 weeks in all HFD sham mice compared with LFD sham controls, and were significantly reduced by transplantation in the 45%–60% fat group at both ages (Figures 5A and 5B). Transplant recipients on 45%–60% fat diets also displayed a significantly lower glucose excursion following a mixed-meal stimulus compared with sham mice at 20 weeks (Figure 5C), and all HFD transplant recipients had significantly improved glucose tolerance at 24 weeks post-transplantation (Figures 5E and S4B). These improvements were not yet evident at 18 weeks (Figures 5D and S4A). Glucose tolerance in the 45%–60% fat group was not completely ameliorated at 24 weeks, but the area under the curve for transplant recipients in the Western group was indistinguishable from that obtained for controls (Figures 5E and S4B). Interestingly, we also observed a modest but statistically significant improvement in insulin sensitivity at 22 weeks in transplanted HFD-fed mice compared with shams (Figures 5F, S4C, and S4D), which may have contributed to the improved glucose tolerance in HFD transplant recipients (Figure 5E).
Figure 5.
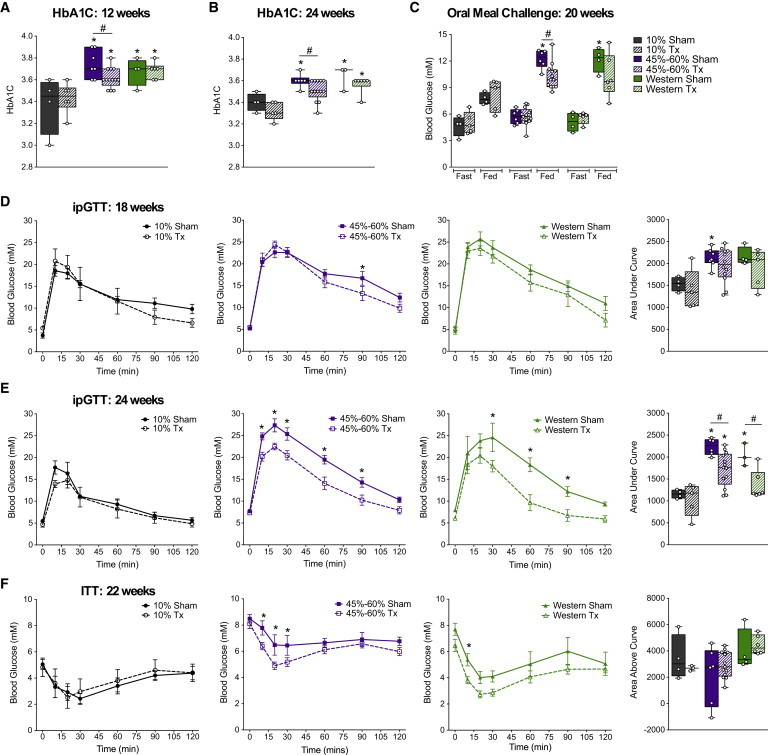
Cell Transplant Recipients on HFDs Show Improved Glucose Homeostasis Compared with Sham-Treated Mice
(A and B) HbA1C levels were measured at 12 (A) and 24 (B) weeks post-transplantation in sham mice (solid bars) and transplant recipients (Tx, striped bars).
(C) At 20 weeks post-transplantation, blood glucose levels were measured after an overnight fast and 40 min following an oral meal challenge (“Fed”).
(D and E) ipGTTs were performed at 18 (D) and 24 (E) weeks post-transplantation in sham mice (solid lines with closed symbols; solid bars) and transplant recipients (Tx, dashed lines with open symbols; striped bars) on 10% fat (gray; 18 weeks: Tx/Sham, n = 4 mice; 24 weeks: Tx, n = 5 and Sham, n = 4 mice), 45% or 60% fat (purple; 18 weeks: Tx, n = 12 and Sham, n = 7 mice; 24 weeks: Tx, n = 13 and Sham, n = 6 mice), or Western (green; 18 weeks: Tx, n = 5 and Sham, n = 4 mice; 24 weeks: Tx, n = 6 and Sham, n = 3 mice) diets. The area under the curve is shown to the right for each ipGTT.
(F) An ITT was performed at 22 weeks post-transplantation in sham and transplant recipients from each diet group (10% Tx, n = 5 mice; 10% Sham, n = 4 mice; 45%–60% Tx, n = 12 mice; 45%–60% Sham, n = 6 mice; West Tx, n = 6 mice; West Sham, n = 4 mice). The area above the curve (right panel) was calculated using the fasting glucose level (100%) for each animal as the baseline. For clarity, all glucose curves (GTTs and ITTs) from sham and transplanted mice are shown separately for each diet group. See Figure S4 for glucose curves combined on the same plots. Data are represented as mean ± SEM (line graphs) or as box-and-whisker plots showing individual mice as separate data points. For all box-and-whisker plots: ∗p < 0.05, one-way ANOVA versus 10% sham; #p < 0.05, two-tailed t test (Tx versus sham). For all line graphs: ∗p < 0.05, two-way ANOVA, Tx versus sham. See Figure S3 for long-term body weight and blood glucose tracking, and Figure S5 for the effect of cell transplants on endogenous pancreas, liver, fat, and circulating leptin levels.
See also Table S2.
We assessed beta and alpha cell mass in the endogenous pancreas from mice fed 10% or 60% fat diets to determine whether the improved glucose tolerance in HFD-fed transplant recipients could be accounted for by expansion of the endogenous endocrine pancreas. Beta cell mass was significantly higher in all mice on 60% fat diets compared with LFD sham controls, and there was no difference between sham and transplanted mice in either diet group (Figures S5A and S5D). Interestingly, there was no effect of HFDs on alpha cell mass, but a significant reduction in alpha cell mass was observed in LFD transplant recipients compared with LFD shams (Figures S5B and S5D). There were no significant differences in the ratio of insulin-positive to glucagon-positive area in the pancreas of mice on either diet (Figure S5C).
Cell Therapy Had No Effect on the Obesity Phenotype, but Improved Liver Weight in HFD Mice
Although hESC-derived cells improved glucose homeostasis in HFD-fed mice, there was no apparent effect on the obesity phenotype. At the end of our study (29 weeks post-transplantation and 36 weeks post-diet), mice on the 45%–60% fat diets (sham and Tx) had significantly higher BW, adiposity (epididymal fat pad weight as a proportion of BW) and circulating leptin levels than LFD shams (Figures S5E–S5G). The obesity phenotype was more subtle in Western-diet mice during the first 7 weeks (Figure 1A) and by the end of the study there were no significant differences in BW, adiposity, or leptin levels between Western-fed mice (sham and Tx) and LFD sham controls (Figures S5E–S5G). All HFD groups had significantly higher liver weight (Figure S5H) and evidence of cytoplasmic vacuolation, consistent with dietary lipidosis in the liver (Figure S5I; Western and 45% fat groups not shown) compared with LFD controls. Interestingly, transplant recipients fed 45%–60% fat diets had significantly reduced liver weight relative to shams (Figure S5H), although a pathology assessment did not reveal differences in cytoplasmic vacuolation in H&E-stained liver sections (Figure S5I). Similarly, vacuolation of renal tubular epithelium was observed in kidney sections from all HFD groups (consistent with dietary lipidosis) and there was no effect of cell transplantation on this phenotype (data not shown). Other tissue pathologies (Table S1) were consistent with spontaneous age- and sex-related events and considered to be unrelated to exposure to diets or cell transplants (data not shown).
Combined Treatment of HFD-Fed Mice with Progenitor Cell Transplants and an Antidiabetic Drug Rapidly Improved Diet-Induced Obesity and Glucose Intolerance
Since the progenitor cell therapy resulted in improved glucose tolerance in HFD-fed mice at 24 weeks, but not full reversal of the diabetes phenotype, we next investigated a combination therapy with known antidiabetic drugs. A second cohort of SCID-beige mice was placed on either a LFD (10% fat) or HFD (60% fat) for 6 weeks, followed by long-term administration of either sitagliptin (4 g/kg in 60% fat diet), rosiglitazone (18 mg/kg in 60% fat diet), or metformin (1.25 mg/ml in drinking water) in HFD-fed mice (treatment groups are summarized in Table S2). Similar to our first cohort, SCID-beige mice in this second cohort rapidly developed attributes of type 2 diabetes following HFD administration (Figure S6).
At the time of transplantation (1 week after drug administration), all HFD-fed mice were significantly heavier than LFD controls (Figure 6F). Interestingly, weight loss was observed within the first 2 weeks following transplantation in HFD-fed mice on antidiabetic drugs (Figures 6C–6E). In contrast, no change in BW was observed during this time in either HFD transplant recipients without drug treatment (Figure 6B) or sham mice on any drug (Figures 6A–6E). All transplant recipients that received antidiabetic drugs had significantly lower BW on day 75 (Figure 6F) and reduced epididymal fat pad weight (relative to BW; Figure 6G) compared with sham mice, such that neither parameter was different from LFD-fed sham controls. As in our previous cohort (Figures S3 and S5E), there was no effect of transplantation on BW (Figures 6B and 6F) or circulating leptin levels (Figure 6H) in HFD-fed mice without drug treatment, although we did observe a reduction in relative epididymal fat pad weight in this cohort (Figure 6G). Interestingly, the combination of a cell transplant with either metformin or sitagliptin resulted in significantly reduced circulating leptin levels in the transplant recipients compared with their respective sham controls (Figure 6G). The cell therapy had no effect on restoring leptin levels in the rosiglitazone group (Figure 6G).
Figure 6.
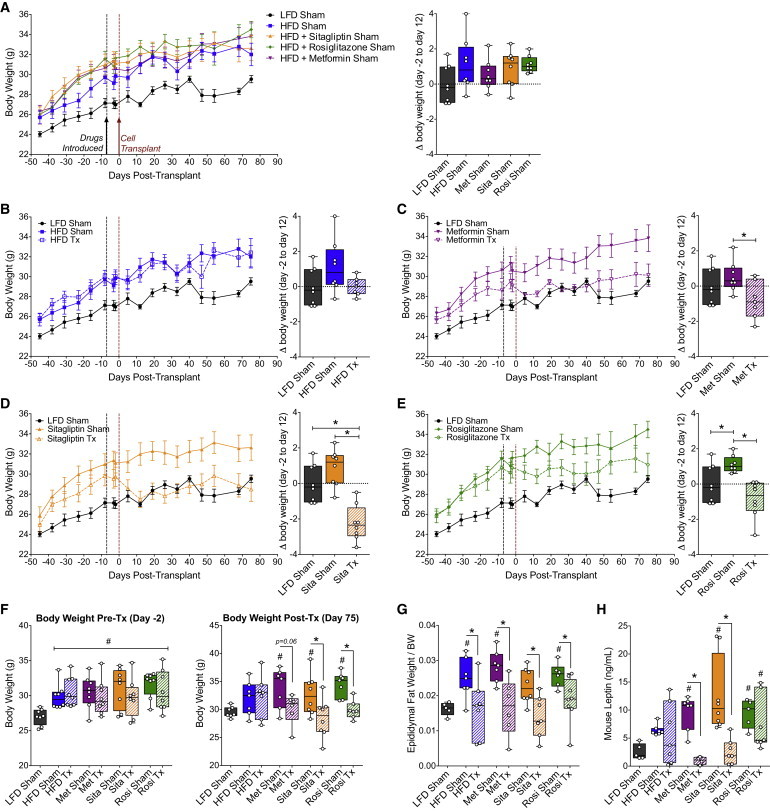
Diet-Induced Obesity Was Reversed Following Progenitor Cell Transplantation Combined with an Antidiabetic Drug
(A–F) Fasting body weight was assessed in mice fed a 10% fat diet without drug (black/gray; all panels; n = 8 mice), 60% fat diet without drug (A and B, blue; n = 7–8 mice per group), 60% fat diet plus metformin (A and C, purple; n = 7–8 mice per group), 60% fat diet plus sitagliptin (A and D, orange; n = 8 mice per group), or 60% fat diet plus rosiglitazone (A and E, green; n = 8 mice per group). Body weight tracking for sham mice from all treatment groups is shown in (A). Sham mice (solid lines, closed symbols) and transplant recipients (Tx; dashed lines, open symbols) from each treatment group are shown together with the LFD control as a reference (B–E). The change in body weight from day −2 to day 12 is shown in box-and-whisker plots to the right of each line graph, with each data point representing an individual mouse. Data on line graphs are represented as mean ± SEM. (F) Body weight pre-transplant (day −2) and post-transplantation (day 75).
(G and H) Epididymal fat pad weight relative to body weight (G) and plasma mouse leptin levels (H) were assessed at 20 weeks post-transplantation.
(A–E) ∗p < 0.05, one-way ANOVA for comparisons of all groups (LFD versus HFD Sham, LFD versus HFD Tx, HFD Sham versus HFD Tx); (F–H) #p < 0.05, one-way ANOVA for comparison of each group to LFD control; ∗p < 0.05, two-tailed t test, Tx versus Sham. See Figure S6 for the phenotype of mice on HFDs in cohort 2 (pre-transplant) and Figure S7 for long-term blood glucose tracking following administration of drugs with or without cell transplants.
Fasting blood glucose levels were not affected by any of the combination therapies throughout the study duration (Figure S7). At 12 weeks post-transplantation, mice in all HFD sham groups were glucose intolerant compared with LFD controls, regardless of the drug treatment used (Figure 7A). As expected based on our previous cohort, we found no effect of the cell therapy on glucose tolerance at 12 weeks post-transplantation in the HFD-fed mice without drug treatment (Figure 7B), and likewise, the combination with rosiglitazone was also ineffective at this time (Figure 7E). Interestingly, the cell therapy significantly improved glucose tolerance at 12 weeks post-transplantation when combined with either metformin (Figure 7C) or sitagliptin (Figure 7D) treatment. In fact, glycemic control during an oral glucose challenge was indistinguishable between the LFD controls and HFD-fed mice receiving sitagliptin with the cell therapy, with the exception of a marginally higher peak glucose level at 15 min post-gavage (Figure 7D). The improved glucose tolerance in cell transplant recipients from the metformin- and sitagliptin-treated groups was associated with significantly reduced fasting mouse C-peptide levels compared with their respective sham controls at 16 weeks post-transplantation (Figure 7G), an effect that was not yet evident at 4 weeks (Figure 7F). Interestingly, the improvements in glucose tolerance were not associated with differences in glucose-stimulated C-peptide secretion by hESC-derived grafts. All transplant recipients showed robust glucose-responsive human C-peptide secretion at 16 weeks, and there were no differences in human C-peptide levels between HFD-fed mice treated with different antidiabetic drugs (Figure 7H).
Figure 7.
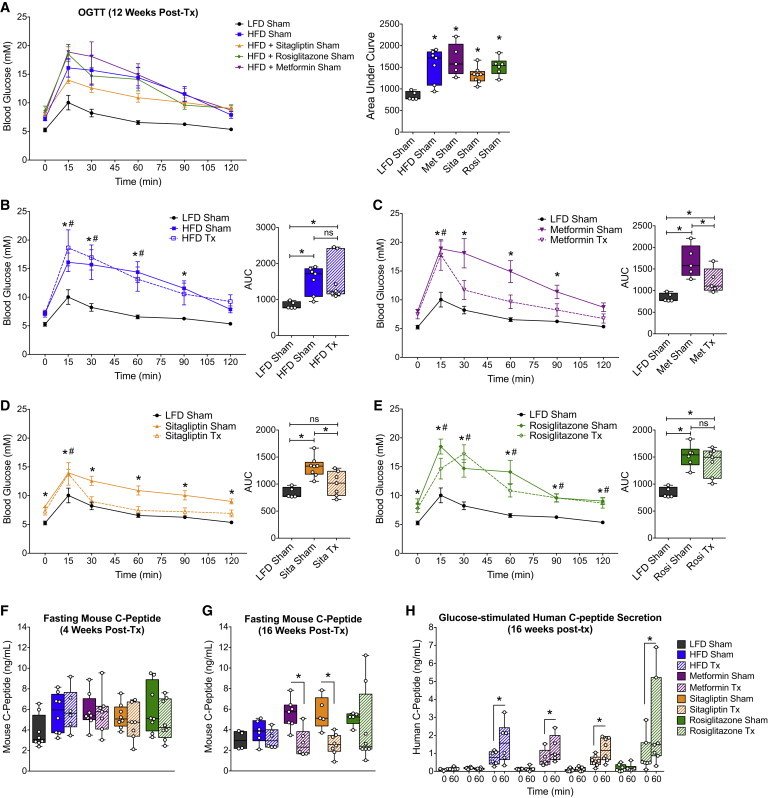
The Combination of Either Sitagliptin or Metformin with a Cell Transplant Resulted in Improved Glucose Tolerance within 12 Weeks
(A–E) OGTTs were performed at 12 weeks post-transplantation in mice fed a 10% fat diet without drug (black/gray; all panels; Sham: n = 5 mice), 60% fat diet without drug (A and B, blue; Tx/Sham: n = 7 mice), 60% fat diet plus metformin (A and C, purple; Tx/Sham: n = 5 mice), 60% fat diet plus sitagliptin (A and D, orange; Tx: n = 7 mice, Sham: n = 8 mice), or 60% fat diet plus rosiglitazone (A and E, green; Tx: n = 7 mice, Sham: n = 6 mice). Blood glucose levels for sham mice from all treatment groups are shown together in (A). Sham mice (solid lines, closed symbols) and transplant recipients (Tx; dashed lines, open symbols) from each treatment group are shown together with LFD controls as a reference (B–E). The area under the curve (AUC) is shown in box-and-whisker plots to the right of each line graph, with separate data points representing individual mice. Data on line graphs are represented as mean ± SEM. (A) ∗p < 0.05 (one-way ANOVA versus LFD).
(B–E) For glucose curves, two-way ANOVA was used to examine differences at each time point during the OGTT (∗p < 0.05, HFD Sham versus LFD; #p < 0.05, HFD Tx versus LFD). For the AUC, one-way ANOVA was used compare all groups (LFD versus HFD Sham, LFD versus HFD Tx, HFD Sham versus HFD Tx; ∗p < 0.05; ns, not significant).
(F and G) Fasting mouse C-peptide levels at 4 weeks (F) or 16 weeks (G) post-transplantation. ∗p < 0.05, two-tailed t test (Tx versus Sham).
(H) Human C-peptide levels were measured after an overnight fast and 60 min following an intraperitoneal glucose challenge. ∗p < 0.05, paired two-tailed t test (0 versus 60 min).
Discussion
Our first goal in these studies was to establish an immunodeficient mouse model of hyperglycemia associated with insulin resistance and obesity that would resemble a type 2 diabetes phenotype. It was not feasible to transplant human progenitor cells into traditional models of type 2 diabetes, such as db/db or ob/ob mice, because macroencapsulation devices are unlikely to protect human cells from xenograft immune rejection (Brauker et al., 1996; Mckenzie et al., 2001). Although macroencapsulation was not required to protect the cells in immunodeficient mice, we opted to transplant within immunoisolation devices so that our findings would be more clinically relevant (macroencapsulated hESC-derived progenitor cells are currently being evaluated in patients with diabetes by ViaCyte [ClinicalTrials.gov, Identifier: NCT02239354]). We used SCID-beige mice for our studies because the maturation of hESC-derived pancreatic progenitor cells has been well characterized in this strain (Rezania et al., 2012, 2013; Bruin et al., 2013). SCID-beige mice fed 45%–60% fat diets had many characteristics of type 2 diabetes, including increased BW and adiposity, fasting hyperglycemia, and glucose intolerance, insulin resistance, hyperleptinemia, and hepatic lipidosis. However, unlike humans with type 2 diabetes, mice fed 45% or 60% fat diets had reduced circulating free fatty acid and triglyceride levels. We speculate that excess lipids are likely taken up by peripheral tissues, which may explain the severe ectopic lipid deposition observed in the liver and kidneys of these mice. Western-diet-fed mice were similar with respect to glucose intolerance and hepatic/kidney lipidosis, but showed a more mild phenotype in terms of obesity, fasting hyperglycemia, insulin resistance, and leptin levels. We also observed pancreatic beta cell dysfunction in the 60% fat and Western-diet groups, as indicated by the lack of glucose-stimulated insulin secretion. Therefore, although no one diet produced a phenotype that perfectly modeled the human condition, all three HFDs resulted in characteristics that mirrored important features of type 2 diabetes, most notably including hyperglycemia associated with obesity and insulin resistance. The mechanism underlying the rapid HFD-induced metabolic dysregulation in mice lacking T cells, B cells, and fully functioning macrophages remains to be determined and is intriguing given that immune cells reportedly play a central role in the development of obesity-associated inflammation and subsequent insulin resistance (Feuerer et al., 2009; Nishimura et al., 2009; Winer et al., 2009, 2011). Notably, in contrast to ob/ob mice, we did not observe infiltration of F4/80-positive macrophages in the epididymal fat tissue of HFD-fed SCID-beige mice, suggesting that other mechanisms may be involved in the development of insulin resistance in this animal model.
Transplantation of human pancreatic progenitor cells into HFD-fed mice resulted in improved glycemic control over time. At 12 weeks post-transplantation, mice on 45%–60% fat diets and with hESC-derived grafts had significantly lower HbA1C levels than sham mice on the same diet, indicative of improved long-term glycemic control, although these mice remained glucose intolerant at 18 weeks. As the progenitor cells matured over time, circulating human C-peptide levels increased, engrafted cells became glucose/arginine responsive, and significant improvements were observed in glucose excursions following mixed-meal and glucose challenges in HFD transplant recipients compared with HFD shams between 20 and 24 weeks post-transplantation. We suspect that the addition of an alternative source of appropriately regulated insulin secretion provided sufficient compensation for peripheral insulin resistance, a feat that the endogenous pancreas was incapable of achieving on its own. Transplantation did not affect the pancreatic beta cell mass, so it is unlikely that expansion of the endogenous pancreas contributed to the observed improvements in glucose homeostasis in HFD-fed mice. Interestingly, transplant recipients from the 45%–60% diet group had enhanced insulin sensitivity compared with shams, which may also have contributed to their improved glucose tolerance. An improvement in insulin sensitivity as a result of higher circulating insulin levels is counterintuitive, but consistent with previous reports in patients with type 1 diabetes following human islet transplantation (Rickels et al., 2013) and patients with type 2 diabetes following short-term intensive insulin therapy (Kramer et al., 2013).
Based on these data, we predicted that co-treating HFD transplant recipients with antidiabetic drugs, particularly insulin sensitizers, would improve the efficacy of the progenitor cell therapy. Although co-treatment with metformin, an insulin sensitizer from the biguanide class, improved BW and glucose intolerance after just 12 weeks, the combination with rosiglitazone, an insulin sensitizer from the TZD class, did not reverse hyperglycemia. Interestingly, the most effective antidiabetic drug tested in our study was the DPP4 inhibitor sitagliptin, which acts to increase circulating incretin levels by preventing inactivation by DPP4. When sitagliptin was combined with the cell therapy, HFD-fed mice had glucose tolerance and BW that was similar to LFD-fed control mice at 12 weeks post-transplantation. Furthermore, transplanted mice on either sitagliptin or metformin had reduced fasting mouse C-peptide levels, which may suggest that the endogenous pancreas has been relieved of the demand to compensate for insulin resistance. The reversal of hyperleptinemia in both sitagliptin- and metformin-treated transplant recipients also suggests a more robust reversal of the obesity phenotype in these groups as compared with the HFD transplant recipients treated with rosiglitazone or without drugs. Taken together, our data suggest that a cell-based insulin replacement therapy could be considered as a future treatment option for type 2 diabetes, particularly if combined with antidiabetic drugs.
Many of the same pathways that trigger obesity-associated beta cell dysfunction in patients with type 2 diabetes may also contribute to graft failure following islet transplantation (Potter et al., 2014). Therefore, a potential caveat in replacing dysfunctional beta cells with healthy insulin-secreting cells is the risk of subsequent graft failure in an obesogenic and hyperglycemic host environment. Indeed, in previous studies, both human and rodent islet grafts displayed beta cell dysfunction following exposure to HFDs (Hiramatsu and Grill, 2001; Gargani et al., 2013) and chronic hyperglycemia (Eizirik et al., 1997; Biarnés et al., 2002). In contrast, the development of hESC-derived pancreatic progenitor cells into insulin-secreting cells was accelerated by exposure to chronic hyperglycemia compared to normoglycemia (Bruin et al., 2013). However, the potential effect of an obesogenic environment on hESC-derived progenitor cells was not previously examined. We also demonstrated previously that short-term exposure to exendin-4, a GLP-1 mimetic, for 4 weeks post-transplantation did not affect the development of progenitor cells (Bruin et al., 2013), but the possible effects of long-term treatment with antidiabetic medications has not been reported. In the current studies there was a tendency for increased basal glucagon levels and arginine-induced human insulin secretion in HFD-fed compared with LFD-fed transplant recipients, but overall the results showed that neither an obesogenic environment nor exposure to antidiabetic drugs had a major impact on the function of hESC-derived insulin-secreting cells in vivo. However, there are several important caveats to our immunocompromised mouse model that should be considered: first, we cannot exclude the possibility that HFDs or antidiabetic drugs may impact the function of hESC-derived endocrine cells in an immunocompetent host environment; second, human progenitor cells may respond differently to mouse versus human diabetogenic/obesogenic stimuli; and third, the potential long-term impact of HFDs or antidiabetic drugs on hESC-derived grafts cannot be examined in mice because of their relatively short lifespan. Regardless, hESC-derived cells secreted similar levels of human C-peptide in response to secretagogue challenges in all diet groups (with or without antidiabetic drugs), and qPCR demonstrated no significant differences in expression of several islet-related genes within grafts from different diet or drug treatment groups. The only sign that an obesogenic environment may have impacted progenitor cell maturation was a small but significant increase in the percentage of insulin/glucagon bihormonal cells in grafts from mice fed 45%–60% fat versus 10% fat diets. These cells may represent a subpopulation of immature, fetal-like endocrine cells (Riedel et al., 2012; Bruin et al., 2014), but without conducting lineage-tracing studies, we cannot know their source or fate. Regardless, the ability of hESC-derived progenitor cells to thrive and mature into functional pancreatic endocrine cells in a chronic obesogenic and hyperglycemic (albeit immunocompromised) environment further supports the feasibility of a stem cell-based therapy for type 2 diabetes.
In our previous studies, a dose of 5 million progenitor cells was sufficient to normalize hyperglycemia in mice with severe insulin deficiency caused by streptozotocin-induced beta cell destruction (Rezania et al., 2012, 2013; Bruin et al., 2013). With diet-induced hyperglycemia, the same dose of progenitor cells generated higher levels of human C-peptide, yet the graft-derived insulin (combined with endogenous mouse insulin) was insufficient to completely normalize glycemia. Therefore, it is likely that a larger mass of insulin-producing cells would be required to ameliorate hyperglycemia in insulin-resistant patients with type 2 diabetes compared with insulin-deficient patients with type 1 diabetes. Alternatively, our studies also suggest that a progenitor-based cell therapy may be more effective in a type 2 diabetic setting if it is combined with an antidiabetic drug. These findings are in line with another report by our lab, in which leptin therapy was shown to enhance the performance of transplanted islets in diabetic mice (Denroche et al., 2013), likely due to the insulin-sensitizing effects of leptin (Denroche et al., 2011).
Conclusions
In conclusion, we have demonstrated that hESC-derived pancreatic progenitor cells may be a feasible option for treating patients with type 2 diabetes, in addition to those with type 1 diabetes. Our data suggest that transplanted hESC-derived insulin-producing cells thrive following chronic exposure to HFDs, at least in immunodeficient mice. Thus, stem cells are a candidate for restoring functional beta cells in an insulin-resistant, obese setting.
Experimental Procedures
In Vitro Differentiation of hESCs and Assessment of Pancreatic Progenitor Cells
The H1 hESC line was obtained from the WiCell Research Institute. All experiments at the University of British Columbia (UBC) with H1 cells were approved by the Canadian Stem Cell Oversight Committee and the UBC Clinical Research Ethics Board. Pluripotent H1 cells were differentiated into pancreatic progenitor cells according to our 14-day, four-stage protocol as previously described (Bruin et al., 2013). Expression of key pancreatic progenitor cell markers was assessed prior to transplantation using custom Taqman qPCR Arrays (Applied Biosystems) and flow cytometry as described in Supplemental Experimental Procedures.
Animals
Male SCID-beige mice (C.B-Igh-1b/GbmsTac-Prkdcscid-LystbgN7, 8–10 weeks old; Taconic) were maintained on a 12 hr light/dark cycle throughout the study. All experiments were approved by the UBC Animal Care Committee and carried out in accordance with the Canadian Council on Animal Care guidelines.
Diets and Drug Administration
All mice were given ad libitum access to a standard irradiated diet (Harlan Laboratories, Teklad Diet #2918) for 2 weeks to allow for acclimatization following their arrival at UBC. In the first cohort, mice were placed on one of four different diet regimens (Research Diets) for the 36-week study (n = 11 per diet): (1) “10% fat” control diet (D12450K, 10 kcal% fat, 70 kcal% carbohydrate [no sucrose]), (2) “45% fat” diet (D12451, 45 kcal% fat [primarily lard], 35 kcal% carbohydrate), (3) “60% fat” diet (D12492, 60 kcal% fat [primarily lard], 20 kcal% carbohydrate), or (4) “Western” diet (D12079B, 41 kcal% fat [primarily milk fat], 43 kcal% carbohydrate [primarily sucrose]).
In the second cohort, mice were placed on either the 10% fat control diet (D12450K; n = 8) for the duration of the study or 60% fat diet (D12492; n = 64) for 6 weeks, followed by one of the following treatment regimens for the remainder of the study (n = 16 per group): (1) 60% fat diet with no drug (D12492), (2) 60% fat diet containing rosiglitazone (18 mg/kg diet or ∼3 mg/kg BW per day; Cayman Chemical; Research Diets custom diet formulation D08121002), (3) 60% fat diet containing sitagliptin (4 g/kg diet or ∼750 mg/kg BW per day; sitagliptin phosphate monohydrate, BioVision; Research Diets custom diet formulation D08062502R), or (4) 60% fat diet (D12492) and metformin (1,1-dimethylbiguanide hydrochloride) in drinking water (1.25 mg/ml or ∼250 mg/kg BW per day).
Transplantation of hESC-Derived Pancreatic Progenitor Cells
The procedure used for transplantation of macro-encapsulated pancreatic progenitor cells is described in Supplemental Experimental Procedures. In the first cohort, mice were randomly assigned to receive either a cell transplant (Tx, n = 7 per diet) or sham surgery (sham, n = 4 per diet) after 7 weeks of LFD or HFD feeding. In the second cohort, HFD-fed mice (+/− drug treatment) received either a transplant (n = 8 per group) or sham surgery (n = 8 per group) 1 week after administration of the antidiabetic drugs. LFD controls all received sham surgery. The treatment groups are summarized in Table S2.
Metabolic Assessments
All metabolic analyses were performed in conscious, restrained mice and blood samples were collected via the saphenous vein. BW and blood glucose levels were assessed regularly throughout each study following a 4-hr morning fast. For all other metabolic tests, blood was collected after fasting (time 0) and at the indicated time points following administration of various secretagogues. Details about the fasting time, route of administration, and dose used for each metabolic test are provided in Supplemental Experimental Procedures along with information about the assays used to measure circulating hormones, lipids, and HbA1C levels.
Dual-Energy X-Ray Absorptiometry
Body composition was determined using dual-energy X-ray absorptiometry (DEXA) with a PIXImus Mouse Densitometer (Inside Outside Sales). Data are expressed as % fat.
qRT-PCR
Theracyte devices were harvested at 29 weeks post-transplantation from cohort 1 and stored for qPCR analysis. Details regarding the qPCR analysis, human islet donors, and the procedure used to isolate RNA from engrafted tissue are described in Supplemental Experimental Procedures. Primers are listed in Table S3.
Immunofluorescent Staining and Image Quantification
Prior to transplantation, a portion of differentiated pancreatic progenitor cells were fixed overnight in 4% paraformaldehyde (PFA) and then embedded in 1% agarose prior to paraffin embedding. In cohort 1, the Theractye devices and a variety of tissues (listed in Table S1) were harvested at 29 weeks post-transplantation, fixed in 4% PFA, and stored in 70% EtOH prior to paraffin embedding. All paraffin sections (5 μm thick) were prepared by Wax-it Histology Services. Immunofluorescent staining was performed as previously described (Rezania et al., 2011) and details about the primary antibodies are provided in Table S4. The procedures used for image analysis and quantification are described in Supplemental Experimental Procedures. H&E staining was performed according to standard procedures and tissue analysis was performed in a blinded fashion by an independent pathologist (Nova Pathology PC).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software (GraphPad Software). Details about specific statistical tests are described in Supplemental Experimental Procedures. For all analyses, p < 0.05 was considered statistically significant. Data are presented as the mean ± SEM (line graphs) or as box-and-whisker plots showing individual data points.
Author Contributions
J.E.B. wrote the manuscript. J.E.B., A.R., and T.J.K. contributed to the conception and design of experiments. J.E.B., N.S., N.B., M.M., J.K.F., A.A., S.O., C.D., D.S.R., V.A.S., and A.R. were responsible for acquisition, analysis, and interpretation of data. All authors contributed to manuscript revisions and approved the final version of the manuscript.
Acknowledgments
We thank Subashini Karunakaran and Dr. Susanne Clee for their technical assistance with DEXA. This work was funded by the Canadian Institutes of Health Research (CIHR) Regenerative Medicine and Nanomedicine Initiative, Stem Cell Network (SCN), Juvenile Diabetes Research Foundation (JDRF), and Stem Cell Technologies. J.E.B. and M.M. were funded by JDRF postdoctoral fellowships and the CIHR Transplantation Training Program. J.E.B. was also the recipient of a CIHR postdoctoral fellowship and L’Oréal Canada for Women in Science Research Excellence Fellowship. N.S. and C.D. received funding from the Stem Cell Network, and N.B. was the recipient of a DAAD RISE worldwide international research internship. A.R. is an employee and shareholder of Janssen R&D, LLC. D.S.R. and V.A.S. are employees of Janssen R&D, LLC. T.J.K. received financial support from Janssen R&D, LLC for the research described in this article.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
References
- Biarnés M., Montolio M., Nacher V., Raurell M., Soler J., Montanya E. Beta-cell death and mass in syngeneically transplanted islets exposed to short- and long-term hyperglycemia. Diabetes. 2002;51:66–72. doi: 10.2337/diabetes.51.1.66. [DOI] [PubMed] [Google Scholar]
- Brauker J., Martinson L.A., Young S.K., Johnson R.C. Local inflammatory response around diffusion chambers containing xenografts. Nonspecific destruction of tissues and decreased local vascularization. Transplantation. 1996;61:1671–1677. doi: 10.1097/00007890-199606270-00002. [DOI] [PubMed] [Google Scholar]
- Bruin J.E., Rezania A., Xu J., Narayan K., Fox J.K., O’Neil J.J., Kieffer T.J. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia. 2013;56:1987–1998. doi: 10.1007/s00125-013-2955-4. [DOI] [PubMed] [Google Scholar]
- Bruin J.E., Erener S., Vela J., Hu X., Johnson J.D., Kurata H.T., Lynn F.C., Piret J.M., Asadi A., Rezania A., Kieffer T.J. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. (Amst.) 2014;12:194–208. doi: 10.1016/j.scr.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Butler A.E., Janson J., Bonner-Weir S., Ritzel R., Rizza R.A., Butler P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Denroche H.C., Levi J., Wideman R.D., Sequeira R.M., Huynh F.K., Covey S.D., Kieffer T.J. Leptin therapy reverses hyperglycemia in mice with streptozotocin-induced diabetes, independent of hepatic leptin signaling. Diabetes. 2011;60:1414–1423. doi: 10.2337/db10-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denroche H.C., Quong W.L., Bruin J.E., Tudurí E., Asadi A., Glavas M.M., Fox J.K., Kieffer T.J. Leptin administration enhances islet transplant performance in diabetic mice. Diabetes. 2013;62:2738–2746. doi: 10.2337/db12-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik D.L., Jansson L., Flodström M., Hellerström C., Andersson A. Mechanisms of defective glucose-induced insulin release in human pancreatic islets transplanted to diabetic nude mice. J. Clin. Endocrinol. Metab. 1997;82:2660–2663. doi: 10.1210/jcem.82.8.4150. [DOI] [PubMed] [Google Scholar]
- Ferrannini E., Gastaldelli A., Miyazaki Y., Matsuda M., Mari A., DeFronzo R.A. beta-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J. Clin. Endocrinol. Metab. 2005;90:493–500. doi: 10.1210/jc.2004-1133. [DOI] [PubMed] [Google Scholar]
- Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A., Lee J., Goldfine A.B., Benoist C., Shoelson S., Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargani S., Thévenet J., Yuan J.E., Lefebvre B., Delalleau N., Gmyr V., Hubert T., Duhamel A., Pattou F., Kerr-Conte J. Adaptive changes of human islets to an obesogenic environment in the mouse. Diabetologia. 2013;56:350–358. doi: 10.1007/s00125-012-2775-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halban P.A. Cell therapy for type 2 diabetes: is it desirable and can we get it? Diabetes Obes. Metab. 2008;10(Suppl 4):205–211. doi: 10.1111/j.1463-1326.2008.00957.x. [DOI] [PubMed] [Google Scholar]
- Hariri N., Thibault L. High-fat diet-induced obesity in animal models. Nutr. Res. Rev. 2010;23:270–299. doi: 10.1017/S0954422410000168. [DOI] [PubMed] [Google Scholar]
- Hiramatsu S., Grill V. Influence of a high-fat diet during chronic hyperglycemia on beta-cell function in pancreatic islet transplants to streptozotocin-diabetic rats. Eur. J. Endocrinol. 2001;144:521–527. doi: 10.1530/eje.0.1440521. [DOI] [PubMed] [Google Scholar]
- International Diabetes Federation . 6th ed. International Diabetes Federation; Brussels, Belgium: 2014. IDF Diabetes Atlas update poster. [Google Scholar]
- Kahn S.E., Hull R.L., Utzschneider K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kahn S.E., Cooper M.E., Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383:1068–1083. doi: 10.1016/S0140-6736(13)62154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalupahana N.S., Moustaid-Moussa N., Claycombe K.J. Immunity as a link between obesity and insulin resistance. Mol. Aspects Med. 2012;33:26–34. doi: 10.1016/j.mam.2011.10.011. [DOI] [PubMed] [Google Scholar]
- Kramer C.K., Zinman B., Retnakaran R. Short-term intensive insulin therapy in type 2 diabetes mellitus: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2013;1:28–34. doi: 10.1016/S2213-8587(13)70006-8. [DOI] [PubMed] [Google Scholar]
- Markan K.R., Naber M.C., Ameka M.K., Anderegg M.D., Mangelsdorf D.J., Kliewer S.A., Mohammadi M., Potthoff M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. 2014;63:4057–4063. doi: 10.2337/db14-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckenzie A.W., Georgiou H.M., Zhan Y., Brady J.L., Lew A.M. Protection of xenografts by a combination of immunoisolation and a single dose of anti-CD4 antibody. Cell Transplant. 2001;10:183–193. [PubMed] [Google Scholar]
- Nishimura S., Manabe I., Nagasaki M., Eto K., Yamashita H., Ohsugi M., Otsu M., Hara K., Ueki K., Sugiura S. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- Osborn O., Olefsky J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012;18:363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- Potter K.J., Westwell-Roper C.Y., Klimek-Abercrombie A.M., Warnock G.L., Verchere C.B. Death and dysfunction of transplanted β-cells: lessons learned from type 2 diabetes? Diabetes. 2014;63:12–19. doi: 10.2337/db12-0364. [DOI] [PubMed] [Google Scholar]
- Rezania A., Riedel M.J., Wideman R.D., Karanu F., Ao Z., Warnock G.L., Kieffer T.J. Production of functional glucagon-secreting α-cells from human embryonic stem cells. Diabetes. 2011;60:239–247. doi: 10.2337/db10-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A., Bruin J.E., Riedel M.J., Mojibian M., Asadi A., Xu J., Gauvin R., Narayan K., Karanu F., O’Neil J.J. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61:2016–2029. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A., Bruin J.E., Xu J., Narayan K., Fox J.K., O’Neil J.J., Kieffer T.J. Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells. 2013;31:2432–2442. doi: 10.1002/stem.1489. [DOI] [PubMed] [Google Scholar]
- Rickels M.R., Kong S.M., Fuller C., Dalton-Bakes C., Ferguson J.F., Reilly M.P., Teff K.L., Naji A. Improvement in insulin sensitivity after human islet transplantation for type 1 diabetes. J. Clin. Endocrinol. Metab. 2013;98:E1780–E1785. doi: 10.1210/jc.2013-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel M.J., Asadi A., Wang R., Ao Z., Warnock G.L., Kieffer T.J. Immunohistochemical characterisation of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia. 2012;55:372–381. doi: 10.1007/s00125-011-2344-9. [DOI] [PubMed] [Google Scholar]
- Ryan E.A., Lakey J.R., Rajotte R.V., Korbutt G.S., Kin T., Imes S., Rabinovitch A., Elliott J.F., Bigam D., Kneteman N.M. Clinical outcomes and insulin secretion after islet transplantation with the Edmonton protocol. Diabetes. 2001;50:710–719. doi: 10.2337/diabetes.50.4.710. [DOI] [PubMed] [Google Scholar]
- Shapiro A.M., Lakey J.R., Ryan E.A., Korbutt G.S., Toth E., Warnock G.L., Kneteman N.M., Rajotte R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- Srinivasan K., Ramarao P. Animal models in type 2 diabetes research: an overview. Indian J. Med. Res. 2007;125:451–472. [PubMed] [Google Scholar]
- Svenson K.L., Von Smith R., Magnani P.A., Suetin H.R., Paigen B., Naggert J.K., Li R., Churchill G.A., Peters L.L. Multiple trait measurements in 43 inbred mouse strains capture the phenotypic diversity characteristic of human populations. J. Appl. Physiol. 2007;102:2369–2378. doi: 10.1152/japplphysiol.01077.2006. [DOI] [PubMed] [Google Scholar]
- Tibell A., Rafael E., Wennberg L., Nordenström J., Bergström M., Geller R.L., Loudovaris T., Johnson R.C., Brauker J.H., Neuenfeldt S., Wernerson A. Survival of macroencapsulated allogeneic parathyroid tissue one year after transplantation in nonimmunosuppressed humans. Cell Transplant. 2001;10:591–599. [PubMed] [Google Scholar]
- Weisberg S.P., McCann D., Desai M., Rosenbaum M., Leibel R.L., Ferrante A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J., Li Y., Xu W., Shi L., Zhang Q., Zhu D., Hu Y., Zhou Z., Yan X., Tian H. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet. 2008;371:1753–1760. doi: 10.1016/S0140-6736(08)60762-X. [DOI] [PubMed] [Google Scholar]
- Weyer C., Bogardus C., Mott D.M., Pratley R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer S., Chan Y., Paltser G., Truong D., Tsui H., Bahrami J., Dorfman R., Wang Y., Zielenski J., Mastronardi F. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer D.A., Winer S., Shen L., Wadia P.P., Yantha J., Paltser G., Tsui H., Wu P., Davidson M.G., Alonso M.N. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K.H., Ko S.H., Cho J.H., Lee J.M., Ahn Y.B., Song K.H., Yoo S.J., Kang M.I., Cha B.Y., Lee K.W. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J. Clin. Endocrinol. Metab. 2003;88:2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.