

Summary
Inflammasomes are protein complexes that promote the maturation and release of pro-inflammatory cytokines and danger signals as well as pyroptosis in response to infections and cellular stress. Inflammasomes consist of a sensor, an adapter, and the effector caspase-1, which interact through homotypic interactions of caspase recruitment domains (CARDs) or PYRIN domains (PYDs). Hence, decoy proteins encoding only a CARD or PYD, COPs and POPs, respectively, are assumed to inhibit inflammasome assembly. Sensors encoding a PYD belong to the families of NOD-like receptors containing a PYD (NLRPs) or AIM2-like receptors (ALRs), which interact with the PYD and CARD-containing adapter ASC through homotypic PYD interactions. Subsequently, ASC undergoes PYD-dependent oligomerization, which promotes CARD-mediated interactions between ASC and caspase-1, resulting in caspase-1 activation. POPs are suggested to interfere with the interaction between NLRPs/ALRs and ASC to prevent nucleation of ASC and therefore prevent an oligomeric platform for caspase-1 activation. Similarly, COPs are suggested to bind to the CARD of caspase-1 to prevent its recruitment to the oligomeric ASC platform and its activation. Alternatively, the adapter ASC may regulate inflammasome activity by expressing different isoforms, which are either capable or incapable of assembling an oligomeric ASC platform. The molecular mechanism of inflammasome assembly has only recently been elucidated, but the effects of most COPs and POPs on inflammasome assembly have not been investigated. Here we discuss our model of COP and POP-mediated inflammasome regulation.
Keywords: inflammasome, ASC, pyrin domain (PYD), PYD only proteins (POPs), caspase recruitment domain (CARD), CARD only proteins (COPs)
Introduction
Caspase recruitment domains (CARDs)
There are 1724 CARD domains within 1626 proteins registered in the SMART non-redundant database (1, 2). Humans have at least 28, mice have 24, and C. elegans have 2 confirmed CARD-encoding genes. The CARD belongs to the death domain fold (DDF) superfamily, which also includes the death domain (DD), death effector domain (DED), and pyrin domain (PYD) families. Like all DDFs, CARDs are characterized by encoding 6 antiparallel α-helices with a hydrophobic core and an outer surface that is composed of charged residues. There are variations in the length and orientation of these α-helices, and the specificity of protein-protein interactions largely depends on the charged and hydrophobic pockets on the surface (3). Proteins can contain only a CARD or a CARD in combination with up to 4 different other domains of the NACHT, PYD, leucine rich repeat (LRR), WD repeat, Src homology3 (SH3), PDZ, RING, BIR, kinase, helicase, and DD domain families (4, 5). The CARD is commonly implicated in the regulation of caspases containing a CARD in their N-terminal pro-domains, including human caspases-1, -2, -4, -5, and -9, -12, mouse caspases-1, -2, -9, -11, and -12 and the nematode caspase Ced3. However, CARDs are also involved in the regulation of NF-κB, which is also a crucial regulator in apoptosis and inflammation (5). The CARD functions as protein-protein interaction domain and is mostly involved in homotypic interactions, forming dimers or trimers, which triggers the formation of multi-protein activation complexes (6, 7).
PYRIN domains (PYDs)
The pyrin domain (PYD) was initially referred to as ‘domain in apoptosis and interferon response’ (DAPIN) or ‘pyrin, AIM, ASC and death domain-like’ (PAAD) but is now named after the protein that it was originally discovered in: Pyrin (Marenostrin, MEFV) (8, 9). It is usually about 90 amino acids long and is exclusively located at the amino terminal end of proteins. There are 1623 PYD domains within 1580 proteins registered in the SMART non-redundant database (1, 2). Humans have 23 and mice at least 29 PYD-encoding genes, but C. elegans and Drosophila have none, suggesting that this domain may have evolved more recently (10, 11). The PYD also belongs to the DDF superfamily encoding 6 antiparallel α-helices, which are organized around a highly conserved hydrophobic core into a Greek key motif (12–17). Residues from all α-helices except helix α3 are involved in the formation of the hydrophobic core, which stabilizes the overall PYD structure. As with all DDFs, the electrostatic surface patches and hydrophobic residues are also critical for PYD-PYD protein interactions. However, PYDs differ from other DDFs by displaying a shorter or unorganized α3 helix and the α2-α3 loop region is therefore extended. Even within PYDs the length and organization of this loop is highly variable and may contribute to PYD binding specificities (18, 19). Proteins can contain only a PYD or a PYD in combination with various other domains of the CARD, NACHT, LRR, hematopoietic interferon-inducible nuclear protein with a 200 amino acid repeat (HIN-200), B-Box zinc finger and sprouty (SPRY) domain families (4, 11). PYDs function as homotypic protein-protein interaction domains between proteins that are involved in the regulation of inflammatory caspases, particularly caspase-1, and NF-κB (11, 20). The best-characterized function of PYD containing proteins is their ability to assemble a high molecular weight activation complex for caspase-1, called the inflammasome (21).
Inflammasomes
Innate immune responses are triggered by the detection of pathogens or danger signals by pattern recognition receptors (PRRs). The subsequent maturation and release of pro-inflammatory cytokines and danger signals then promotes the recruitment of immune cells that perform pathogen clearance and wound healing. In 2002, the group of Jürg Tschopp (21) first described an oligomeric multi-protein signaling platform, called the inflammasome, which is crucial for the release of pro-inflammatory cytokines and danger signals during innate immune responses. The inflammasome is assembled through PYD- and CARD-mediated interactions and facilitates the activation of pro-inflammatory caspases, including caspase-1, caspase-4, and caspase-5, which belong to the family of cysteine-dependent aspartate-directed endoproteases. In particular, the proteolytic activity of caspase-1 results in the cleavage of its substrates, the pro-inflammatory cytokines pro-IL-1β and pro-IL-18, which causes their maturation and release. Active IL-1β and IL-18 are potent mediators of inflammation that stimulate fever, the recruitment and activation of immune cells, and the production of secondary cytokines (22). In addition, danger signals, including high mobility group box 1 (HMGB1) (23, 24), lactate dehydrogenase (LDH) (25), and polymerized ASC aggregates (26, 27), are released through caspase-1 and caspase-4 dependent pyroptosis (28, 29). Together, the release of pro-inflammatory cytokines and danger signals triggers a potent anti-microbial innate immune response.
Germline-encoded PRRs sense extracellular and intracellular pathogen-associated molecular patterns (PAMPs) and self-derived damage-associated molecular patterns (DAMPs) or danger signals. PRRs are grouped into membrane-bound receptors, including Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and intracellular sensors, including NOD-like receptors (NLRs), Aim2-like receptors (ALRs), RIG-I-like receptors (RLRs), and an increasing number of nucleic acid sensors (30–32). So far inflammasome-forming PRRs have been described for ALRs and NLRs. NLRs are subgrouped into NLRs containing a PYD (NLRPs) and NLRs containing a CARD (NLRCs), and inflammasome formation and function has been described for both groups. While most inflammasomes, including NLRP3-, NLRP7- and Aim2- inflammasomes, require the recruitment of the adapter protein apoptosis-associated speck-like protein containing a CARD (ASC), some PRRs, namely NLRP1 and NLRC4, can directly activate caspase-1 (33, 34). ASC encodes a PYD and a CARD (35). While the PYD of ASC interacts with the PYD of PRRs, its CARD binds to the CARD of pro-caspase-1 (36–38). Recent studies showed that the recruitment of ASC to PRRs acts as a nucleation event for the subsequent PYD-mediated oligomerization of ASC, which is a prerequisite for bringing pro-caspase-1 monomers into close proximity, as illustrated in Fig. 1. Subsequently, self-cleavage facilitates the formation of a highly active tetrameric caspase-1 complex consisting of p20 and p10 subunits (39, 40), which performs the proteolytic cleavage and activation of pro-IL-1β and pro-IL-18 (39, 41–45). Other caspases, including human caspase-4 and caspase-5 and mouse caspase-11, can be involved in particular inflammasomes and promote certain inflammasome functions (21, 29, 46–48).
Fig. 1. Schemata of the inflammasome complex and the potential mechanism of POP-, COP-, and ASC-c-mediated disruption of the inflammasome complex.
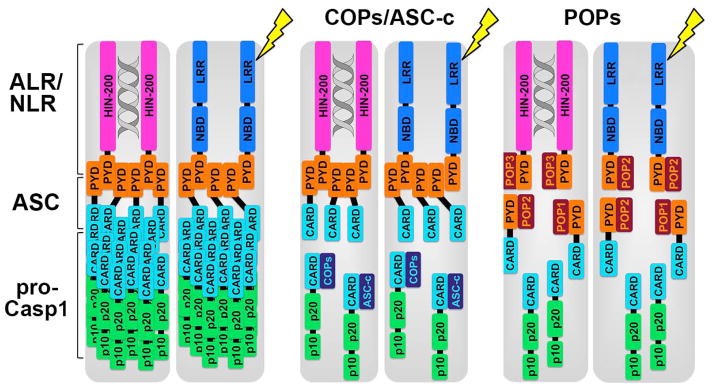
ALRs and NLRs nucleate the adapter ASC by PYD-PYD interaction, which polymerizes and in turn nucleates pro-caspase-1 through CARD-CARD interaction, resulting in pro-caspase-1 polymerization, clustering, and consequently its activation. Hence, activation of few sensor molecules can promote a dramatically amplified response. COPs, including Pseudo-ICE/COP, INCA, and ICEBERG as well as ASC-c, can bind to the CARD of pro-caspase-1, thereby occupying and sequestering the binding platform for ASC, preventing recruitment of pro-caspase-1 to ASC and consequently inhibiting pro-caspase-1 polymerization and activation. COPs have not been shown to bind to other CARD proteins, besides pro-caspase-1. On the other hand, POPs have been shown to bind either the ASCPYD, which is the case for POP1 and POP2, or the PYD of sensors, including AIM2 and IFI16, which is the case for POP3. POP4 has not been shown to bind to any PYD.
PYD and CARD-containing proteins frequently show a filamentous or punctate localization within cells. In particular, inflammasomes have been shown to form these structures (35, 49, 50). An oligomeric structure of inflammasomes has been initially shown for a reconstituted recombinant NLRP1 inflammasome (33). Using a similar approach, but reconstituting AIM2 and NLRP3 inflammasomes in cells, the molecular mechanism for inflammasome assembly has been elucidated using cryoelectron microscopy at near atomic resolution, revealing that these filaments are actually comprised of helical polymers (51). Assembly of these polymers requires all three types of interactions described for DDFs and referred to as type I, type II, and type III, which each require distinct interaction points in the individual α-helices (3, 37, 52). The central component of the inflammasome is the ASCPYD filament, which is formed by a three-start helical strands with a C3 symmetry with type I interactions in each of the strands and type II and type III interactions between the strands (51). ASCPYD polymerization uses a prion mechanism, which enables converting monomeric proteins into self-perpetuating polymerized forms, which adopt a very stable fibrous state (53). Nucleation of prions is necessary due to the high energy barrier and the ASCPYD polymerization is driven by oligomerized AIM2PYD and NLRP3PYD nucleation. Polymerized ASCPYD can therefore induce conversion of monomeric ASCPYD into a self-perpetuating polymer (53). The ASCPYD is connected by a flexible linker to the ASCCARD, which the recruits pro-caspase-1 by CARD-CARD interaction (51, 54). Here, ASC filaments act as activation platforms for pro-caspase-1 through nucleating in turn branched filaments of pro-caspase-1, and therefore ASC is capable able to nucleate an excess of pro-caspase-1 (51). Hence, activation of a limited number of receptor molecules can therefore promote the nucleation of many ASC molecules and even more pro-caspase-1 molecules, thus dramatically amplifying this response. Similar observations have been made in response to Salmonella infection, although inflammasomes showed concentric, spherical, rather than filamentous organization (55). A biological relevance for prion-like ASC particles was revealed, as these are present in sera of inflammatory disease patients, are phagocytized by neighboring cells and trigger polymerization of endogenous ASC without the need of inflammasome activation by a sensor (26, 27). Thus, polymerized ASC particles act as danger signals to propagate inflammasome responses to by-stander cells (56). Thus, due to the central involvement of PYDs and CARDs in inflammasome assembly, and the observation that, once nucleated, this process is self-perpetuating, these steps therefore require a tight control to prevent unwanted initiation of this response and increasing evidence supports that POPs and COPs have a central role in this process.
PYD-only proteins (POPs)
Three POP proteins have been identified in humans, namely POP 1, POP2, and POP3, and a truncated, partial POP4 (NLRP2P). The HUGO Gene Nomenclature Committee now assigned the names pyrin domain containing 1 (PYDC1) and PYDC2 for POP1 and POP2, respectively. Although there are no orthologs for human POPs in mice, there are 2 murine POP proteins predicted in mouse, namely Pydc3, which is 588 amino acids long and Pydc4, which is 135 amino acids long. They are localized on chromosome 1H3 in close proximity to Pyhin1 (Ifi209), which is the ortholog of human interferon-inducible protein X (IFIX). However, both murine POPs are less than 15% identical to POP3 or any other POP, and their function has not yet been described. Interestingly, virus-encoded POPs (vPOPs) have been identified to provide an anti-inflammatory strategy.
POP1 (PYDC1)
POP1 encodes a protein of 89 amino acids, which was first discovered in 2003 (57). It displays high homology to the ASCPYD with 64% identity in their amino acid sequence and is therefore expected to have originated from gene duplication of the ASCPYD encoding exon. In addition, POP1 is localized in close proximity to ASC on chromosome 16p11.2. POP1 is expressed in several tissues and particularly in monocytes, macrophages, and granulocytes. The NMR structure for POP1, but none of the other POPs, has been resolved (58–60). It adopts a typical DD fold and has high structural homology to the ASCPYD, as expected from their high degree of sequence similarity. Also, their hydrophobic residues that populate the inter-helical interaction surface, and thereby define the overall topology, are conserved. On the surface, negatively charged residues from helices 1 and 4 form a negative patch and positively charged residues from helices α2 and α3 form a basic patch (Fig. 2). Although the residues responsible for ASCPYD oligomerization and filament formation are conserved in POP1, it does not form these characteristic filaments but localizes uniformly throughout the cell (51, 57, 61). However, POP1 co-localizes with ASC in perinuclear specks, and the ASCPYD specifically binds POP1 (57). Particularly, the negatively charged residues of the ASCPYD (D6, E13, D48, D54) and the positively charged residues of POP1 (K21, R41) are essential for this interaction (60). A second group confirmed that mutation of residues E13 and D48 in the ASCPYD reduce or abolish interactions with POP1, but these mutations also interfere with ASCPYD self-association and ASCPYD interactions with NLRP3 (37). In addition, residues K21, R41, and D51, which have previously been identified as crucial residues for ASC oligomerization (51), are also crucial for ASCPYD self-association and ASCPYD interactions with the NLRP3PYD and POP1 (37). Interestingly, mutation of R3, D10, R38, D54, and Q84 diminished the interaction between the ASCPYD and POP1, but not the ASCPYD interaction with itself or the NLRP3PYD, suggesting that additional charged residues are required for strong ASCPYD-POP1 interactions (37). Since the same core residues are utilized for ASCPYD self-association and ASC interactions with the NLRP3PYD and POP1, it is important to address which interactions are preferred and possibly occur in vivo. While ASCPYD-ASCPYD complexes are still able to interact with the NLRP3PYD, they compete with POP1 interactions. Hence, the ASCPYD can simultaneously interact with other ASCPYDs and the NLRP3PYD, which allows ASCCARD-mediated recruitment, clustering, and activation of pro-caspase-1. Although POP1 was initially assumed to act as a decoy to disrupt PYD-PYD mediated inflammasome formation (Fig. 1), its ability to interact with both surfaces of the ASCPYD in the same manner as ASCPYD filaments makes it more likely that POP1 is incorporated into ASC filaments. Since ASCPYD self-association involves the same residues as its interaction with the NLRP3PYD and POP1, it was surprising that POP1 is unable to inhibit ASC-NLRP3 interactions, which questions its ability to function as a negative regulator of the NLRP3 inflammasome. Also, in an in vitro inflammasome assay POP1 enhanced inflammasome activity rather than disrupting it (57). Hence, the POP1 participation in ASCPYD polymers could possibly have a positive effect on inflammasome activity. However, its function may be affected by posttranslational modifications during inflammation since POP1 and ASCPYD are both phosphorylated in response to TNFα, which warrants further investigations (57). Likewise, additional studies will have to address the ability of POP1 to inhibit NF-κB activation in vitro by interacting with IKKα and IKKβ (57).
Fig. 2. The structure of the ASCPYD, POP1, POP,2 and POP3 is shown as ribbon diagram and the electrostatic surface is displayed on a scale of -4 kT/e (red) to 4kT/e (blue).

The structure of the ASCPYD (1UCP) (97) and POP1 (2HM2) (59) has been resolved. Homology modeling (SWISS-MODEL) (65) is shown for POP2, based on the NLRP7PYD (2KM6) (64) and for POP3, based on the AIM2PYD (3VD8) (70). Helices are colored in green (α1), red (α2), blue (α3), yellow (α4), purple (α5) and turquoise (α6).
POP2 (PYDC2)
POP2 is localized on human chromosome 3q28 with no recognizable PYD proteins in close proximity, and it encodes a protein of 97 amino acids, which was discovered by two independent groups in 2007 (62, 63). It has high similarity to the PYDs of NLRP2 (69% similarity) and NLRP7 (50% similarity), but only 37% and 40% similarity to ASC and POP1, respectively (62). POP2 interacts with ASC, NLRP1, NLRP2, NLRP4, and NLRP12 but not with NLRP3, NLRP10, and NLRP11 in a yeast-two-hybrid assay (62). Although the structure for POP2 has not been resolved, its similarity to the NLRP7PYD, whose structure has already been determined (Protein Data Bank accession code 2KM6) (64), allows homology modelling (SWISS-MODEL) (65) (Fig. 2). Here, POP2 adopts a typical DD fold with six α-helices. The strong positive electrostatic surface patch that is present in the ASCPYD and POP1, which is involved in ASC oligomerization and ASC-POP1 interactions, is missing in POP2. However, the negative surface patch comprised by residues E16 and E57 from helices α1 and α4 of POP2 is conserved between the ASCPYD, POP1, and POP2. Hence, interactions of POP2 with ASC most likely involve the negatively charged surface patch of POP2 and the positively surface patch of ASC. Similarly, we predict that interactions between POP2 and NLRPs engage a positively charged surface patch within the PYD of NLRPs. For instance, the POP2 binding partner NLRP4 has a strong positively charged patch (66). However, other POP2 binding partners, including NLRP1 and NLRP12, do not have a strong positively charged surface patch (19, 64, 66, 67). Hence, it is possible that the determining factors for homotypic PYD interactions are more complex than the currently accepted model, which proposes that the specificity of PYD-PYD interactions is determined by complementary charged surfaces (66). Also, more detailed studies will have to confirm all suggested POP2 interactions and their physiological consequences. So far, POP2 has been determined to prevent NLRP1, NLRP3, and NLRP12-mediated ASC-speck formation and blocks IL-1β release from NLRP2 and NLRP3 inflammasomes (62, 63). Hence, POP2 has a broad range of blocking inflammasomes by interacting with ASC and/or NLRPs (Fig. 1). However, reminiscent to POP1, POP2 is also able to inhibit NF-κB activation, which is independent of ASC (68). In addition, the α1 helix proofed to be necessary and sufficient to inhibit NF-κB and inflammasome activation. Specifically, the acidic residues D6, E8, and D16 within the α1 helix are involved in blocking inflammasome activation but are not involved in the inhibition of NF-κB activation (68). Hence, as suggested above, negatively charged residues of POP2 likely interact with the positive surface patch from the α2 and α3 helices of ASC to facilitate PYD-PYD interactions in the inflammasome, but since these residues are not involved in the NF-κB inhibition, these 2 functions are most likely mechanistically uncoupled (68). The importance of negatively charged residues in the α1 helix of POP2 for its interaction with NLRPs still needs to be determined.
POP3
POP3 is the most recently discovered POP and was characterized in 2014 (69). It is localized on human chromosome 1q23 within an interferon-inducible gene cluster containing interferon gamma (IFNγ) inducible protein 16 (IFI16), absent in melanoma 2 (AIM2), myeloid cell nuclear differentiation antigen (MNDA), and PYD- and HIN domain-containing protein 1 (PYHIN1), which is also called IFIX (69). The 113 amino acid protein is 60.9% identical to the AIM2PYD and probably originated from an exon duplication event of the AIM2-PYD, reminiscent to POP1, which is derived from the PYD of ASC. In addition, POP3 also encodes a unique sequence motif found within all PYHINPYDs (69, 70). The structure of POP3 has not been resolved yet, but due to its high similarity to the AIMPYD, whose structure has already been determined (Protein Data Bank accession code 3VD8) (70), homology modelling (SWISS-MODEL) (65) is possible (Fig. 2). POP3 is predicted to have five α-helices instead of the typical six α-helices found in most DDFs and in the AIM2PYD. The third α-helix in POP3 seems to be unstructured compared to the AIM2PYD. However, the PYD of NLRP1 also forms a flexible loop instead of a third α-helix (19). The electrostatic potential surface patches (EPSPs) of the AIM2PYD are formed by acidic residues of the α1 and α2 helices and by basic residues of the α5 and α6 helices (70). The first 3 α-helices, which equal the first 44 amino acids, are the most conserved between POP3 and the AIM2PYD with 90% identity. Hence, the acidic EPSP formed by negatively charged residues from the α1 and α2 helices is conserved between POP3 and the AIM2PYD, but from the 10 amino acid basic EPSP of the AIM2PYD only 3 residues (R67, K71 and K79) remain in POP3. Interestingly, the acidic residues of the α1 and α2 helices are crucial for the interaction between the AIM2PYD and the ASCPYD as well as for intramolecular interactions between PYD and HIN domains of AIM2 (70). Since these residues are very conserved between POP3 and the AIM2PYD it is surprising that, unlike POP1 and POP2, POP3 does not interact with the ASCPYD. POP3 however binds to the AIM2PYD and IFI16PYD, thereby abrogating their interaction with ASC (69) (Fig. 1). Subsequently, the AIM2 inflammasome assembly and activation is disrupted and the release of IL-1β and IL-18 is blocked. To date it is unclear whether POP3 interferes with the intramolecular PYD-HIN complex of AIM2. However, since intramolecular PYD-HIN interactions are considered to supply an autoinhibitory state of AIM2, which can only be released through the binding of a dsDNA ligand to the HIN domain, it is unlikely that POP3 binds to the HIN domain and thereby releases the autoinhibition, since it has been shown to function as a negative inflammasome regulator rather than an inflammasome activator (69, 71). Also, although the negative EPSP of the AIM2PYD facilitates its interaction with the ASCPYD, the conserved negative EPSP within POP3 is unable to support its interaction with the ASCPYD. In addition to the negative EPSP of the AIM2PYD, hydrophobic residues within the α2 helix of the AIM2PYD (F27 and F28) are also crucial for AIM2-ASC interactions (70). POP3 only retains one of these residues, which might be insufficient to enable ASC-POP3 interactions. It is also unclear whether the strong negative EPSP of POP3 enables its interaction with the positive EPSP of the AIM2PYD or if its residual basic EPSP interacts with the acidic EPSP of the AIM2PYD. Future studies will need to address these question together with the resolution of the detailed POP3 structure. So far evidence exists that POP3 inhibits ALR inflammasomes, which has been demonstrated on endogenous level and in vivo (69).
POP4 (NLRP2P)
The recently identified POP4 is encoded by the pseudogene NLRP2P. It is only 45 amino acids long and shares 80% identity with POP2 (72). POP4 only contains the α1 and α2 helices and thus is unlikely to adopt the characteristic PYD fold. However, a partial PYD fold is possible, which still needs to be determined. Although the α1 helix of POP4 is highly similar to the one in POP2, it lacks the acidic residues at positions 6 and 16, which prevents ASC-POP4 interactions and thereby inflammasome inhibition (72). However, POP4 is still able to inhibit NF-κB activation, which also in POP2 only requires the α1 helix (68, 72).
vPOPs
Analogous to human POPs, vPOPs are utilized by viruses as an immune evasive strategy. Specifically, the myxoma virus (MV) M013L, the Shope fibroma virus (SFV) S013L, the Swinepox virus SPV14L, the Yaba-like disease virus YLDV18L, and the mule deer poxvirus DPV83gp024 show significant homologies to POP1 and the ASCPYD (73, 74). M013L and S013L specifically bind to the PYD but not to the CARD of ASC, signifying a homotypic PYD-PYD interaction (73). Also, while S013L and M013L show a diffuse or punctate localization in uninfected cells, S013L aggregates into one perinuclear speck upon SFV infection. However, disruption of the M013L-PYD alters its localization and is unable to form punctate structures (74). In addition, vPOPs co-localize with ASC in perinuclear specks. Importantly, S013L and M013L inhibit the NLRP3-ASC-Caspase-1 mediated processing and release of IL-1β in inflammasomes (73). Hence, vPOPs are viral immune evasive proteins that impair the host response. Consistently, mutated MV, which has a disrupted M013LPYD, displayed attenuated virulence in rabbits, since the virus was unable to dampen the host immune response. In that case, myxomatosis was non-lethal, and the infection was rapidly resolved. Also, the acute inflammatory response to the virus infection was enhanced. The loss of virulence was attributed to decreased virus spread and dissemination from primary sites of infection due to the abortion of the infection in monocytes and lymphocytes. Further, while caspase-1 activation and IL-1β and IL-18 secretion was abrogated in MV infections, infection with the MV mutant lacking the PYD resulted in caspase-1 activation and the processing and secretion of pro-inflammatory cytokines (74). Hence, some viruses adopted the cellular mechanism of inflammasome regulation, by utilizing vPOPs as decoy molecules to inhibit inflammasome assembly similar to cellular POPs, to circumvent host responses.
CARD-only proteins (COPs)
Three COP proteins have been identified, namely Pseudo-IL-1β converting enzyme (ICE)/COP, INCA, and ICEBERG. The HUGO Gene Nomenclature Committee now assigned the names CARD16, CARD17, and CARD18, respectively. Interestingly, all COPs are highly homologous to the caspase-1CARD and are all located on chromosome 11q22 in humans, where they cluster with the inflammatory caspase-1, caspase-4, caspase-5, and caspase-12. They are most likely the result of recent gene duplication events. However, these caspase-1-like genes have acquired a premature nonsense mutation that limits their expression to the CARD. All 3 COPs are absent in the mouse or rat genome. However, putative Pseudo-ICE/COP and INCA orthologues exist on chromosome 11 of chimpanzees and Pseudo-ICE/COP, INCA, and ICEBERG orthologues exist on chromosome 14 of Rhesus monkeys. In addition, the ASC-c splice variant of ASC could also be classified as a COP, but with high homology to ASC.
Pseudo-ICE/COP (CARD16)
Pseudo-ICE/COP was first described in 2001 by two separate groups (75, 76). Meanwhile, two isoforms have been identified. The longer variant encodes a 197 amino acid and the shorter a 97 amino acid protein. In each case amino acids 1-91 encode a CARD. So far only the shorter isoform has been characterized. Compared to other COPs, the short isoform of Pseudo-ICE has the highest homology to caspase-1 with 92% identity to the procaspase-1CARD. The CARD of Pseudo-ICE/COP alone (amino acid 1-91) is even 97% identical to the procaspase-1CARD. Pseudo-ICE/COP can self-associate and binds to the pro-caspase-1CARD and RIP2CARD. RIP2 interactions with caspase-1 trigger proteolytic processing and activation of caspase-1 (77), but in the presence of Pseudo-ICE/COP, the interaction between caspase-1 and RIP2 and the RIP2-mediated oligomerization of pro-caspase-1 were inhibited (75). Subsequently also the RIP2- and caspase-1-mediated IL-1β secretion was decreased in the presence of Pseudo-ICE/COP in a dose-dependent manner. Interestingly, Pseudo-ICE/COP was also able to block caspase-1-mediated IL-1β secretion in the absence of RIP2. Hence, Pseudo-ICE/COP seems to block the RIP-2 mediated nucleation of caspase-1 oligomerization as well as the subsequent polymerization of caspase-1. Interestingly, Pseudo-ICE/COP is able to interact with ICEBERG, which may provide an additional level of inflammasome regulation, by potentially sequestering two inhibitors. However, their expression profile during inflammation is unknown, and it is unclear if they are expressed under the same physiological conditions in the same cell type. In most human tissues and human cell lines, Pseudo-ICE/COP expression coincides with caspase-1 expression, suggesting that their transcription is regulated similarly. One might even speculate that Pseudo-ICE/COP expression prevents caspase-1 activation in the absence of an inflammatory stimulus. However, while Pseudo-ICE/COP and caspase-1 are strongly expressed in bone marrow, lymph nodes, and spleen, there is no ICEBERG expression, suggesting that the transcriptional regulation of ICEBERG is distinct from that of Pseudo-ICE/COP and caspase-1. Only in placenta Pseudo-ICE/COP, caspase-1, and ICEBERG are all strongly expressed (76). Furthermore, similar to RIP2 and several other CARD-containing proteins (78–81), Pseudo-ICE/COP also has the capacity to trigger NF-κB activation and enhances TNF-α-mediated NF-κB activation, which may contribute to an effective inflammatory immune response (76).
INCA (CARD17)
The inhibitory CARD, or INCA, was first described in 2004 (82). The 110 amino acid long protein has 81% identity with the pro-domain of caspase-1 and, similarly to caspase-1, is upregulated in response to IFNγ. INCA interacts with pro-caspase-1 and blocks IL-1β release in response to lipopolysaccharide (LPS). However, INCA does not interact with RIP2 and does not induce NF-κB (82). In addition, INCA does also not inhibit the TNFα, caspase-1, Pseudo-ICE/COP, or RIP2-mediated activation of NF-κB (82). Interestingly, INCA also interacts with itself and both other COPs. However, the functional consequences of these interactions have not been determined, but COPs may participate in a complex caspase-1 repression/derepression system. INCA expression can be detected in a wide variety of human tissues but the strongest expression levels are found in heart, brain, and salivary gland. In general, INCA expression is more common than procaspase-1 expression, which suggests that their expression is regulated differently.
ICEBERG (CARD18)
ICEBERG, which was first described in 2000 (83), encodes a 90 amino acid protein, which in its entirety comprises a CARD with 52% identity to the procaspase-1CARD. It interacts with itself, Pseudo-ICE/COP and caspase-1 but contrary to Pseudo-ICE/COP ICEBERG does not interact with RIP2 (76, 83). The interaction of ICEBERG with caspase-1 occurs through a homotypic CARD-CARD interaction and interferes with the interaction of caspase-1 and RIP2. Caspase-1 preferentially binds to ICEBERG and is even able to displace RIP2 from caspase-1/RIP2 complexes. As expected, the ICEBERG-mediated disruption of the caspase-1/RIP2 complex prevents RIP2-mediated caspase-1 oligomerization, processing, and activation (83). Accordingly, IL-1β secretion was inhibited in response to LPS stimulation in ICEBERG-expressing cells (76, 83). Based on the ability of ICEBERG to shut off cytokine generation and inflammation it is expected to be regulated in a way to allow initial immune responses before returning to homeostasis. Indeed, ICEBERG expression is upregulated after 7 h of LPS and TNFα stimulation, which coincided with declining caspase-1 activity and IL-1β secretion after an initial rapid burst. Hence, ICEBERG seems to function as a negative feedback inhibitor, which regulates IL-1β production. Interestingly, ICEBERG is strongly expressed in the human placenta in the absence of inflammatory stimuli (76, 83). However, contrary to Pseudo-ICE/COP, ICEBERG does not trigger NF-κB activation and cannot enhance TNF-α-mediated NF-κB activation (76). ICEBERG is the only COP for which the structure has been determined. Three dimensional heteronuclear NMR spectra revealed a typical DD structure with an antiparallel six α-helical bundle with Greek-key topology and a hydrophobic core (Fig. 3). Helices 1-5 are α-helices, but helix 6 is a 310 helix. The structure of ICEBERG is most similar to the CARDs of RAIDD (84), Apaf-1 (85, 86), and caspase-9 (87) with conserved hydrophobic residues that make up the core. The surface of ICEBERG contains 3 highly charged patches. Helices 1, 4, and 6 provide basic residues that form a positively charged patch, Helices 2 and 5 provide acidic residues that form a negatively charged patch, and the helix 3-4 turn together with helix 4 provide acidic residues that form another negatively charged patch.
Fig. 3. The structure of the caspase-1CARD, Pseudo-ICE/COP, INCA, and ICEBERG is shown as ribbon diagram and the electrostatic surface is displayed on a scale of -4 kT/e (red) to 4kT/e (blue).

The structure of the ICEBERG (1DGN) (83) has been resolved. Homology modeling (SWISS-MODEL) (65) is shown for the caspase-1CARD, Pseudo-ICE/COP, and INCA based on ICEBERG. Helices are colored in green (α1), red (α2), blue (α3), yellow (α4), purple (α5) and turquoise (α6).
Based on the ICEBERG structure, we performed protein structure homology modeling (SWISS-MODEL) (65) to determine the structures for Pseudo-ICE/COP, INCA, and the caspase-1CARD (Fig. 3). As previously observed for ICEBERG and the caspase-1CARD, also Pseudo-ICE/COP and INCA display a positively charged patch that is formed by residues from helices 1 and 4 and a negatively charged patch on the opposite surface formed by residues from helices 2 and 5 (83). We speculate that charge-charge interactions are important for mediating COP-caspase-1 interactions, similar to RAIDD-caspase-2 (84) and Apaf-1-caspase-9 (85, 87).
ASC isoforms
The adapter protein ASC is critical for inflammasome assembly and activation. Interestingly, cells are able to express 4 differentially spliced ASC isoforms (88, 89). Full length ASC encodes a PYD, a linker region, and a CARD. ASC-b lacks the linker region, ASC-c lacks most of the PYD, and ASC-d lacks most of the PYD, the linker, and the CARD, and only retains the first 35aa of ASC, comprising the α1 and α2 helix of the PYD. ASC localizes to the nucleus or to perinuclear aggregates. Surprisingly, the absence of the linker region changes this localization pattern, and ASC-b localizes diffusely to the cytoplasm, thus evading the regulatory retention mechanism within the nucleus (49, 90, 91). ASC-c forms long filamentous structures in the cytoplasm, which is consistent with the localization pattern of the ASC-CARD (38, 92). The least ASC related isoform, ASC-d, localizes to the cytoplasm. Inflammasome assembly is initiated by the recruitment of ASC to activated receptors of the NLRP and ALR families through PYD-PYD interactions. Hence, only the isoforms encoding a PYD, ASC, and ASC-b are able to co-localize with NLRP3. However, while ASC and NLRP3 form a characteristic perinuclear aggregate, ASC-b and NLRP3 co-localize in a filamentous cytosolic aggregate. Activation of caspase-1 in the inflammasome requires the ASC-CARD to interact with the CARD of pro-caspase-1. Consistently, all isoforms containing a CARD (ASC, ASC-b, and ASC-c) co-localize with pro-caspase1 in perinuclear aggregates (ASC), in the cytoplasm (ASC-b), or in cytosolic filamentous aggregates (ASC-c), but ASC-d did not co-localize with pro-caspase-1. Most importantly, the ASC isoforms have different functions in regulating inflammasome activity. Particularly, ASC and ASC-b but not ASC-c and ASC-d function as NLRP3 inflammasome adapters that activate caspase-1 and promote IL-1β secretion (88). However, while ASC-b causes enhanced IL-1β secretion compared to ASC, in the absence of an NLRP (89), there is decreased IL-1β secretion upon co-expression of dominant active NLRP3 or upon stable expression of ASC-b and LPS/ATP treatment (88). Interestingly, ASC-c functions similarly to COPs. In the presence of a functional inflammasome consisting of dominant active NLRP3, ASC, pro-caspase1, and pro-IL-1β, only ASC-c but not ASC, ASC-b, or ASC-d was able to disrupt inflammasome activation and IL-1β secretion in an inflammasome reconstitution assay and in ASC-c expressing stable cell lines (88). Overall, ASC isoforms seem to differentially regulate inflammasome function, but their physiological role in regulating inflammatory immune responses remains unclear. Interestingly, ASC-b and ASC-c expression is upregulated upon LPS stimulation of macrophages, suggesting that the presence of distinct combinations of ASC splice variants may affect inflammasome activity at different stages of inflammatory responses (88). In fact, ASC-c is only upregulated after 24 h of LPS activation, thus may have a role in resolving this response (88). It is unclear how the expression of different ASC isoforms affects the oligomerization of ASC. Since ASC-b and ASC-c are able to co-localize with ASC, one might speculate that they can be incorporated into ASC complexes and alter their structure and caspase-1 activating function, thereby fine-tuning inflammasome activity. One hypothesis is that the absence of the flexible linker in ASC-b, which is required for proper inflammasome complex organization (51), may somehow attenuate inflammasome activity compared to ASC (88).
Other COPs
Similar to the splicing of ASC, a short isoform of NOD2, NOD2-S, which essentially is comprised of only the first of its CARDs, results from skipping exon 3 (93). NOD2-S inhibits muramyl dipeptide (MDP)-induced and NOD2-mediated activation of NF-κB and inflammatory cytokine release, and is in turn itself downregulated by pro-inflammatory cytokines but upregulated by anti-inflammatory cytokines, including IL-10 (93). NOD2-S binds to both NOD2 and the adapter receptor-interacting protein kinase 2 (RIP2K) and thereby inhibits NODosome assembly, reminiscent to the mechanism of POPs. Another splice form of NOD2, NOD2-C2, encodes the truncated NOD2 comprised of only the tandem CARDs of NOD2 (94). Contrary to NOD2-S, NOD2C2 activates NF-κB independent of MDP but nevertheless competes with NOD2 for MDP-mediated NF-κB activation (94). Another example is caspase-12. The majority of the human population express a truncated form of caspase-12, caspase-12-S, caused by a premature stop codon, and only about 20% of individuals of African descent bear a single nucleotide polymorphism in the CASP12 gene, which results in a read-through expression of caspase-12 (95). Caspase-12 expression results in a dampened endotoxin response and increased risk of developing severe sepsis. Accordingly, caspase-12-deficient mice are more resistant to sepsis, resulting from an inhibitory function of caspase-12 on caspase-1 (96). Caspase-12-S is less efficient in blocking caspase-1 than caspase-12 (95).
Future perspectives
Although inflammasomes have been identified over a decade ago, the precise molecular mechanism responsible for inflammasome assembly, and caspase-1 activation has only been recently described. However, it is evident that PYDs and CARDs play an essential and central role in this process through mediating homotypic protein-protein interactions. Therefore, the single PYD and CARD proteins POPs and COPs may likely have important regulatory mechanism to fine-tune or completely abolish inflammasome responses. A compelling scenario could be that due to the late response expression by pro-inflammatory stimuli of many of these proteins, POPs and COPs contribute to the resolution phase of inflammasome responses, which is still poorly understood. However, it is quite astonishing that humans encode at least six proteins that may regulate just two steps in inflammasome assembly, namely recruitment of ASC and recruitment of pro-caspase-1, while mice lack these proteins altogether. As numerous NLRPs/ALRs exist, POPs may provide some form of selectivity for specific sensors. However, since recruitment of caspase-1 is also regulated by three COPs, this level of complexity is less obvious but may provide a redundant safeguard mechanism regulating this key step. However, equally possible is that COPs have a much broader function beyond the canonical inflammasome, since CARD-containing proteins are involved in diverse immune-regulatory pathways, including non-canonical inflammasomes, RIG-I signaling, apoptosome, NODosome, PIDDosome, and CARMA signaling. This is supported by the existence of NOD2-S, which inhibits the NODosome, and likely many more CARD- or PYD-containing proteins may exist, where differential splicing may produce COPs and POPs. Since the effector caspases of the non-canonical inflammasomes function also simultaneously as the sensors for LPS (29), this pathway probably lacks many check-points present in the canonical inflammasomes, and therefore the potential regulation by a COP may be of importance to prevent the unwanted occurrence of pyroptotic cell death. However, the precise function of these small proteins is largely elusive, in spite of being in most cases discovered over a decade ago.
Acknowledgments
This work was supported by the National Institutes of Health (GM071723, AI099009 and AR064349 to C.S., and AR057532 to A.D.) and the American Heart Association (12GRNT12080035 to C.S).
Footnotes
The authors declare that they have no conflict of interest.
References
- 1.Letunic I, Doerks T, Bork P. SMART 6: Recent updates and new developments. Nucleic Acids Res. 2009 Jan;37(Database issue):D229–32. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–64. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park HH, Lo Y-C, Lin S-C, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–86. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reed JC, Doctor KS, Godzik A. The domains of apoptosis: A genomics perspective. Sci STKE. 2004 Jun 29;2004(239):re9. doi: 10.1126/stke.2392004re9. [DOI] [PubMed] [Google Scholar]
- 5.Kao W-P, Yang C-Y, Su T-W, Wang Y-T, Lo Y-C, Lin S-C. The versatile roles of cards in regulating apoptosis, inflammation, and nf-κb signaling. Apoptosis. 2014 Nov 25; doi: 10.1007/s10495-014-1062-4. [DOI] [PubMed] [Google Scholar]
- 6.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, et al. A unified model for apical caspase activation. Mol Cell. 2003 Feb;11(2):529–41. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 7.Jin T, Xiao TS. Activation and assembly of the inflammasomes through conserved protein domain families. Apoptosis. 2014 Nov 15; doi: 10.1007/s10495-014-1053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Consortium TFF. A candidate gene for familial mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 9.Consortium TIF. Ancient missense mutations in a new member of the roret gene family are likely to cause familial mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 10.Reed JC, Doctor K, Rojas A, Zapata JM, Stehlik C, Fiorentino L, et al. Comparative analysis of apoptosis and inflammation genes of mice and humans. Genome Res. 2003 Jun;13(6B):1376–88. doi: 10.1101/gr.1053803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu LH, Gangopadhyay A, Dorfleutner A, Stehlik C. An updated view on the structure and function of PYRIN domains. Apoptosis. 2014 Dec 2; doi: 10.1007/s10495-014-1065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.[NO STYLE for: Martinon 2001].
- 13.Bertin J, DiStefano PS. The PYRIN domain: A novel motif found in apoptosis and inflammation proteins. Cell Death and Differentiation. 2000 Dec;7(12):1273–4. doi: 10.1038/sj.cdd.4400774. [DOI] [PubMed] [Google Scholar]
- 14.Pawłowski K, Pio F, Chu Z, Reed JC, Godzik A. PAAD - a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem Sci. 2001 Feb;26(2):85–7. doi: 10.1016/s0968-0004(00)01729-1. [DOI] [PubMed] [Google Scholar]
- 15.Staub E, Dahl E, Rosenthal A. The DAPIN family: A novel domain links apoptotic and interferon response proteins. Trends Biochem Sci. 2001 Feb;26(2):83–5. doi: 10.1016/s0968-0004(00)01717-5. [DOI] [PubMed] [Google Scholar]
- 16.Fairbrother WJ, Gordon NC, Humke EW, O’Rourke KM, Starovasnik MA, Yin J-P, Dixit VM. The PYRIN domain: A member of the death domain-fold superfamily. Protein Sci. 2001 Sep;10(9):1911–8. doi: 10.1110/ps.13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber CH, Vincenz C. The death domain superfamily: A tale of two interfaces? Trends Biochem Sci. 2001 Aug;26(8):475–81. doi: 10.1016/s0968-0004(01)01905-3. [DOI] [PubMed] [Google Scholar]
- 18.Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends Biochem Sci. 2011 Oct;36(10):541–52. doi: 10.1016/j.tibs.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Hiller S, Kohl A, Fiorito F, Herrmann T, Wider G, Tschopp J, et al. NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure. 2003 Oct;11(10):1199–205. doi: 10.1016/j.str.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Stehlik C. The PYRIN domain in signal transduction. Curr Protein Pept Sci. 2007 Jun;8(3):293–310. doi: 10.2174/138920307780831857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol Cell. 2002 Aug;10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 22.Dinarello CA. Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann N Y Acad Sci. 1998 Sep 29;856:1–11. doi: 10.1111/j.1749-6632.1998.tb08307.x. [DOI] [PubMed] [Google Scholar]
- 23.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-Dependent release of the alarmin HMGB1 in endotoxemia. The Journal of Immunology. 2010;185(7):4385–92. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu B, Wang H, Andersson U, Tracey KJ. Regulation of HMGB1 release by inflammasomes. Protein Cell. 2013 Mar;4(3):163–7. doi: 10.1007/s13238-012-2118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rayamajhi M, Zhang Y, Miao EA. Detection of pyroptosis by measuring released lactate dehydrogenase activity. Methods Mol Biol. 2013;1040:85–90. doi: 10.1007/978-1-62703-523-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014 Jun 22;15(8):727–37. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014 Jun 22;15(8):738–48. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 28.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006 Nov;8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 29.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014 doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 30.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–44. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 31.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006 Feb 24;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Ablasser A, Hertrich C, Waßermann R, Hornung V. Nucleic acid driven sterile inflammation. Clin Immunol. 2013 Jun;147(3):207–15. doi: 10.1016/j.clim.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007 Mar 9;25(5):713–24. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 34.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and ipaf. Nature. 2004 Jul 8;430(6996):213–8. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 35.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, et al. ASC, a novel 22-kda protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–8. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 36.Srinivasula SM, Poyet J-L, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. Journal of Biological Chemistry. 2002 Jun 14;277(24):21119–22. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 37.Vajjhala PR, Mirams RE, Hill JM. Multiple binding sites on the ASC pyrin domain allow self-association and interaction with NLRP3. J Biol Chem. 2012 Oct 12;287(50):41732–43. doi: 10.1074/jbc.M112.381228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol. 2003 Dec 1;171(11):6154–63. doi: 10.4049/jimmunol.171.11.6154. [DOI] [PubMed] [Google Scholar]
- 39.Yamin TT, Ayala JM, Miller DK. Activation of the native 45-kda precursor form of interleukin-1-converting enzyme. Journal of Biological Chemistry. 1996 May 31;271(22):13273–82. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]
- 40.Elliott JM, Rouge L, Wiesmann C, Scheer JM. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. Journal of Biological Chemistry. 2009 Mar 6;284(10):6546–53. doi: 10.1074/jbc.M806121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Black RA, Kronheim SR, Cantrell M, Deeley MC, March CJ, Prickett KS, et al. Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. Journal of Biological Chemistry. 1988 Jul 5;263(19):9437–42. [PubMed] [Google Scholar]
- 42.Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. 1997 Jan 10;275(5297):206–9. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- 43.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes ifn-gamma-inducing factor and regulates lps-induced ifn-gamma production. Nature. 1997 Apr 10;386(6625):619–23. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 44.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995 Mar 31;267(5206):2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 45.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. Mice deficient in il-1b-converting enzyme are defective in production of mature il-1b and resistant to endotoxic shock. Cell. 1995;80:401–11. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 46.Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011 Oct 16;479(7371):117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 47.Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, Buxbaum JD. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol. 2014 May 30;193(1):335–43. doi: 10.4049/jimmunol.1303424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sollberger G, Strittmatter GE, Kistowska M, French LE, Beer HD. Caspase-4 is required for activation of inflammasomes. The Journal of Immunology. 2012;188(4):1992–2000. doi: 10.4049/jimmunol.1101620. [DOI] [PubMed] [Google Scholar]
- 49.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol. 2009 Mar 1;182(5):3173–82. doi: 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007 Sep;14(9):1590–604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of asc-dependent inflammasomes. Cell. 2014 Mar 13;156(6):1193–206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weber CH, Vincenz C. A docking model of key components of the DISC complex: Death domain superfamily interactions redefined. FEBS Letters. 2001 Mar;492(3):171–6. doi: 10.1016/s0014-5793(01)02162-7. [DOI] [PubMed] [Google Scholar]
- 53.Cai X, Chen J, Xu H, Liu S, Jiang Q-X, Halfmann R, Chen ZJ. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014 Mar 13;156(6):1207–22. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC) J Biol Chem. 2009 Nov 20;284(47):32932–41. doi: 10.1074/jbc.M109.024273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Man SM, Hopkins LJ, Nugent E, Cox S, Glück IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A. 2014 May 6; doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Broderick L, Hoffman HM. CASCading specks. Nat Immunol. 2014 Aug;15(8):698–700. doi: 10.1038/ni.2942. [DOI] [PubMed] [Google Scholar]
- 57.Stehlik C, Krajewska M, Welsh K, Krajewski S, Godzik A, Reed JC. The PAAD/pyrin-only protein POP1/ASC2 is a modulator of asc-mediated nf-kb and pro-caspase-1 regulation. Biochem J. 2003;373:101–13. doi: 10.1042/BJ20030304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Espejo F, Green M, Preece NE, Assa-Munt N. NMR assignment of human ASC2, a self contained protein interaction domain involved in apoptosis and inflammation. J Biomol NMR. 2002 Jun;23(2):151–2. doi: 10.1023/a:1016398403157. [DOI] [PubMed] [Google Scholar]
- 59.Natarajan A, Ghose R, Hill JM. Structure and dynamics of ASC2, a pyrin domain-only protein that regulates inflammatory signaling. Journal of Biological Chemistry. 2006 Oct 20;281(42):31863–75. doi: 10.1074/jbc.M605458200. [DOI] [PubMed] [Google Scholar]
- 60.Srimathi T, Robins SL, Dubas RL, Chang H, Cheng H, Roder H, Park YC. Mapping of pop1-binding site on pyrin domain of ASC. Journal of Biological Chemistry. 2008 Mar 24;283(22):15390–8. doi: 10.1074/jbc.M801589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moriya M, Taniguchi S, Wu P, Liepinsh E, Otting G, Sagara J. Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry. 2005 Jan 18;44(2):575–83. doi: 10.1021/bi048374i. [DOI] [PubMed] [Google Scholar]
- 62.Dorfleutner A, Bryan NB, Talbott SJ, Funya KN, Rellick SL, Reed JC, et al. Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infection and Immunity. 2007 Mar;75(3):1484–92. doi: 10.1128/IAI.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bedoya F, Sandler LL, Harton JA. Pyrin-only protein 2 modulates nf-kappab and disrupts ASC:CLR interactions. Journal of Immunology. 2007 Mar 15;178(6):3837–45. doi: 10.4049/jimmunol.178.6.3837. [DOI] [PubMed] [Google Scholar]
- 64.Pinheiro AS, Proell M, Eibl C, Page R, Schwarzenbacher R, Peti W. Three-dimensional structure of the NLRP7 pyrin domain: INSIGHT INTO PYRIN-PYRIN-MEDIATED EFFECTOR DOMAIN SIGNALING IN INNATE IMMUNITY. Journal of Biological Chemistry. 2010;285(35):27402–10. doi: 10.1074/jbc.M110.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006 Jan 15;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 66.Eibl C, Grigoriu S, Hessenberger M, Wenger J, Puehringer S, Pinheiro AS, et al. Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry. 2012 Sep 18;51(37):7330–41. doi: 10.1021/bi3007059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinheiro AS, Eibl C, Ekman-Vural Z, Schwarzenbacher R, Peti W. The NLRP12 pyrin domain: Structure, dynamics, and functional insights. J Mol Biol. 2011 Nov 4;413(4):790–803. doi: 10.1016/j.jmb.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Atianand MK, Harton JA. Uncoupling of pyrin-only protein 2 (POP2)-mediated dual regulation of nf-κb and the inflammasome. J Biol Chem. 2011 Nov 25;286(47):40536–47. doi: 10.1074/jbc.M111.274290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khare S, Ratsimandresy RA, de Almeida L, Cuda CM, Rellick SL, Misharin AV, et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat Immunol. 2014 Feb 16;15(4):343–53. doi: 10.1038/ni.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jin T, Perry A, Smith P, Jiang J, Xiao TS. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem. 2013 May 10;288(19):13225–35. doi: 10.1074/jbc.M113.468033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin T, Perry A, Jiang J, Smith P, Curry J, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012 Apr;36(4):561–71. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porter KA, Duffy EB, Nyland P, Atianand MK, Sharifi H, Harton JA. The CLRX.1/NOD24 (NLRP2P) pseudogene codes a functional negative regulator of nf-κb, pyrin-only protein 4. Genes Immun. 2014 May 29;15(6):392–403. doi: 10.1038/gene.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dorfleutner A, Talbott SJ, Bryan NB, Funya KN, Reed JC, Shi X, et al. A shope fibroma virus pyrin-only protein modulates the host immune response. Virus Genes. 2007 Dec;35(3):685–94. doi: 10.1007/s11262-007-0141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnston JB, Barrett JW, Nazarian SH, Goodwin M, Ricuttio D, Wang G, McFadden G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity. 2005 Dec;23(6):587–98. doi: 10.1016/j.immuni.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 75.Lee S-H, Stehlik C, Reed JC. Cop, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. Journal of Biological Chemistry. 2001 Sep 14;276(37):34495–500. doi: 10.1074/jbc.M101415200. [DOI] [PubMed] [Google Scholar]
- 76.Druilhe A, Srinivasula SM, Razmara M, Ahmad M, Alnemri ES. Regulation of il-1beta generation by pseudo-ice and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ. 2001 Jun;8(6):649–57. doi: 10.1038/sj.cdd.4400881. [DOI] [PubMed] [Google Scholar]
- 77.Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, Tschopp J. Identification of CARDIAK, a rip-like kinase that associates with caspase-1. Current Biology. 1998 Jul 16;8(15):885–8. doi: 10.1016/s0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- 78.McCarthy JV, Ni J, Dixit VM. RIP2 is a novel nf-kappab-activating and cell death-inducing kinase. Journal of Biological Chemistry. 1998;273(27):16968–75. doi: 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- 79.Koseki T, Inohara N, Chen S, Carrio R, Merino J, Hottiger MO, et al. CIPER, a novel NF kappab-activating protein containing a caspase recruitment domain with homology to herpesvirus-2 protein E10. Journal of Biological Chemistry. 1999;274(15):9955–61. doi: 10.1074/jbc.274.15.9955. [DOI] [PubMed] [Google Scholar]
- 80.Bertin J, Nir WJ, Fischer CM, Tayber OV, Errada PR, Grant JR, et al. Human CARD4 protein is a novel CED-4/apaf-1 cell death family member that activates nf-kappab. Journal of Biological Chemistry. 1999 May 7;274(19):12955–8. doi: 10.1074/jbc.274.19.12955. [DOI] [PubMed] [Google Scholar]
- 81.Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, et al. Nod1, an apaf-1-like activator of caspase-9 and nuclear factor-kappab. J Biol Chem. 1999 May 21;274(21):14560–7. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 82.Lamkanfi M, Denecker G, Kalai M, D’Hondt K, Meeus A, Declercq W, et al. INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1beta generation. J Biol Chem. 2004 Dec 10;279(50):51729–38. doi: 10.1074/jbc.M407891200. [DOI] [PubMed] [Google Scholar]
- 83.Humke EW, Shriver SK, Starovasnik MA, Fairbrother WJ, Dixit VM. ICEBERG: A novel inhibitor of interleukin-1beta generation. Cell. 2000 Sep 29;103(1):99–111. doi: 10.1016/s0092-8674(00)00108-2. [DOI] [PubMed] [Google Scholar]
- 84.Chou JJ, Matsuo H, Duan H, Wagner G. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell. 1998 Jul 24;94(2):171–80. doi: 10.1016/s0092-8674(00)81417-8. [DOI] [PubMed] [Google Scholar]
- 85.Zhou P, Chou J, Olea RS, Yuan J, Wagner G. Solution structure of apaf-1 CARD and its interaction with caspase-9 CARD: A structural basis for specific adaptor/caspase interaction. Proc Natl Acad Sci USA. 1999 Sep 28;96(20):11265–70. doi: 10.1073/pnas.96.20.11265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vaughn DE, Rodriguez J, Lazebnik Y, Joshua-Tor L. Crystal structure of apaf-1 caspase recruitment domain: An alpha-helical greek key fold for apoptotic signaling. J Mol Biol. 1999 Oct 29;293(3):439–47. doi: 10.1006/jmbi.1999.3177. [DOI] [PubMed] [Google Scholar]
- 87.Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999 Jun 10;399(6736):549–57. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- 88.Bryan NB, Dorfleutner A, Kramer SJ, Yun C, Rojanasakul Y, Stehlik C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflamm (Lond) 2010;7:23. doi: 10.1186/1476-9255-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Matsushita K, Takeoka M, Sagara J, Itano N, Kurose Y, Nakamura A, Taniguchi S. A splice variant of ASC regulates il-1beta release and aggregates differently from intact ASC. Mediators Inflamm. 2009;2009:287387. doi: 10.1155/2009/287387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dowling JK, Becker CE, Bourke NM, Corr SC, Connolly DJ, Quinn SR, et al. Promyelocytic leukemia protein (PML) interacts with ASC to limit inflammasome activation. J Biol Chem. 2014 Jan 9; doi: 10.1074/jbc.M113.539692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin BN, Wang C, Willette-Brown J, Herjan T, Gulen MF, Zhou H, et al. IKKα negatively regulates asc-dependent inflammasome activation. Nat Commun. 2014;5:4977. doi: 10.1038/ncomms5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Masumoto J, Taniguchi S, Sagara J. Pyrin n-terminal homology domain- and caspase recruitment domain-dependent oligomerization of ASC. Biochem Biophysical Res Commun. 2001;280:652–5. doi: 10.1006/bbrc.2000.4190. [DOI] [PubMed] [Google Scholar]
- 93.Rosenstiel P, Huse K, Till A, Hampe J, Hellmig S, Sina C, et al. A short isoform of NOD2/CARD15, NOD2-S, is an endogenous inhibitor of NOD2/receptor-interacting protein kinase 2-induced signaling pathways. Proc Natl Acad Sci U S A. 2006 Feb 28;103(9):3280–5. doi: 10.1073/pnas.0505423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kramer M, Boeck J, Reichenbach D, Kaether C, Schreiber S, Platzer M, et al. NOD2-C2 - a novel NOD2 isoform activating nf-kappab in a muramyl dipeptide-independent manner. BMC Res Notes. 2010;3:224. doi: 10.1186/1756-0500-3-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004 May 6;429(6987):75–9. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 96.Saleh M, Mathison JC, Wolinski MK, Bensinger SJ, Fitzgerald P, Droin N, et al. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature. 2006 Apr 20;440(7087):1064–8. doi: 10.1038/nature04656. [DOI] [PubMed] [Google Scholar]
- 97.Liepinsh E, Barbals R, Dahl E, Sharipo A, Staub E, Otting G. The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J Mol Biol. 2003 Oct 3;332(5):1155–63. doi: 10.1016/j.jmb.2003.07.007. [DOI] [PubMed] [Google Scholar]