

Abstract
Objective
A risk factor for cardiovascular disease, hyperhomocystinemia (HHcy), is associated with endothelial dysfunction. In this study, we examined the mechanistic role of HHcy in endothelial dysfunction.
Methods and Results
Through the use of 2 functional models, aortic rings and intravital video microscopy of the cremaster, we found that arterial relaxation in response to the endothelium-dependent vessel relaxant, acetylcholine or the nitric oxide synthase (NOS) activator (A23187), was significantly impaired in cystathionine β-synthase null (CBS−/−) mice. However, the vascular smooth muscle cell (VSMC) response to the nitric oxide (NO) donor (SNAP) was preserved in CBS−/− mice. In addition, superoxide dismutase and catalase failed to restore endothelium-dependent vasodilatation. Endothelial nitric oxide synthase (eNOS) activity was significantly reduced in mouse aortic endothelial cells (MAECs) of CBS−/− mice, as well as in Hcy-treated mouse and human aortic endothelial cells (HAECs). Hcy-mediated eNOS inhibition—which was not rescued by adenoviral transduction of superoxide dismutase and glutathione peroxidase, or by tetrahydrobiopterin, sepiapterin, and arginine supplementations in MAEC—was associated with decreased protein expression and increased threonine 495 phosphorylation of eNOS in HAECs. Ultimately, a protein kinase C (PKC) inhibitor, GF109203X (GFX), reversed Hcy-mediated eNOS inactivation and threonine 495 phosphorylation in HAECs.
Conclusions
These data suggest that HHcy impairs endothelial function and eNOS activity, primarily through PKC activation.
Keywords: homocysteine, endothelial function, eNOS, protein kinase C
It is now recognized that hyperhomocystinemia (HHcy) is a common risk factor for cardiovascular disease (CVD) in the general population, similar to hyperlipidemia, hypertension, and smoking.1,2 Endothelial dysfunction is an early event in the development of atherosclerosis, occurring before morphological changes in the endothelium can be detected.3 Hcy-related endothelial dysfunction has been reported in mild HHcy in human and animal models. Hcy impairs brachial artery endothelial-dependent, flow-mediated dilatation (FMD) in elderly HHcy subjects.4 Moreover, short-term Hcy-lowering therapy improves FMD in children with chronic renal failure5 and in patients with coronary heart disease.6 Dietary-induced HHcy is associated with endothelial dysfunction in animal models, including mice.7,8
We have previously reported that pathophysiological concentrations of Hcy inhibit endothelial cell (EC) growth via hypomethylation and cyclin A transcriptional inhibition- related mechanisms;9,10 we have also demonstrated that HHcy accelerates spontaneous atherosclerosis in CBS/apolipoprotein E-deficient mice.11 Studies of cellular and dietary animal models have suggested that Hcy-educed endothelial dysfunction is mediated by oxidative inactivation of nitric oxide (NO) through the generation of superoxide via inhibition of the antioxidant enzyme glutathionine peroxidase (GPX-1)12 and the depletion of intracellular glutathionine,13 or by uncoupling endothelial nitric oxide synthase (eNOS) through the formation of asymmetrical dimethylarginine (ADMA).14 Notwithstanding, Hcy-mediated endothelial dysfunction has not been studied in a genetic animal model of severe HHcy.
Because endothelial dysfunction plays a significant role in the early development of CVD, it is vital to characterize the effect of Hcy on vascular function and ascertain the related mechanisms. Previously, the only genetic model of severe HHcy—CBS−/− mice—has not been viable for functional study, because of poor survivability.15 In the current study, we were able to obtain adult CBS−/− mice by enhancing maternal care. Subsequently, we assessed vascular reactivity and examined the mechanisms contributing to endothelial dysfunction in CBS−/− mice in MAECs and HAECs.
Methods
CBS-Deficient Mice and Hcy Measurement
CBS mice from The Jackson Laboratory (Bar Harbor, Me) were backcrossed 12 generations in a C57BL/B6 genetic background and genotyped by polymerase chain reaction (PCR).15 To improve maternal care, CBS−/− mice were identified before 2 weeks of age, and their nonexperimental littermates were euthanized. Mice were weaned at 6 weeks of age. At the age of 10 weeks, CBS littermates were selected for vascular reactivity studies. Mouse plasma was collected for Hcy and ADMA measurement.
Hcy and ADMA Measurement
Hcy concentrations were measured via liquid chromatography-electrospray tandem mass spectrometry methods, as previously described.11 Concentrations of ADMA were measured by a pre-column derivatization with dabsyl chloride (DABS-Cl), and derivatives were separated by high-performance liquid chromatography on reverse-phase columns.16
Organ Chamber and Aortic Ring Model
Aortic vasomotor responses were evaluated in mouse thoracic aortas. Three 3-mm aortic rings from each mouse were mounted in horizontal stirrups connected to a transducer and then an ETH-400 bridge amplifier (CB Sciences, Inc) to measure isometric tension. Vessel relaxation was induced by 10−5 M A23187, a calcium ionophore that activates eNOS, and the NO donor S-nitroso-N-acetylpenicillamine (SNAP) (10−6 M). In some experiments, vessels were preincubated with the nitric oxide synthase (NOS) inhibitor, N-nitro-L-arginine methyl ester hydrochloride (L-NAME) (3×10−5 mol/L), or superoxide dismutase (SOD) (200 U/mL) and catalase (140 U/mL) before the addition of acetylcholine.
Intravital Microscopy and Cremaster Microcirculation Model
Vascular function of the cremaster microvasculature was measured in sedated mice.17 The cremaster microvasculature was superfused with cumulative increments of acetylcholine (10−7 to 10−5 M) at 3-minute intervals. Vasomotor response was determined with a video analysis system through measurement of the inner lumen diameter of the arteriole immediately before the next dose of acetylcholine.
MAEC Isolation and HAEC Culture
Primary MAECs were isolated by modifying Matrigel18 and endothelial cell (EC) antibody purification19 procedures. Thoracic aortas from both CBS and control C57/B6 mice were divided into segments of ≈3 mm in length, cultured in mouse EC medium (DMEM supplemented with 20% fetal bovine serum [FBS], 50 μg/mL EC growth supplement, and 50 μg/mL heparin) for 6 days on BD Matrigel coated plates (BD Biosciences/Discovery Labware). Vessel rings were removed after MAEC outgrowth was observed. MAEC were purified with platelet-endothelial cell adhesion molecule-1 antibody (BD Biosciences/Pharmingen) (10 μg/mL for 15 minutes) and a magnetic anti-IgG microbead column (Miltenyi Biotec Inc). MAECs were further cultured on 0.2% gelatin-coated dishes, analyzed for endothelial specificity by flow cytometry assay using anti–platelet-endothelial cell adhesion molecule-1 antibody, and used for experiments at passage 3. HAEC (Clonetics Corporation) were cultured in human EC medium10 and used from passages 6 to 8. DL-Hcy was directly added to cell culture media in all experiments.
Western Blot Analysis
Whole cell extract (50 μg) from each sample was fractionated analyzed with antibodies against eNOS, eNOS phosphothreonine 495, or phosphoserine 1177 (BD Transduction Laboratories, BD Biosciences/Pharmingen).
eNOS Activity in MAECs and HAECs
eNOS activity was determined by measuring the conversion of 3H-Arg to 3H-citrulline in intact cells, and by assessing nitrite concentration in media using the Griess method. Citrulline assay: MAEC or HAEC were grown to confluence in 6-well plates, treated with Hcy for 24 hours, equilibrated with Krebs buffer for 5 minutes, and incubated with 3 μCi 3H-Arg and 10 mmol/L Arg for 15 minutes. Reactions were terminated by adding 2 mL stop solution (5 mmol/L Arg and 5 mmol/L EDTA in PBS). Cell lysates were applied to a cation exchange resin column (Dowex 50W-X8; Bio-Rad Laboratories). The neutrally charged citrulline was eluted, measured for radioactivity, and normalized via protein concentration. Griess method: Freshly collected (200 mL) cell culture supernatants were mixed with 200 μL Griess reagent (1:1 mixture of 1% sulfanilamide in 5% H3PO4 and 0.1% naphthylethylenediamine dihydrochloride in water) and incubated in 96-well plates for 10 minutes at room temperature. Absorbance at 543 nm was recorded. Nitrite concentration was calculated using sodium nitrite standards and normalized by cellular protein concentration.
Aortic Cyclic Guanosine Monophosphate Levels
Cyclic guanosine monophosphate (cGMP) content was measured as an indicator of eNOS activity in mouse aortas.8 The vessel was homogenized with 6% ice-cold trichloroacetic acid. cGMP content in aorta extracts was measured using a commercial immunoassay (Cayman Chemical Company).
Arg Transport Activity
Arg transport activity was measured by incubating confluent MAEC in 12-well plates with 2 μCi/mL 3H-Arg and 50 μmol/L cold L-Arg, in addition to 330 μmol/L of L-Arg endogenous to the medium. Arg transport activity was expressed as pmol intracellular 3H-Arg, per mg protein, per minute.20
Rescue of eNOS Activity in EC by Adenoviral Transduction of Extracellular SOD and GPX-1 by Supplementation of Arg, Tetrahydrobiopterin, Sepiapterin, or GF109203X
Adenoviruses expressing 2 antioxidant enzymes, human extracellular SOD (ecSOD) and GPX-1, were obtained from the Gene Transfer Vector Core, University of Iowa. MAEC at 90% confluence were infected with purified adenovirus9 at indicated multiplicity of infection for 24 hours. Infected MAECs were treated with Hcy for an additional 24 hours and subjected to the citrulline assay. Western blotting with anti-GPX-1 antibody (Medical & Biological Laboratories Co, Ltd, Nagoya, Japan) or anti-ecSOD antibody (provided gratis by Dr James D. Crapo, National Jewish Medical and Research Center) was used to confirm ectopic gene expression. Arg (1 mmol/L, 24 hours), sepiapterin (10 μmol/L, 24 hours), or tetrahydrobiopterin (BH4) (10 μmol/L, 5 minutes) were added to the culture medium before performing the citrulline assay. PKC inhibitor GF109203X (GFX) (2 μmol/L, 30 minutes) or PKC activator phorbol-12-myristate-13-acetate (PMA) (100 nmol/L, 10 minutes) (EMD Biosciences, Inc/Calbiochem), respectively, were added to cells 30 minutes or 10 minutes before citrulline assay.
SOD and GPX-1 Activities
SOD and GPX-1 activities were measured in MAECs infected with adenoviral ecSOD or GPX-1,21,22 through the use of a SOD or GPX-1 assay kit (EMD Biosciences, Inc/Calbiochem).
Statistics
Results are expressed as the mean±SEM. Statistical comparisons between 2 groups were performed via Student t test. One-way ANOVA was used to compare the means of multiple groups. P≤0.01 was considered significant.
Results
CBS Mice and Plasma Hcy/ADMA Levels
With improved mouse maternal care, the survival rate of CBS-null mice was increased from 1% to 10%. However, 90% of the CBS null mice died between 2 and 3 weeks of age; the survivors died at ≈6 to 8 months of age. The weight of CBS-null mice was 70.5% of the control mice (Figure 1B). Consistent with previous observations, plasma Hcy levels were increased ≈56-fold in CBS−/− mice (277±110.2 μmol/L) compared with CBS+/+ mice (4.9±1.3 μmol/L). CBS−/+ mice (7.5±2.1 μmol/L) had ≈2-fold increase in Hcy (Figure 1A). Plasma ADMA levels were similar between CBS+/+ (4.5±2.0 μmol/L) and CBS−/+ mice (4.6±1.7 μmol/L), and slightly decreased in CBS−/− mice (3.5±1.3 μmol/L) (Figure 1A).
Figure 1.
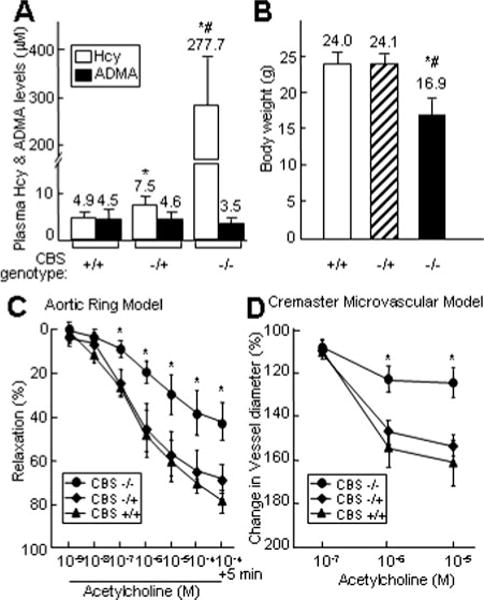
Plasma Hcy/ADMA levels, body weight, and vasorelaxation to acetylcholine in CBS mice. A. Plasma levels of Hcy and ADMA. Hcy concentrations were measured by liquid chromatography-electrospray tandem mass spectrometry. ADMA levels were measured by high-performance liquid chromatography (n = 12). B. Bodyweight of CBS mice at 10 weeks of age. C. Aortic vasomotor response. Mouse thoracic aortic rings were precontracted with KCl and contracted with phenylephrine. Dose-response relaxation was measured for cumulative increments of acetylcholine at 1 minute intervals (n=12). D. Cremaster microvascular vasomotor response. Mouse cremaster microvasculature was superfused with acetylcholine at 3 minutes intervals. The inner lumen diameter of the arteriole was measured before and after superfusion (n=10). Values are mean±SEM; *P<0.01 vs CBS+/+ mice; #P<0.01 vs CBS−/+ mice.
HHcy Impaired Endothelium-Dependent Arterial Relaxation in CBS−/− Mice
Acetylcholine produced a dose-dependent relaxation in precontracted aortic rings from the mice of all 3 CBS genotypes. However, vasorelaxation to acetylcholine was significantly impaired in CBS−/− mice, compared with CBS−/+ and CBS+/+ mice (Figure 1C). After incubation with 10−4 M acetylcholine, the CBS+/+ aortic segment relaxed by 81%±6% and that of the CBS−/+ relaxed by 68%±7%, whereas the CBS−/− aortic segment relaxed by only 42%±9%. Vasorelaxation in CBS−/+ mice was noticeably attenuated, but did not achieve statistical significance. Similar impairment was observed in the in vivo cremaster microcirculation model (Figure 1D). Cremaster arteriolar relaxation—as reflected by an increase in the inner lumen diameter of the arteriole, resulting from acetylcholine superfusion—was significantly impaired in CBS−/− mice, but not in CBS−/+ or CBS+/+ mice. Data from these 2 independent functional models supported one another, confirming that severe HHcy impairs endothelium-dependent vasodilatation in CBS−/− mice.
Antioxidants Failed to Restore Impaired Endothelium-Dependent Vasodilatation in CBS−/− Mice
We then assessed the potential mechanism of impaired endothelium-dependent relaxation in CBS−/− mice. CBS−/− mice exhibited a diminished capacity for aortic relaxation in response to the receptor-independent NOS activator, A23187 (Figure 2A), suggesting an impairment of the eNOS signaling pathway that is not specific to acetylcholine. To determine whether endothelial dysfunction in CBS−/− mice is caused by the impaired response of vascular smooth muscle cells (VSMC), we treated aortic rings with an NO donor, SNAP, and found that aortic rings from mice of all 3 genotypes relaxed similarly in response to increasing concentrations of SNAP (Figure 2B). The preserved response to SNAP indicated intact VSMC function in the CBS−/− mice, thereby demonstrating that VSMC response to NO is not altered by HHcy. Pretreatment of CBS vessels with L-NAME, a NOS-specific inhibitor, abolished the relaxation response to acetylcholine in all 3 genotypes (Figure 2C), suggesting that eNOS activity is required for acetylcholine dilatation. In addition, we examined whether oxidant stress contributes to endothelial dysfunction in CBS−/− mice. Before acetylcholine exposure, aortic rings were incubated with SOD and catalase to reduce superoxide. Acetylcholine dilatation remained suppressed in CBS−/− mice after SOD and catalase pretreatment (Figure 2D). Thus, NO-mediated endothelium-dependent vasorelaxation is impaired in CBS−/− mice, independent of changes in SMC response to NO or oxidant stress.
Figure 2.
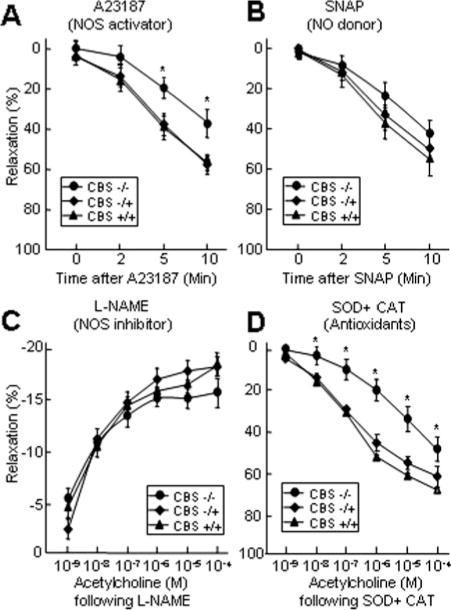
eNOS mediates HHcy impaired endothelium-dependent vasorelaxation. A and B. Vasorelaxation with A23187 and SNAP. Aortic rings were precontracted with KCl and contracted with phenylephrine. Vasorelaxation was measured in response to the addition of 10−5 M A23187 or 10−6 M SNAP, at the indicated times (n=9). C and D. Vasorelaxation to acetylcholine following L-NAME and OD+CAT. Aortic rings were precontracted with KCl, treated with 3×10−5 mol/L L-NAME for 20 minutes, or 200 U/mL SOD plus 140 U/mL CAT for 2 minutes, and contracted with phenylephrine. Dose-response relaxation was measured for cumulative increments of acetylcholine at 1 minute intervals (n=9). *P≤0.01 vs CBS−/+ or CBS+/+ mice, with identical treatment.
Hcy Inhibited eNOS Activity in MAECs and Mouse Aortas
Next, we assayed eNOS activity in MAECs by monitoring citrulline conversion in intact cells and measuring nitrite, the stable end product of NO, in the culture media. This direct measurement of eNOS activity in EC does not require the addition of any cofactors for reaction and it averts multiple confounding variables related to the actions of Hcy. We found that eNOS activity was significantly reduced in EC from CBS−/+ and CBS−/− mice, as well as in control MAECs treated with Hcy (Figure 3A, 3B, 3D, and 3E). Because NO causes vascular smooth muscle relaxation via cGMP formation—leading to the phosphorylation of cGMP-dependent protein kinases and calcium sequestration—we examined the contents of cGMP in the aortas from CBS mice as another method of measuring eNOS activity. Consistent with results from the aortic ring model (Figure 2), cGMP levels were not changed in the aortas from CBS−/+ mice, compared with CBS+/+ mice; however, cGMP levels were significantly decreased in CBS−/− aortas (Figure 3C). These data suggest that severe HHcy inhibits eNOS activity, resulting in endothelial dysfunction in CBS−/− mice.
Figure 3.
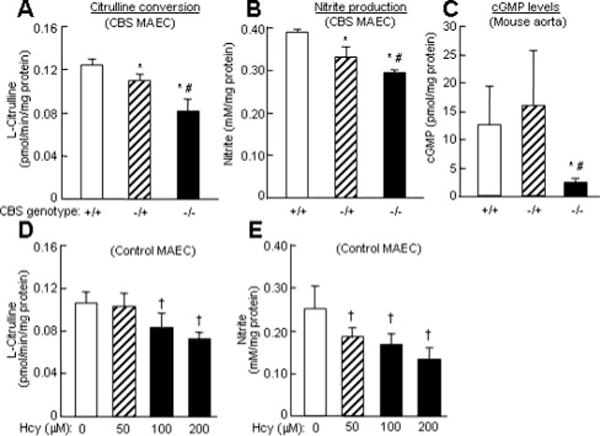
eNOS activity in CBS MAEC and aortas. A and D. Citrulline conversion in MAEC. B and E. Nitrite production in MAEC. Confluent CBS MAEC or control MAEC were incubated with DL-Hcy for 24 hour. eNOS activity was determined by measuring citrulline conversion and by nitrite production (n=9). C. cGMP levels in CBS aortas. Aortic cGMP accumulation with acetylcholine for 3 minutes in 8 week old CBS+/+ (n=8), CBS−/+ (n=8), and CBS−/− (n=4) mice. *P≤0.01 vs CBS+/+ mice; #P≤0.01 vs CBS−/+ mice; †P≤0.05 vs control MAEC without Hcy treatment; ‡P≤0.01 vs MAEC without Hcy treatment.
Hcy-Mediated eNOS Inactivation Was Not Corrected by the Overexpression of SOD and GPX-1 in MAECs
To examine oxidative stress hypothesis in Hcy-induced eNOS inhibition and endothelial dysfunction, we overexpressed antioxidant enzymes, SOD and GPX-1, via adenoviral transduction and assessed their effect on eNOS activity in MAECs. We found that adenoviruses expressing ecSOD and GPX-1 resulted in a corresponding dose-dependent increase in enzymatic activity of each protein in control MAECs (Figure 4B and 4D); however, this did not correct Hcy-suppressed eNOS activity in control MAEC (Figure 4A and 4C).
Figure 4.
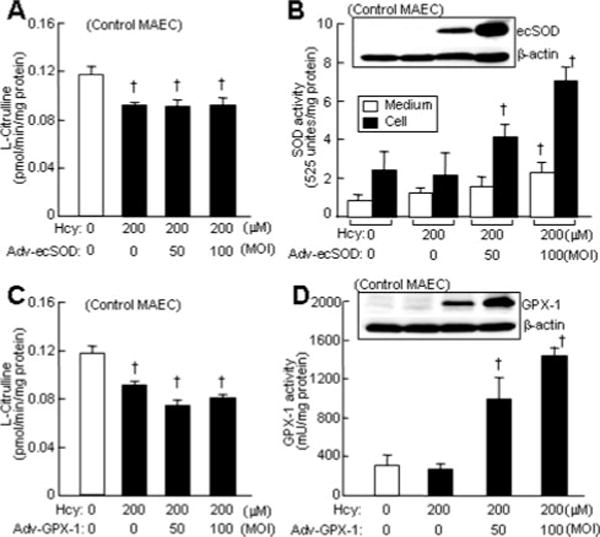
Effect of SOD or GPX-1 overexpression on eNOS activity in MAEC. Control MAEC at 90% confluence were infected with adenovirus vector (Adv-vector) or adenoviruses expressing ecSOD (Adv-ecSOD) or GPX-1 (Adv-GPX-1) at indicated MOI for 24 hour, and then treated with Hcy for 24 hour. eNOS activity was determined by measuring citrulline conversion. Ectopic gene expression was confirmed by Western blotting with antibodies against SOD or GPX-1. Enzymatic activities of SOD and GPX-1 were examined using commercial kits (n=9).
Hcy Did Not Change Arg Transport Activity in MAECs
eNOS activity can be regulated through the uptake of Arg, and it has been reported that Hcy inhibits eNOS activity by decreasing Arg transport activity in bovine aortic ECs.23 We examined the effect of Hcy on Arg transport activity and found that it was unchanged in Hcy-treated MAECs (Figure 5A).
Figure 5.
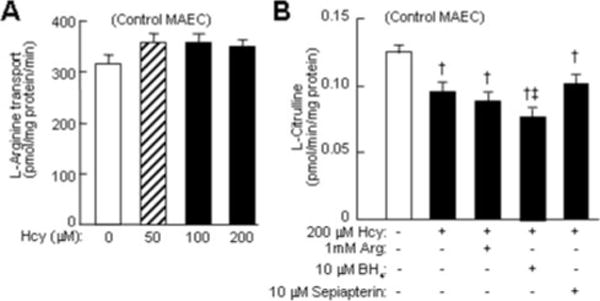
A. Arg transport activity. Confluent MAEC were incubated with DL-Hcy for 24 hour, and then with 2 μCi/mL 3H-L-Arg for 5 minutes. Arg transport activity is expressed as pmol intracellular 3H-L-Arg per mg protein per min (n=9). B. Effect of Arg, H4, and sepiapterin on eNOS activity. Confluent MAEC were incubated with DL-Hcy, and with Arg, BH4, or sepiapterin. eNOS activity was determined by measuring citrulline conversion (n=9). †P≤0.01 vs MAEC control; ‡P≤0.05 vs Hcy-treated MAEC (n=9).
Hcy-Mediated eNOS Inactivation in MAECs Was Not Corrected by Supplementation of Arg, BH4, and Sepiapterin
Arg is the exclusive substrate for eNOS. It has been suggested that extracellular Arg availability limits intracellular eNOS activity, and that Arg supplementation improves endothelium-dependent vasorelaxation.24 Furthermore, BH4 a cofactor of NOS, was recently reported to attenuate Hcy-mediated impairment of endothelium-dependent vasorelaxation in rat aortic rings.25 Notwithstanding, we found that the addition of Arg (1 mmol/L), BH4 (10 μmol/L), or sepiapterin (BH4 precursor, 10 μmol/L) to the culture media did not correct eNOS activity in Hcy-treated MAECs (Figure 5B).
Hcy Inhibited eNOS Activity and Protein Expression and Increased eNOS Threonine 495 Phosphorylation in HAECs
Regulation of NOS has been described at all levels, from gene expression to covalent modification. To address the relevance of Hcy/eNOS regulation to human endothelial biology, we studied eNOS activity and protein regulation in early passage HAEC. Consistent with our observations in MAECs (Figure 3B and 3C), pathophysiologically relevant concentrations of Hcy reduced eNOS activity in HAECs in a dose-sensitive manner (Figure 6A and 6B). Protein levels of eNOS were decreased by Hcy in a similar fashion (Figure 6C). In contrast, Hcy increased eNOS phosphorylation at threonine 495 (Thr-495), a negative regulatory site phosphorylated by PKC. Hcy did not cause a significant change in phosphorylation levels of eNOS at serine 1177 (Ser-1177), a positive regulatory site phosphorylated by Akt. Furthermore, a general PKC inhibitor, GFX, completely reversed eNOS Thr-495 phosphorylation by Hcy but did not affect eNOS Ser-1177 phosphorylation; also, it did not restore eNOS protein levels in the presence of HHcy. A PKC activator, PMA, induced eNOS Thr-495 phosphorylation, and suppressed eNOS Ser-1177 phosphorylation (Figure 6D).
Figure 6.
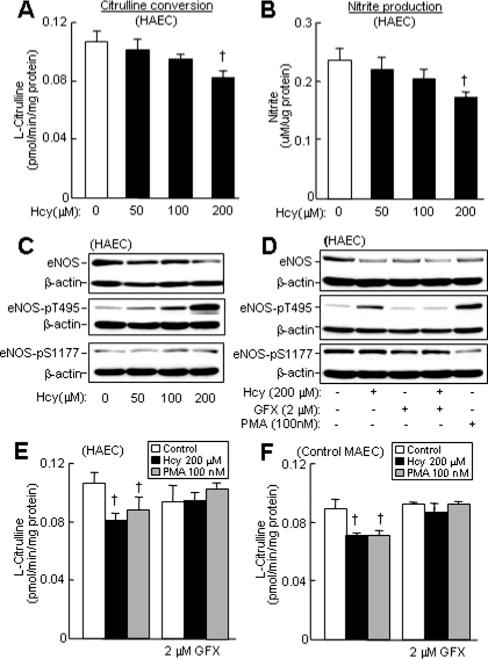
eNOS activity, expression, phosphorylation, and PKC activation in HAEC and MAEC. Confluent HAEC or MAEC were incubated with DL-Hcy for 24 hour. PMA were added for the last 10 minutes. GFX were added for the last 30 minutes. eNOS activity was determined by measuring citrulline conversion (A, E, and F) and by nitrite production (B) (n=9). †P≤0.01 vs HAEC or MAEC control. eNOS protein expression and phosphorylation were examined by Western blotting with antibodies against eNOS, eNOS-pT1177, or eNOS-pS495, and reblotted with β-actin antibody (C and D).
To determine the role of PKC activation in Hcy-induced eNOS inactivation, we incubated Hcy-treated HAECs or MAECs with GFX and assessed eNOS activity. Hcy-induced eNOS inactivation was largely restored by GFX, in both HAECs and MAECs (Figure 6E and 6F). GFX obviated the inhibition of eNOS activity in HAECs and MAECs through the PKC activator, PMA.
Discussion
In this study, we used CBS−/− mice as a genetic model of severe HHcy and found that severe HHcy impairs endothelium-dependent vasorelaxation through eNOS inhibition. We also found that eNOS activity is significantly reduced in both CBS-deficient MAECs and in Hcy-treated control MAECs. Hcy-induced eNOS inactivation is restored neither by Arg and BH4 supplementation nor by overexpression of the antioxidant enzymes SOD and GPX-1. Arg transport activity is not affected by Hcy treatment in MAECs. We further demonstrated that Hcy inhibits eNOS activity and protein expression in HAECs, which associates with increased eNOS phosphorylation at Thr-495. Finally, a PKC inhibitor, GFX, reverses Hcy-mediated eNOS inactivation in HAECs and MAECs, as well as Thr-495 phosphorylation in HAECs.
During the past few years, different HHcy animal models have been generated via dietary means. Animals develop moderate HHcy (plasma Hcy 20 to 30 μmol/L) with the addition of methionine to chow (2%) or drinking water (0.5%). Moreover, dietary-induced HHcy has been associated with vascular dysfunction7 and increased intimal hyperplasia26 in mice. The CBS−/− mouse is the only genetic model of severe human HHcy (plasma Hcy 300 μmol/L). However, until recently, the poor survivability of the CBS−/− mouse prevented its use in functional studies. Having ameliorated mouse maternal care through various methods, we have obtained sufficient numbers of CBS−/− mice for this and previous studies.11,27 It is our belief that the adult CBS−/− mouse is a valid model for mechanistic studies as its use averts dietary manipulations that may elicit broad physiological changes and complicate data interpretation.
We measured the vasomotor response of CBS mice ex vivo, using excised aortas in organ chamber, and in vivo, with arterioles from the cremaster microvascular bed. Data derived from these 2 independent functional models complement one another and confirm that severe HHcy impairs endothelium-dependent vasorelaxation. Although we observed mild impairment of endothelial vasorelaxation in CBS−/+ mice, it did not attain statistical significance. In this study we now demonstrate, for the first time to our knowledge, that severe HHcy significantly inhibits endothelial function in a patho-physiologically relevant animal model of human severe HHcy that does not use dietary manipulation.
Our vascular reactivity data suggest that defective eNOS signaling, but not altered VSMC responsiveness, is responsible for impaired endothelial function in CBS−/− mice, because VSMC response to the NO donor (SNAP) was preserved in these animals. In addition, we found that cGMP levels, another measurement of eNOS activity, were significantly reduced in CBS−/− aortas, and that eNOS activity was significantly decreased in the isolated ECs of CBS−/− mice. Furthermore, we demonstrated that Hcy—at physiologically relevant concentrations observed in CBS−/− mice and severe HHcy patients (100 to 200 μmol/L)—significantly inhibited eNOS activity in HAECs and MAECs. Thus, we have shown, for the first time to our knowledge, consistent eNOS inhibition in CBS−/− aortas, in CBS−/− MAECs, and in Hcy-treated human and mouse EC—which is associated with impaired endothelial function mediated by eNOS inactivation in CBS−/− mice.
It has been suggested that Hcy inhibits eNOS bioactivity by generating superoxide through autooxidation or by inhibiting the antioxidant enzyme GPX-1.12 However, we found that SOD and catalase failed to rescue the impaired endothe-lium-dependent vasodilatation in CBS−/− mice. In addition, we found that ectopic expression of ecSOD and GPX-1 by adenoviral transduction does not restore Hcy-inhibited eNOS activity in MAEC. Cumulatively, these data suggest that oxidative stress may not be the primary mechanism mediating Hcy-induced eNOS inhibition.
Because Arg transport activity was not changed by Hcy, and Arg and BH4 or BH4 precursor sepiapterin supplementation did not rescue eNOS inhibition in Hcy-treated MAECs, we believe that intracellular Arg or BH4 deficiencies do not play a significant role in Hcy-mediated eNOS inhibition. Also, because ADMA concentration is not increased in CBS−/− mice, our data do not support a primary role for ADMA in HHcy-mediated endothelial dysfunction and eNOS inactivation. Furthermore, Arg supplementation, which has been shown to reverse endothelium-dependent vascular function in subjects with high ADMA levels,28 does not overcome the inhibition of eNOS activity by Hcy in ECs.
Through our efforts to understand the molecular basis of eNOS regulation, we ascertained that Hcy decreases eNOS protein and increases eNOS phosphorylation at Thr-495, but not at Ser-1177. Ser-1177, in the reductase domain, and Thr-495, in the CaM-binding domain, are key regulatory sites for eNOS activity. eNOS Thr-495 (human) can be phosphorylated by AMP-activated kinase and PKC, resulting in reduced eNOS catalytic activity. In contrast, Ser-1177 (human) can be phosphorylated by phosphatidylinositol 3-kinase/Akt activation, which leads to eNOS activation.29 We found that a general PKC inhibitor, GFX, reversed Hcy-induced eNOS Thr-495 phosphorylation in HAECs and largely restored eNOS activity, but did not alter reduced eNOS protein levels in the presence of Hcy in both HAECs and MAECs. These data lead us to conclude that Hcy inhibits eNOS activity, primarily via activation of PKC in EC. Moreover, in HHcy, a reduction in eNOS protein levels may also contribute to a decrease in NO bioavailability in ECs. Because Hcy-induced eNOS inactivation is consistent in mouse arteries, as well as in HAECs and MAECs, PKC activation may constitute a primary mechanism by which Hcy impairs eNOS and endothelial function. Studies are currently underway to determine the role of PKC activation, in vivo, in Hcy-mediated endothelial dysfunction.
The activation of PKC by Hcy has been shown to stimulate spleen B lymphocyte proliferation30 and activate both macrophages31 and NFκB signaling in VSMCs.32 Thus, PKC activation may represent a common signaling pathway by which HHcy exerts its pathogenic actions in the vasculature. A substantial body of literature supports the importance of PKC activation in the development of various forms of CVD. Recent findings from clinical trials suggest that isoform-specific PKC inhibitors may be effective in the prevention or treatment of diabetic vascular complications.33 The identification of PKC isoforms in Hcy-mediated PKC activation may permit specific targeting in the development of pharmacological therapies that will reverse HHcy-induced endothelial dysfunction and CVD.
In summary, we have demonstrated that severe HHcy in CBS−/− mice causes endothelial dysfunction by eNOS inactivation and that Hcy inhibits eNOS activity via PKC activation in EC. Both PKC activation and eNOS inhibition have been implicated in the pathophysiology of several vascular disorders. Elucidating the molecular basis of PKC-mediated eNOS inactivation in Hcy signaling may provide important insight into the role of Hcy in CVD, and facilitate the identification of new therapeutic approaches in the treatment of cardiovascular disease.
Acknowledgments
This work was supported, in part, by the following National Institutes of Health grants: HL67033, HL74925, and HL77288 (H.W.), HL36045 (A.I.S.), HL59976 (W.D.), and HL64721 (R.R.).
Footnotes
Arterioscler Thromb Vasc Biol. is available at http://www.atvbaha.org
References
- 1.Wang H, Tan H, Yang F. Mechanisms in homocysteine-induced vascular disease. Drug Discovery Today (Disease mechanisms) 2005;2:25–31. [Google Scholar]
- 2.Yang F, Tan HM, Wang H. Hyperhomocysteinemia and atherosclerosis. Acta Physiologica Sinica. 2005;57:103–114. [PubMed] [Google Scholar]
- 3.Schulz E, Anter E, Keaney JF., Jr Oxidative stress, antioxidants, and endothelial function. Curr Med Chem. 2004;11:1093–1104. doi: 10.2174/0929867043365369. [DOI] [PubMed] [Google Scholar]
- 4.Tawakol A, Omland T, Gerhard M, Wu JT, Creager MA. Hyperhomocyst(e)inemia is associated with impaired endothelium-dependent vasodilation in humans. Circulation. 1997;95:1119–1121. doi: 10.1161/01.cir.95.5.1119. [DOI] [PubMed] [Google Scholar]
- 5.Bennett-Richards K, Kattenhorn M, Donald A, Oakley G, Varghese Z, Rees L, Deanfield JE. Does oral folic acid lower total homocysteine levels and improve endothelial function in children with chronic renal failure? Circulation. 2002;105:1810–1815. doi: 10.1161/01.cir.0000014417.95833.1d. [DOI] [PubMed] [Google Scholar]
- 6.Chambers JC, Ueland PM, Obeid OA, Wrigley J, Refsum H, Kooner JS. Improved vascular endothelial function after oral B vitamins: An effect mediated through reduced concentrations of free plasma homocysteine. Circulation. 2000;102:2479–2483. doi: 10.1161/01.cir.102.20.2479. [DOI] [PubMed] [Google Scholar]
- 7.Dayal S, Bottiglieri T, Arning E, Maeda N, Malinow MR, Sigmund CD, Heistad DD, Faraci FM, Lentz SR. Endothelial dysfunction and elevation of S-adenosylhomocysteine in cystathionine beta-synthase-deficient mice. Circ Res. 2001;88:1203–1209. doi: 10.1161/hh1101.092180. [DOI] [PubMed] [Google Scholar]
- 8.Eberhardt RT, Forgione MA, Cap A, Leopold JA, Rudd MA, Trolliet M, Heydrick S, Stark R, Klings ES, Moldovan NI, Yaghoubi M, Goldschmidt-Clermont PJ, Farber HW, Cohen R, Loscalzo J. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J Clin Invest. 2000;106:483–491. doi: 10.1172/JCI8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Jiang X, Yang F, Chapman GB, Durante W, Sibinga NE, Schafer AI. Cyclin A transcriptional suppression is the major mechanism mediating homocysteine-induced endothelial cell growth inhibition. Blood. 2002;99:939–945. [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Yoshizumi M, Lai K, Tsai JC, Perrella MA, Haber E, Lee ME. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J Biol Chem. 1997;272:25380–25385. doi: 10.1074/jbc.272.40.25380. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Jiang X, Yang F, Gaubatz JW, Ma L, Magera MJ, Yang X, Berger PB, Durante W, Pownall HJ, Schafer AI. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood. 2003;101:3901–3907. doi: 10.1182/blood-2002-08-2606. [DOI] [PubMed] [Google Scholar]
- 12.Weiss N, Keller C, Hoffmann U, Loscalzo J, van Guldener C, Nanayakkara PW, Stehouwer CD, De Bree A, Verschuren WM, Kromhout D, Kluijtmans LA, Blom HJ. Endothelial dysfunction and atherothrombosis in mild hyperhomocysteinemia. Vasc Med. 2002;7:227–239. doi: 10.1191/1358863x02vm428ra. [DOI] [PubMed] [Google Scholar]
- 13.Weiss N, Heydrick S, Zhang YY, Bierl C, Cap A, Loscalzo J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22:34–41. doi: 10.1161/hq1201.100456. [DOI] [PubMed] [Google Scholar]
- 14.Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98:1842–1847. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang JY, Knecht R, Braun DG. Amino acid analysis at the picomole level. Application to the C-terminal sequence analysis of polypeptides. Biochem J. 1981;199:547–555. doi: 10.1042/bj1990547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rumbaut RE, Randhawa JK, Smith CW, Burns AR. Mouse cremaster venules are predisposed to light/dye-induced thrombosis independent of wall shear rate, CD18, ICAM-1 or P-selectin. Microcirculation. 2004;11:239–247. doi: 10.1080/10739680490425949. [DOI] [PubMed] [Google Scholar]
- 18.Shi W, Haberland ME, Jien ML, Shih DM, Lusis AJ. Endothelial responses to oxidized lipoproteins determine genetic susceptibility to atherosclerosis in mice. Circulation. 2000;102:75–81. doi: 10.1161/01.cir.102.1.75. [DOI] [PubMed] [Google Scholar]
- 19.Marelli-Berg FM, Peek E, Lidington EA, Stauss HJ, Lechler RI. Isolation of endothelial cells from murine tissue. J Immunol Methods. 2000;244:205–215. doi: 10.1016/s0022-1759(00)00258-1. [DOI] [PubMed] [Google Scholar]
- 20.Durante W, Liao L, Iftikhar I, O’Brien WE, Schafer AI. Differential regulation of L-arginine transport and nitric oxide production by vascular smooth muscle and endothelium. Circ Res. 1996;78:1075–1082. doi: 10.1161/01.res.78.6.1075. [DOI] [PubMed] [Google Scholar]
- 21.Dayal S, Brown KL, Weydert CJ, Oberley LW, Arning E, Bottiglieri T, Faraci FM, Lentz SR. Deficiency of glutathione peroxidase-1 sensitizes hyperhomocysteinemic mice to endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2002;22:1996–2002. doi: 10.1161/01.atv.0000041629.92741.dc. [DOI] [PubMed] [Google Scholar]
- 22.Fennell JP, Brosnan MJ, Frater AJ, Hamilton CA, Alexander MY, Nicklin SA, Heistad DD, Baker AH, Dominiczak AF. Adenovirus-mediated overexpression of extracellular superoxide dismutase improves endothelial dysfunction in a rat model of hypertension. Gene Ther. 2002;9:110–117. doi: 10.1038/sj.gt.3301633. [DOI] [PubMed] [Google Scholar]
- 23.Jin L, Abou-Mohamed G, Caldwell RB, Caldwell RW. Endothelial cell dysfunction in a model of oxidative stress. Med Sci Monit. 2001;7:585–591. [PubMed] [Google Scholar]
- 24.Tousoulis D, Antoniades C, Tentolouris C, Goumas G, Stefanadis C, Toutouzas P. L-arginine in cardiovascular disease: dream or reality? Vasc Med. 2002;7:203–211. doi: 10.1191/1358863x02vm434ra. [DOI] [PubMed] [Google Scholar]
- 25.Dhillon B, Badiwala MV, Maitland A, Rao V, Li SH, Verma S, Powers RW, Majors AK, Cerula SL, Huber HA, Schmidt BP, Roberts JM, Haynes WG, McGregor DO, Buttimore AL, Lynn KL, Yandle T, Nicholls MG, Holven KB, Haugstad TS, Holm T, Aukrust P, Ose L, Nenseter MS. Tetrahydrobiopterin attenuates homocysteine induced endothelial dysfunction. Mol Cell Biochem. 2003;247:223–227. doi: 10.1023/a:1024146501743. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ, Kohl B, Rao V, Kisiel W, Stern DM, Schmidt AM. Hyperhomocys-teinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J Clin Invest. 2001;107:675–683. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vitvitsky V, Dayal S, Stabler S, Zhou Y, Wang H, Lentz SR, Banerjee R. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol. 2004;287:11. doi: 10.1152/ajpregu.00036.2004. [DOI] [PubMed] [Google Scholar]
- 28.Boger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004;134 doi: 10.1093/jn/134.10.2842S. [DOI] [PubMed] [Google Scholar]
- 29.Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299:818–824. [PubMed] [Google Scholar]
- 30.Zhang Q, Zeng X, Guo J, Wang X. Effects of homocysteine on murine splenic B lymphocyte proliferation and its signal transduction mechanism. Cardiovasc Res. 2001;52:328–336. doi: 10.1016/s0008-6363(01)00376-5. [DOI] [PubMed] [Google Scholar]
- 31.Beauchamp MC, Renier G. Homocysteine induces protein kinase C activation and stimulates c-Fos and lipoprotein lipase expression in macrophages. Diabetes. 2002;51:1180–1187. doi: 10.2337/diabetes.51.4.1180. [DOI] [PubMed] [Google Scholar]
- 32.Wang G, Woo CW, Sung FL, Siow YL, Karmin O. Increased monocyte adhesion to aortic endothelium in rats with hyperhomocysteinemia: role of chemokine and adhesion molecules. Arterioscler Thromb Vasc Biol. 2002;22:1777–1783. doi: 10.1161/01.atv.0000035404.18281.37. [DOI] [PubMed] [Google Scholar]
- 33.Shen GX. Selective protein kinase C inhibitors and their applications. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:301–307. doi: 10.2174/1568006033481375. [DOI] [PubMed] [Google Scholar]