

Summary
Recently developed reprogramming and genome editing technologies make possible the derivation of corrected patient-specific pluripotent stem cell sources—potentially useful for the development of new therapeutic approaches. Starting with skin fibroblasts from patients diagnosed with cystic fibrosis, we derived and characterized induced pluripotent stem cell (iPSC) lines. We then utilized zinc-finger nucleases (ZFNs), designed to target the endogenous CFTR gene, to mediate correction of the inherited genetic mutation in these patient-derived lines via homology-directed repair (HDR). We observed an exquisitely sensitive, homology-dependent preference for targeting one CFTR allele versus the other. The corrected cystic fibrosis iPSCs, when induced to differentiate in vitro, expressed the corrected CFTR gene; importantly, CFTR correction resulted in restored expression of the mature CFTR glycoprotein and restoration of CFTR chloride channel function in iPSC-derived epithelial cells.
Highlights
-
•
We corrected the mutant CFTR gene in cystic fibrosis iPSCs
-
•
Correction restored protein expression and function in iPSC-derived epithelial cells
-
•
We observed an exquisitely sensitive, homology-dependent, allele-preferred targeting
In this article, Davis and colleagues demonstrate site-specific correction of the mutant CFTR gene in cystic fibrosis iPSCs. Interestingly, they observed a highly sensitive, homology-dependent, allele-preferred targeting. When the corrected iPSCs were differentiated in vitro into epithelial cells, CFTR gene correction was shown to result in restoration of the CFTR chloride channel function.
Introduction
Cellular transplantation of lung stem/progenitor cells represents a potential therapeutic approach for a variety of inherited monogenic lung diseases. A patient-specific approach would first involve derivation of autologous induced pluripotent stem cells (iPSCs) from skin or blood cells of affected patients. Utilizing site-specific homology-directed repair (HDR), the disease-causing mutation would then be corrected in the endogenous, chromosomal DNA sequence. Finally, a directed differentiation approach would be employed to obtain highly purified populations of the relevant lung stem/progenitor cells from the corrected iPSCs for the purpose of transplantation.
We focused our initial development of this therapeutic approach on cystic fibrosis (CF). The primary defect in CF, an autosomal recessive disorder, is the regulation of epithelial chloride transport by a chloride channel protein encoded by the CF transmembrane conductance regulator (CFTR) gene (Kerem et al., 1989). Recurrent pulmonary infections are responsible for 80%–90% of the deaths in CF patients. Therefore, transplantation of CFTR-corrected, autologous lung stem/progenitor cells provides an attractive alternative strategy for treating CF.
Here we used zinc-finger nuclease (ZFN)-mediated HDR to edit the endogenous CFTR locus and precisely correct mutations responsible for CF in patient-derived iPSCs.
Results
Correction of CFTR Mutation via ZFN-Mediated HDR in CF iPSCs
Retroviral vectors encoding reprogramming factors (OCT4, SOX2, KLF4, C-MYC, and NANOG) were utilized to reprogram CF primary fibroblasts to iPSCs; the CF fibroblasts and derived iPSCs were compound heterozygous at the CFTR locus, with one allele ΔF508 and the other allele ΔI507 (Figure S1A). CF iPSC clones expressed cellular antigens characteristic of undifferentiated human embryonic stem cells (hESCs), were pluripotent as assayed by teratoma formation, and retained a normal karyotype (Figures S1B–S1D).
The overall strategy for correction of CFTR exon 10 (CFTR legacy exon notation) mutations consisted of delivering CFTR-specific ZFNs together with a selectable CFTR donor DNA (Figure 1A). We designed ZFNs targeting CFTR exon 10, recognizing DNA sequences approximately 110 bp upstream of either the ΔI507 or ΔF508 deletions, to facilitate the correction of either mutant allele by HDR. The CFTR ZFNs were co-delivered with a plasmid encoding the CFTR donor to CF iPSCs. Puromycin-resistant colonies were initially screened via PCR and then sequenced to confirm that CFTR exon 10 was corrected via HDR. Southern blot analysis confirmed that four clones (17-1, 17-9, 17-14, and 17-16) exhibited the expected genomic organization in the corrected CFTR allele without any additional integration of pgk-puroTK sequences (Figure 1B). Sequencing of CFTR genomic DNA exon 10 sequences at targeted (wild-type [WT]) and unmodified (ΔF508) alleles for each of the four corrected clones demonstrated correction of one CFTR allele (ΔI507) per clone (Figure 1C). Transient delivery of a Cre-recombinase expression plasmid resulted in numerous puroTK-excised clones from each of the four successfully edited clones; successful excision was confirmed via PCR analysis with subsequent Cla I digestion (Figures 2A and 2B) and Southern blot analysis (Figure 1B).
Figure 1.
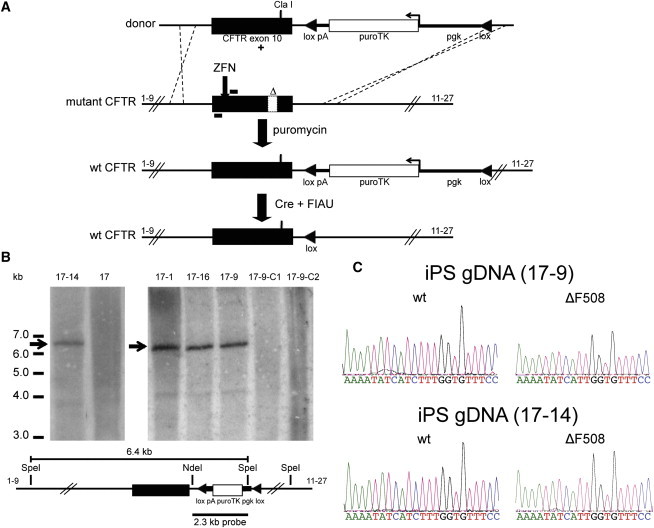
ZFN-Mediated Correction of ΔI507 or ΔF508 CFTR Mutations in CF iPSCs
(A) Outline of methodology involving co-delivery of CFTR-specific ZFNs together with CFTR donor, followed by Cre-recombinase-mediated excision.
(B) The schematic shows the expected genomic organization of a targeted CFTR allele including the WT exon 10 (shown in black) together with the pgk-puroTK selection cassette. A unique 6.4-kb hybridizing band is expected for a correctly modified clone and is apparent in the four corrected clones (17-14, 17-1, 17-16, and 17-9), but absent in the Cre-excised clones and the non-targeted clone 17 CF iPSCs.
(C) Sequence chromatograms of the modified WT and unmodified ΔF508 CFTR alleles from corrected CF iPSC clones.
See also Figure S1.
Figure 2.
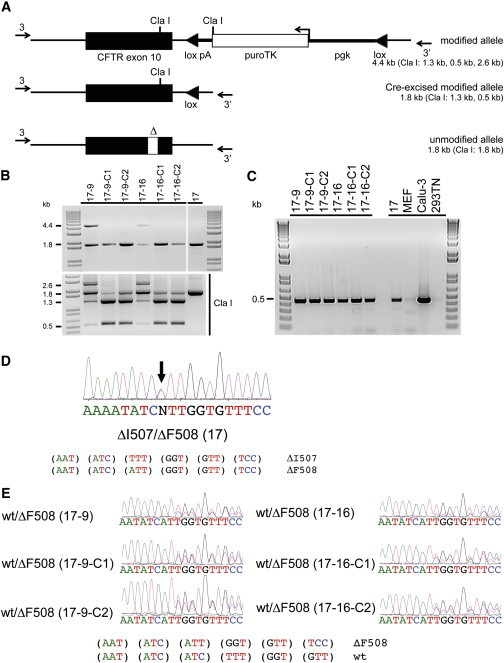
Cre-Mediated Excision of puroTK Cassette from Corrected CF WT/ΔF508 iPSCs
(A) Schematic of the modified allele, before and after Cre-mediated excision, and the unmodified allele. The location of PCR primers (arrows marked 3 and 3′), both located outside of donor sequences, used in verification by amplification are shown. Also indicated are the expected sizes of Cla I digestion products.
(B) (Top) The PCR amplicons for the original targeted clones (17-9 and -16), the Cre-excised clones (17-9-C1 and -C2; 17-16-C1 and -C2), and the original clone 17 CF iPSCs. The presence of only the 1.8-kb band for Cre-excised clones is consistent with successful excision. (Bottom) The results of Cla I digestion of the PCR amplicons. The size of bands for the Cre-excised clones is consistent with successful excision.
(C) RT-PCR analysis of CFTR expression for two targeted CF iPSCs (17-9 and -16) as well as their derived Cre-excised clones. Also shown is CFTR expression by the original clone 17 CF iPSCs and CFTR-expressing Calu-3 cells; mouse embryo fibroblasts (MEFs) and HEK293TN cells are negative controls.
(D) Sequencing of CFTR RT-PCR product from mutant ΔI507/ΔF508 CFTR iPSCs (clone 17) revealed a mixture of ΔI507 and ΔF508 CFTR sequences.
(E) Sequencing of CFTR RT-PCR product from corrected WT/ΔF508 CFTR iPSCs (clones 17-9 and 17-16), together with their Cre-excised derivatives, revealed a mixture of WT and ΔF508 CFTR sequences.
We demonstrated that two of two correctly edited puroTK-excised iPSC clones (17-9-C1 and 17-14-C1) retained pluripotency, as evidenced by teratoma-forming ability (Figures S2A and S2C); furthermore, quantitative transcriptional profiling of 44 genes characteristic of human pluripotent stem cells revealed that both corrected iPSC lines (17-9-C1 and 17-14-C1) exhibited a gene expression pattern highly similar to both the original uncorrected CF iPSCs (clone 17) and hESCs (line WA09) (Figures S2B and S2D).
Karyotypic analysis of 17-9-C1 and 17-14-C1 confirmed retention of a normal karyotype (Figures S2A and S2C). Comparative genomic hybridization (CGH), validated by complete genome sequencing, did not identify any copy-number variations in corrected clones 17-9-C1 and 17-14-C1 not already present in uncorrected clone 17 iPSCs. To assess any off-target effects of ZFN- or Cre-mediated excision methodologies at higher resolution, we also submitted genomic DNA from corrected clones 17-9-C1 and 17-14-C1, as well as from the CF fibroblasts and clone 17 CF iPSCs, for whole-exome and complete genome sequencing. Concordance between exome and whole-genome sequencing was high, validating the variations identified as well as our analysis methodology. We detected only one non-synonymous coding variant (NSCV) unique to clone 17 iPSCs and not present in the original mutant CF fibroblasts. We identified, in comparison with the parental clone 17 iPSCs, two novel NSCVs in 17-9-C1 and eight novel NSCVs in 17-14-C1; these changes consisted entirely of single-base-pair substitutions (Table S1). The DNA sequences flanking these variants were examined to determine whether the DNA variations in the ZFN-corrected iPSCs possibly resulted from non-homologous end joining (NHEJ) at sites of off-target cutting by the ZFNs. Importantly, the NSCV found in the corrected cell lines did not share significant homology to any permutation of ZFN target site (Table S1). Additionally, we did not find any variations that were shared by the two corrected cell lines. Taken together, the type of mutation, the lack of commonality between clones, and the lack of homology to ZFN target sites at/around these changes argue strongly against any ZFN off-target activity in these lines at the whole-exome level. In addition, we interrogated the complete genome sequences of the two corrected iPSC clones (17-9-C1 and 17-14-C1) at the top 100 ranked potential off-target ZFN-binding sequences (representing either homo-dimer or hetero-dimer binding) for any evidence of cleavage (see Supplemental Experimental Procedures). No evidence for NHEJ-induced mutation, either in exon or intron sequences, was identified (Table S2), again arguing against any ZFN off-target activity in these lines.
We similarly applied the ZFN-mediated gene correction methodology to two transgene-free CF iPSC lines homozygous for the ΔF508 mutation. These CF iPSC lines (RC202 and RC204) were originally derived by reprogramming of ΔF508/ΔF508 CF fibroblasts with a Cre-excisable polycistronic lentiviral vector (Somers et al., 2010). A total of six corrected iPSC clones (five from RC202 and one from RC204) were obtained, each of which was corrected at one of the two ΔF508 alleles (i.e., of resulting genotype WT/ΔF508) (data not shown).
Expression of the Corrected CFTR Gene in Gene-Edited iPSCs and iPSC-Derived Cells
We detected low-level CFTR expression in the original, uncorrected ΔI507/ΔF508 clone 17 iPSCs (Figure 2C) and WA09 hESCs (data not shown) by RT-PCR. Similarly RT-PCR analysis for clones 17-1, -9, -14, and -16 yielded a single band of similar size to that seen for clone 17 iPSCs (Figure 2C). Sequencing of the clone 17 iPSC line RT-PCR product demonstrated CFTR mRNA expression arising from both the ΔI507 and ΔF508 alleles (Figure 2D). Sequencing of the RT-PCR product from the four corrected clones, both prior to and following pgk-puroTK excision, confirmed the expected cDNA organization (exons 9–13) and demonstrated CFTR mRNA expression from both the corrected and mutant alleles (ΔF508) (Figure 2E).
We next examined expression of corrected CFTR mRNA and protein under in vitro differentiation conditions previously demonstrated to generate anterior foregut endoderm and primordial lung progenitors from mouse ESC (Longmire et al., 2012) and human ESC/iPSC (Green et al., 2011). In brief, after induction of definitive endoderm, inhibition of BMP/TGF-β signaling with NOGGIN/SB431542 was employed to enrich for anterior foregut endoderm. Subsequent exposure to growth factors implicated in lung development and maturation (WNT3a/KGF/FGF10/BMP4/EGF and retinoic acid) was then used to induce expression of NKX2-1, the earliest marker of commitment of endoderm to either a lung or thyroid epithelial cell fate (Figure 3A). Employing this protocol, mutant CF iPSCs (two independent clones, 17 and 28), corrected CF iPSCs (three clones, 17-9-C1, 17-14-C1, and 17-16-C1, independently obtained from correction of mutant clone 17), and WA09 hESCs efficiently generated definitive endoderm, as evidenced by co-expression of CXCR4 and C-KIT in >90% of cells (Figure S3A) and upregulated expression of endodermal transcriptional regulators, FOXA2 and SOX17 mRNA, by qPCR (data not shown). Further endodermal differentiation, for a total of 19 days in this protocol, subsequently upregulated expression of NKX2-1, SOX9, TP63, FOXP2, and FOXA2, suggesting commitment of at least a sub-population of cells within the endodermal culture to a lung epithelial cell fate (Longmire et al., 2012), and demonstrated increased expression of the CFTR target gene (Figure 3B), as expected from differentiated endodermally derived epithelia. Immunostaining confirmed that a subpopulation of cells co-expressed NKX2-1 and FOXA2 in this directed differentiation protocol (Figure S3B). Importantly, CFTR mRNA expressed on day 19 from differentiated mutant CF iPSCs (clone 17) reflected expression of both mutant ΔI507 and ΔF508 CFTR alleles, whereas differentiation of corrected iPSCs (17-16-C1) revealed co-expression of corrected WT and mutant ΔF508 CFTR mRNAs (Figure S3C). These results indicate appropriately regulated gene expression of the corrected WT allele, in comparison to the mutant ΔF508 allele, in the corrected iPSC-derived cells.
Figure 3.
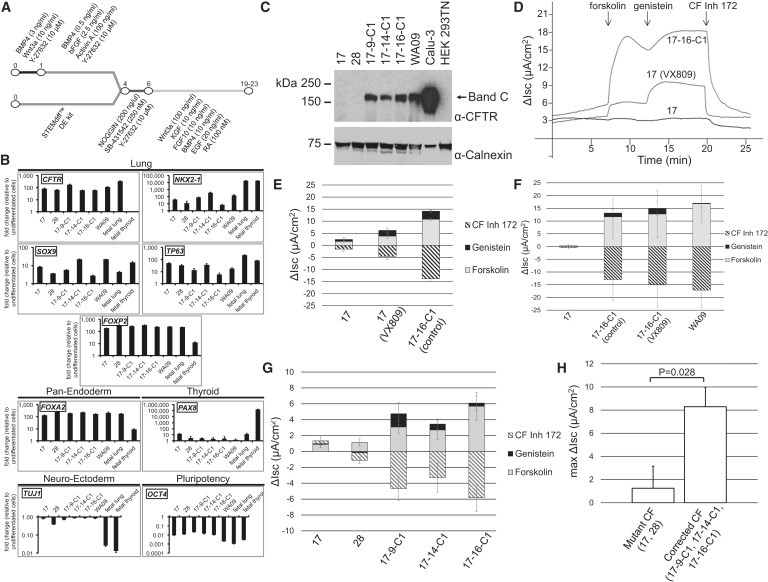
In Vitro Differentiation of Corrected CF WT/ΔF508 iPSCs
(A) Outline of the defined, step-wise differentiation protocol used to generate anterior foregut cell fates.
(B) Gene expression analysis of day 19 differentiated mutant (17 and 28), corrected CF iPSCs (17-9-C1, 17-14-C1, and 17-16-C1), and WA09 hESCs indicates upregulation of lung and pan-endodermal markers. Data (mean ± SD, three well replicates) from a representative experiment further characterized in Figures 3C and 3G. See also Figure S3E.
(C) Western blot analysis of protein lysates from day 19 differentiation cultures probed with a CFTR-specific antibody. Detection of Calnexin demonstrated equal sample loading for differentiated iPSC/hESC samples. See also Figures S3D and S3F.
(D) Representative short-circuit current (Isc) traces of epithelial monolayers differentiated from mutant (17) and corrected (17-16-C1) CF iPSC by Ussing chamber analysis. Cells were treated with either DMSO (0.03%) or VX809 (3 μM) for 24 hr. After establishment of Cl− gradient and the addition of amiloride, monolayers were treated with forskolin and genistein followed by the administration of CFTR inhibitor 172. The change in Isc (μA/cm2) for each perturbation is shown. Only corrected clone 17-16-C1 demonstrates the presence of CFTR channels in the cell membrane, as evidenced by activation of cAMP-dependent short-circuit currents. The addition of CF corrector VX809 to differentiated clone 17 sample partially restores CFTR-mediated chloride activity.
(E) Aggregated data of short-circuit current measurements from an experiment with differentiated mutant (17), with or without CF corrector VX809, and corrected (17-16-C1) CF iPSC (three transwell replicates per sample, mean ± SD.
(F) Aggregated data of short-circuit current measurements from an experiment with differentiated mutant (17) and corrected (17-16-C1), with or without CF corrector VX809, and WA09 hESC control (five transwell replicates per sample, mean ± SD).
(G) Aggregated data of short-circuit current measurements from an experiment including all independent mutant (17 and 28) and corrected (17-9-C1, 17-14-C1, and 17-16-C1) differentiated CF iPSC clones (three to six transwell replicates per sample, mean ± SD).
(H) Aggregated data of short-circuit current measurements; graphed is the maximum change in short-circuit current resulting from the addition of forskolin and genistein. Shown is the mean ± SE. The comparison shown is between two mutant CF clones (17 and 28; total of four independent differentiated experimental samples, three to six transwell replicates per sample) and three corrected CF clones (17-9-C1, 17-14-C1, and 17-16-C1; total of five independent differentiated experimental samples, three to six transwell replicates per sample). Results were clustered by clonal cell line and experiment number; statistical analysis was performed using a linear mixed-effect model by restricted maximum likelihood to account for correlated replicates within the same experiment.
See also Figure S3.
CF patients, either compound heterozygous ΔI507/ΔF508 or homozygous ΔF508/ΔF508, fail to express the mature, fully glycosylated CFTR protein. Correction of either allele, corresponding to carrier state WT/ΔF508 or WT/ΔI507, should result in restored expression of the mature CFTR glycoform. Western blotting of protein lysates from day 19 differentiation cultures, probed with a CFTR-specific antibody, identified the mature 170-kDa CFTR protein in differentiated WT CFTR, WA09 hESCs, and in differentiated corrected iPSCs (17-9-C1, 17-14-C1, and 17-16-C1), but not in differentiated ΔI507/ΔF508 iPSCs (clones 17 and 28; Figure 3C). The identity of the mature glycoform is confirmed by its presence in HEK293TN cells transfected with WT CFTR, but not ΔF508, expression constructs (Figure S3D), as well as in WT CFTR-expressing Calu-3 cells (Figures 3C and S3F). In addition, its identity as the mature CFTR protein is verified by its reduction in molecular weight upon treatment with peptide-N-glycosidase (PNGase F; Figure S3F). These results are consistent with maturation of CFTR protein from the corrected CFTR allele in cells derived from the ZFN-edited iPSC clones.
Restoration of CFTR Chloride Channel Function in Gene-Edited iPSC-Derived Epithelial Monolayers
To examine whether CFTR gene correction resulted in functional rescue of CFTR chloride channel activity, we initially selected the mutant (clone 17) and the corrected CF iPSC line (17-16-C1) for analysis. iPSCs were differentiated for 19 to 20 days as outlined above, replated onto permeable supports, and grown until confluent monolayers were established. Assessment of functional CFTR chloride channel activity in iPSC-derived epithelial monolayers was performed by Ussing chamber analysis. Sodium channel activity was first blocked by amiloride to establish a baseline. Stimulation with forskolin and genistein increased CFTR-dependent short-circuit current (Isc) for corrected 17-16-C1, but not for mutant clone 17 (Figures 3D and 3E). The addition of CFTR inhibitor 172 decreased Isc levels back to baseline and verified the CFTR specificity of the assay (Figures 3D and 3E). Mutant clone 17 monolayers, when treated with VX809 (a CFTR modulator capable of partially correcting the defective folding and aberrant maturation of ΔF508 mutant CFTR), exhibited partial restoration of CFTR-mediated chloride transport (Figures 3D and 3E).
To evaluate the extent of functional correction, in a subsequent experiment we demonstrated that 17-16-C1-derived cells functioned similarly to those of control WA09 hESCs, and they showed no further significant increase in CFTR chloride channel activity when treated with VX809 (Figure 3F). These results establish functional expression of WT CFTR in corrected clone 17-16-C1 samples. A second functional measurement, iodide efflux analysis, confirmed successful CFTR gene correction in clone 17-16-C1. Mutant and corrected CF-iPSC-derived epithelial monolayers were loaded with radioactive 125Iodide and subsequently stimulated with forskolin and genistein to activate cAMP-sensitive CFTR anion transport. In contrast to mutant clone 17 cells, corrected 17-16-C1 cell samples released higher levels of 125Iodide following stimulation (Figure S3G). The suppression of 125I-efflux with CF inhibitor 172 demonstrated specificity for CFTR channels in corrected clone 17-16-C1 (Figure S3G). To ensure that functional correction seen for 17-16-C1 was not simply an anomalous feature of this specific clone, we assayed CFTR functional activity, in addition to the originally assayed 17 and 17-16-C1, in an additional mutant CF iPSC line (28) and two additional corrected CF iPSC lines, each of which were independently derived from mutant clone 17 (17-9-C1 and 17-14-C1). Whereas both mutant clones showed similar non-responsive behavior, all three corrected iPSC clones yielded epithelial monolayers with CFTR functional activity increased with respect to both mutant clones 17 and 28 (Figure 3G). When considered together, the three corrected CF clones (17-9-C1, 17-14-C1, and 17-16-C1) exhibited maximum short-circuit currents significantly greater than mutant CF clones (17 and 28) (Figure 3H, p = 0.028). These functional results exhibit a strong correlation with the expression of fully glycosylated CFTR protein in the corrected clones (Figure 3C).
Allele-Preferred Targeted Correction
In our targeted correction of the ΔI507/ΔF508 clone 17 iPSCs, we observed a strong preference for targeting the ΔI507 allele versus the ΔF508 allele; sequencing of the unmodified allele yielded either the ΔI507 mutant allele (present in 3 of 21 clones) or ΔF508 mutant allele (in 18 of 21 clones) (a ΔI507/ΔF508 targeting ratio of 6:1) (Figure 4A). Although we initially speculated that perhaps a greater level of chromatin accessibility or transcriptional activity for one allele versus the other may have been responsible, our sequencing of CFTR cDNA from the original ΔI507/ΔF508 iPSCs, as shown previously, revealed transcriptional activity from both mutant alleles (Figure 2D).
Figure 4.
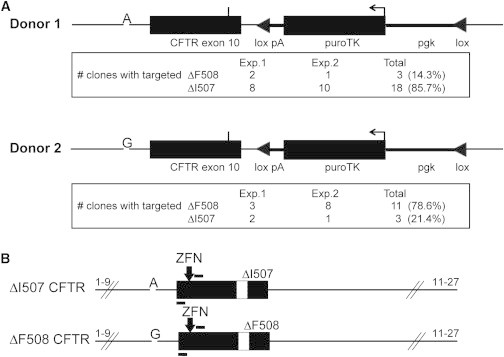
Allele-Preferred Targeting of CF iPSCs
(A) Schematic of donor 1 and donor 2 engineered with respective A or G base change at intron 9; results from a total of two independent targeted-integration experiments with either donor 1 or donor 2 are shown.
(B) Schematic of uncorrected CFTR alleles ΔI507 and ΔF508, highlighting A or G base within intron 9.
To investigate the cause of this allele-specific correction, we fully sequenced the 1.6-kb endogenous CFTR sequences homologous to the donor DNA used in the targeting vector, of each mutant allele (ΔI507 or ΔF508), to determine whether the donor was truly isogenic for both alleles. This analysis revealed a single-base-pair difference (A > G) in intron 9, 76 bp upstream of the ZFN cleavage site, present in the ΔF508 mutant allele, but absent in both the ΔI507 mutant allele and the donor (Figure 4B). In classical homologous recombination, isogenic regions of homology are preferred for donor construction; thus, we speculated that this single-base-pair difference occurring selectively in the ΔF508 allele of the CF iPSCs may have caused this selective behavior. To test this hypothesis, we introduced this A > G mutation into the donor sequences to see whether this donor would now selectively favor targeting of the ΔF508 allele. As shown in Figure 4A, the A > G single-base-pair substitution did in fact skew the allele-specific targeting from the ΔI507 allele to the ΔF508 allele (from a ΔI507:ΔF508 ratio of 6:1 for the A donor to a ΔI507:ΔF508 ratio of 1:3.7 for the G donor, a net 22-fold change resulting from a single-base-pair change). Molecular characterization and sequencing of these ΔF508-targeted, puromycin-resistant iPSC clones now identified three clones with specific correction of the ΔF508 allele. As such, these clones were now of WT/ΔI507 CFTR genotype.
Discussion
We utilized ZFN-mediated gene editing (Urnov et al., 2005; Hockemeyer et al., 2009) to correct, in a site-specific manner, the CFTR mutation in iPSCs derived from CF patients. The generation of iPSCs from CF patients has been reported previously, with subsequent differentiation into epithelial cells (Somers et al., 2010; Mou et al., 2012; Wong et al., 2012; Sargent et al., 2014). The design and assessment of CFTR-specific nucleases also has been reported previously (Maeder et al., 2008; Lee et al., 2012; Sargent et al., 2014), including repair of the mutant CFTR gene. Site-specifically editing the endogenous gene (Garate et al., 2013) offers the potential for physiologically regulated expression of the therapeutic gene, retaining the expression of alternately spliced isoforms and eliminating the potential interfering influence of inherited mutations that may be dominant negative. In vitro differentiation of the mutant CF iPSCs into lung epithelial cells and tissue, controlled for by the parallel differentiation of the otherwise isogenic corrected CF iPSCs, may provide a valuable tool for examining the functional consequence of mutant CFTR expression. Furthermore, corrected CF iPSCs present a potential source of patient-specific cells capable, in vitro, of differentiation into various lung stem/progenitor cells (Weiss et al., 2011), either for transplantation of autologous lung cells or for seeding de-vitalized lung scaffolds ex vivo to generate autologous lungs (Ott et al., 2010).
CF patients, either compound heterozygous ΔI507/ΔF508 or homozygous ΔF508/ΔF508, lacking expression of the normal CFTR protein, manifest various features of CF disease. Individuals bearing either the ΔI507 or ΔF508 CFTR mutations at only one allele (i.e., WT/ΔI507 or WT/ΔF508) are CF carriers and exhibit no defects in lung cell function (Kerem et al., 1990). Thus, correction of CF iPSCs derived from either ΔI507/ΔF508 or ΔF508/ΔF508 CF patients would be achieved by converting to WT either one or both mutant alleles. In this paper we first report targeted correction of ΔI507/ΔF508 iPSCs with the resulting iPSCs, corrected at a single allele, of genotype WT/ΔF508; as such, they correspond to the heterozygous ΔF508 carrier state. We demonstrated restored expression of both WT CFTR mRNA and mature CFTR protein in cells differentiated from a corrected, puroTK-excised clone 17-16-C1. This was not unexpected since the CFTR gene-editing approach simply restores the mutant ΔI507 or ΔF508 sequences in exon 10 to WT, with the only residual footprint being the loxP sequences in intron 10. Starting with the same ΔI507/ΔF508 iPSCs, but utilizing a donor with greater similarity to the ΔF508 CFTR allele (Figure 4), we were then able to obtain corrected iPSCs of genotype WT/ΔI507. Additionally, starting with two transgene-free ΔF508/ΔF508 CF iPSC lines, we were able to obtain corrected iPSCs of genotype WT/ΔF508.
The exquisite sensitivity of the allele-preferred targeting, that only a single-base-pair difference (of a total 1.6 kbp donor) could so dramatically specify the targeting for one allele versus the other was perhaps unanticipated. In subsequent correction of another mutant gene in disease-specific human iPSCs (the PKLR gene in pyruvate kinase deficiency), we again observed a precise allele-specific targeting caused by a single-base-pair difference in 2.0 kb of homology sequences (Z. Garate, A.M.C., B.R.D., and J.C. Segovia, unpublished data). It is possible that mismatches present in the integrated genome sequences, particularly those in proximity to nuclease cleavage sites, may dramatically influence the efficiency of targeting via decreasing the efficiency of strand invasion, recognition of the donor as a homologous template, and/or synthesis-dependent strand annealing. A preference for donor/target similarity also was seen in ZFN-mediated targeting of the histone variant H3.3 gene (Goldberg et al., 2010), although to a lesser extent than seen here. A requirement for exquisite matching between donor sequences and target alleles also recently was observed in AAV-mediated targeting, in which even 1 bp mismatch in 1.8 kb of homology sequence decreased the targeting efficiency for the APP locus by 4.5-fold (Deyle et al., 2014). Although this last report did not include site-specific donor sequence DNA cleavage, it does support the need for close donor-to-target sequence similarity. Taken together, these results suggest that donor sequences utilized for nuclease-mediated HDR should be finely tuned to the targeted gene locus in the recipient cells. Specific rules for the application of allele-preferred targeting remain to be developed (for example, the quantitative effect of mismatch as a function of distance from nuclease cleavage site). Finally, we note that this allele-specific targeting offers the potential for preferential targeting of specific mutant alleles, for example dominant alleles, by sequence-specific nuclease-mediated gene correction.
Having successfully corrected various mutant CFTR alleles in the CF iPSCs, we sought to develop in vitro differentiation conditions allowing us to confirm appropriately regulated expression of the corrected CFTR gene. After a total of 19 days of differentiation, upregulated expression of NKX2-1, SOX9, FOXP2, FOXA2, and CFTR were all consistent with some cells committed to a lung cell fate arising in the culture (Figure 3B). Future work will focus on enriching the NKX2.1-expressing lung progenitors to generate a fully developed lung airway epithelium (Longmire et al., 2012).
We demonstrated that CFTR gene correction resulted in restoration of expression of the mature CFTR glycoprotein and CFTR chloride channel function in iPSC-derived epithelial cells. Mutant clone 17 monolayers, when treated with CFTR corrector VX809, exhibited partial restoration of CFTR-mediated chloride transport. This indicates that the failure of non-drug-treated clone 17 cultures to exhibit CFTR chloride transport was not due to incomplete differentiation. Furthermore, in Figure 3F we show that treatment of the corrected line 17-16-C1 with the corrector VX809 yields no further significant increase in chloride channel activity. Since the 17-16-C1 line is WT/ΔF508, this result suggests that the one corrected allele, corresponding to the carrier CF state, produces sufficient WT CFTR protein to respond normally.
Although in our experience there may be some differences, experiment to experiment, in the efficiency of differentiation, these have not affected the primary observation that, once differentiated, mutant CF iPSCs (17 and 28) yield neither mature CFTR protein nor CFTR-specific chloride channel activity; whereas corrected CF iPSCs (17-9-C1, 17-14-C1, and 17-16-C1) and the normal control WA09 hESCs yield mature CFTR protein and CFTR-specific chloride channel activity. Although prior studies of hESC/iPSC-derived epithelial cells documented CFTR functional activity via patch clamping of individual cells (Firth et al., 2014) or iodide efflux (Wong et al., 2012), our demonstrated ability to evaluate functional CFTR chloride channel activity via the Ussing chamber assay in hESC/iPSC-derived polarized epithelial monolayers should be a valuable tool in CFTR drug screening and analysis of various CFTR mutations.
Experimental Procedures
Human subject and animal subject reviews were performed by University of Texas Health Science Center institutional review committees.
CF iPSC Generation and Characterization
CF fibroblasts were transduced with pMXs retroviruses expressing OCT4, SOX2, KLF4, NANOG, and C-MYC, and reprogrammed iPSC colonies, selected for in hESC media, were subsequently identified based on morphology and live cell staining for Tra-1-60 and Tra-1-81. Pluripotency was assayed by teratoma formation and confirmed by quantitative transcriptional profiling.
ZFN-Mediated Correction
Dissociated iPSCs were nucleofected with ZFNs together with a 1.6-kb donor construct (including WT exon 10, a loxP-flanked pgk-puroTK selection cassette in intron 10, and flanking homology sequences). Colonies exhibiting CFTR correction were identified by PCR and confirmed via Southern blot analysis.
In Vitro Differentiation and Functional Analysis
CF iPSCs, either mutant or corrected, and WA09 hESCs (as control) were differentiated based on Green et al. (2011) and Longmire et al. (2012), with minor modifications. CFTR protein was detected by immunoblotting. Ussing chamber analysis and 125I-efflux experiments were performed to demonstrate restoration of CFTR-mediated chloride channel function.
Assessment of Genome Integrity
The genomic integrity of mutant (clone 17) and two corrected CF iPSC lines (17-9-C1 and 17-14-C1) was assessed by karyotyping, whole-exome and whole-genome sequencing, and CGH.
Data Access
Next-generation sequencing data can be downloaded from http://www.ncbi.nlm.nih.gov/Traces/sra/ using the study accession numbers SRA058070 for the Complete Genomics whole-genome dataset and Illumina exome sequencing data (in .bam file format).
Author Contributions
A.M.C., P.K., J.H.B., and B.R.D designed the experiments and wrote the manuscript with valuable guidance provided by M.C.H., D.N.K., W.J.C., and E.J.S. J.W, H.C.S., D.E.P., D.Y.G., P.D.G., and M.C.H. designed and generated the ZFNs. A.M.C. and J.H.B. performed the targeted correction. A.M.C., P.K., and F.H performed the in vitro differentiation. A.M.C., W.J.C., and E.J.S. performed the functional analysis. X.S.L., P.K., M.L.G.G., and M.C.H. performed the assessment of genome integrity. S.C. performed the statistical analysis. W.L. and D.M. provided valuable technical assistance.
Acknowledgments
We thank Naoki Nakayama (Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center) for helpful discussions; Veronica K. Quiceno for editorial assistance; Kathryn Plath, Connie Cepko, Brian Sauer, and Jeffrey Beekman for providing DNA constructs either directly or through Addgene; and Fyodor Urnov (Sangamo BioSciences, Inc.) for ZFN development through funding from the Cystic Fibrosis Foundation (CFF) Folding Consortium. This study was supported by NIH RC1HL099559 (B.R.D. and R. Wetsel, primary investigators [PIs]); CFF DAVIS12GO (B.R.D., PI); NIH P30 DK072482 (E.J.S., PI); and CFF R464 (E.J.S., PI). J.W., H.C.S., D.E.P., D.Y.G, P.D.G., and M.C.H. are employees of Sangamo BioSciences, Inc.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
References
- Deyle D.R., Li L.B., Ren G., Russell D.W. The effects of polymorphisms on human gene targeting. Nucleic Acids Res. 2014;42:3119–3124. doi: 10.1093/nar/gkt1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth A.L., Dargitz C.T., Qualls S.J., Menon T., Wright R., Singer O., Gage F.H., Khanna A., Verma I.M. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2014;111:E1723–E1730. doi: 10.1073/pnas.1403470111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garate Z., Davis B.R., Quintana-Bustamante O., Segovia J.C. New frontier in regenerative medicine: site-specific gene correction in patient-specific induced pluripotent stem cells. Hum. Gene Ther. 2013;24:571–583. doi: 10.1089/hum.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg A.D., Banaszynski L.A., Noh K.M., Lewis P.W., Elsaesser S.J., Stadler S., Dewell S., Law M., Guo X., Li X. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M.D., Chen A., Nostro M.C., d’Souza S.L., Schaniel C., Lemischka I.R., Gouon-Evans V., Keller G., Snoeck H.W. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 2011;29:267–272. doi: 10.1038/nbt.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem B., Rommens J.M., Buchanan J.A., Markiewicz D., Cox T.K., Chakravarti A., Buchwald M., Tsui L.C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Kerem B.S., Zielenski J., Markiewicz D., Bozon D., Gazit E., Yahav J., Kennedy D., Riordan J.R., Collins F.S., Rommens J.M. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc. Natl. Acad. Sci. USA. 1990;87:8447–8451. doi: 10.1073/pnas.87.21.8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.M., Flynn R., Hollywood J.A., Scallan M.F., Harrison P.T. Correction of the ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. BioRes. Open Access. 2012;1:99–108. doi: 10.1089/biores.2012.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longmire T.A., Ikonomou L., Hawkins F., Christodoulou C., Cao Y., Jean J.C., Kwok L.W., Mou H., Rajagopal J., Shen S.S. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell. 2012;10:398–411. doi: 10.1016/j.stem.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder M.L., Thibodeau-Beganny S., Osiak A., Wright D.A., Anthony R.M., Eichtinger M., Jiang T., Foley J.E., Winfrey R.J., Townsend J.A. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou H., Zhao R., Sherwood R., Ahfeldt T., Lapey A., Wain J., Sicilian L., Izvolsky K., Musunuru K., Cowan C., Rajagopal J. Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell Stem Cell. 2012;10:385–397. doi: 10.1016/j.stem.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott H.C., Clippinger B., Conrad C., Schuetz C., Pomerantseva I., Ikonomou L., Kotton D., Vacanti J.P. Regeneration and orthotopic transplantation of a bioartificial lung. Nat. Med. 2010;16:927–933. doi: 10.1038/nm.2193. [DOI] [PubMed] [Google Scholar]
- Sargent R.G., Suzuki S., Gruenert D.C. Nuclease-mediated double-strand break (DSB) enhancement of small fragment homologous recombination (SFHR) gene modification in human-induced pluripotent stem cells (hiPSCs) Methods Mol. Biol. 2014;1114:279–290. doi: 10.1007/978-1-62703-761-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers A., Jean J.C., Sommer C.A., Omari A., Ford C.C., Mills J.A., Ying L., Sommer A.G., Jean J.M., Smith B.W. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov F.D., Miller J.C., Lee Y.L., Beausejour C.M., Rock J.M., Augustus S., Jamieson A.C., Porteus M.H., Gregory P.D., Holmes M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- Weiss D.J., Bertoncello I., Borok Z., Kim C., Panoskaltsis-Mortari A., Reynolds S., Rojas M., Stripp B., Warburton D., Prockop D.J. Stem cells and cell therapies in lung biology and lung diseases. Proc. Am. Thorac. Soc. 2011;8:223–272. doi: 10.1513/pats.201012-071DW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A.P., Bear C.E., Chin S., Pasceri P., Thompson T.O., Huan L.J., Ratjen F., Ellis J., Rossant J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 2012;30:876–882. doi: 10.1038/nbt.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.