

SUMMARY
Little is known about the role of negative regulators in controlling natural killer (NK) cell development and effector functions. Foxo1 is a multifunctional transcription factor of the forkhead family. Using a mouse model of conditional deletion in NK cells, we found that Foxo1 negatively controlled NK cell differentiation and function. Immature NK cells expressed abundant Foxo1 and little Tbx21 relative to mature NK cells, but these two transcription factors reversed their expression as NK cells proceeded through development. Foxo1 promoted NK cell homing to lymph nodes through upregulating CD62L expression, and impaired late-stage maturation and effector functions by repressing Tbx21 expression. Loss of Foxo1 rescued the defect in late-stage NK cell maturation in heterozygous Tbx21+/− mice. Collectively, our data reveal a regulatory pathway by which the negative regulator Foxo1 and the positive regulator Tbx21 play opposing roles in controlling NK cell development and effector functions.
INTRODUCTION
Natural killer (NK) cells provide early surveillance against malignant transformation and viral clearance (Di Santo, 2006; Vivier et al., 2011). The capacity of NK cells to directly lyse target cells is determined by the balance of activating and inhibitory receptors (Lanier, 2005). Another mechanism by which NK cells eliminate their target cells is through secretion of cytokines, such as the pro-inflammatory cytokine interferon-γ (IFN-γ), which participate in shaping the adaptive immune response (Vivier et al., 2012).
Murine NK cells develop in the bone marrow from common lymphoid progenitor cells through three continuous stages defined as CD122+NK1.1−DX5− precursor NK (pNK) cells, CD122+NK1.1+DX5− immature NK (iNK) cells, and CD122+NK1.1+DX5+ mature NK (mNK) cells, respectively (Colucci et al., 2003; Ramirez and Kee, 2010). After acquisition of NK1.1 surface expression, murine NK cells can be further classified into three stages based on surface expression of CD11b and CD27: CD11b−CD27+, CD11b+CD27+, CD11b+CD27− (Chiossone et al., 2009; Hayakawa and Smyth, 2006; Huntington et al., 2013; Narni-Mancinelli et al., 2011). NK cell development, maturation and effector functions depend on extrinsic and intrinsic factors, and are controlled by both positive and negative regulators (Hesslein and Lanier, 2011). For extrinsic factors, we and others previously showed that interleukin-15 (IL-15) plays a critical role in promoting NK cell development and effector functions (Yu et al., 2013), while transforming growth factor-β (TGF-β) has an important inhibitory role in this respect (Allan et al., 2010; Marcoe et al., 2012; Yu et al., 2006). Intrinsic factors, including the transcription factors Ikaros, Pu.1, Ets1, and VDUP-1, regulate the early stages of NK development by promoting the generation of pNK, while E4BP4, Id2 and MEF function during the iNK stage to promote early NK maturation, and GATA-3, TOX, Tbx21, Eomes and IRF2 mainly modulate late-stage NK maturation (Colucci et al., 2003; Hesslein and Lanier, 2011; Luevano et al., 2012; Ramirez and Kee, 2010). The key role of Tbx21 in regulating late-stage maturation of NK cells is supported by the fact that the final stage CD11b+CD27− subpopulation is almost absent in Tbx21–deficient (Tbx21−/−) mice (Townsend et al., 2004). In addition, Tbx21−/− mice are susceptible to challenge with tumor cells, while infusion of wild-type (WT) NK cells restores anti-tumor capacity in vivo (Werneck et al., 2008).
Until now, at least two fundamental questions regarding NK cell development remain unaddressed. These include the nature of the signaling pathway upstream of Tbx21 that controls late-stage NK cell maturation and function, and also whether any intrinsic checkpoint factors negatively regulate NK cell development. The latter question is important as negative regulators or checkpoints are undoubtedly involved in NK cell development or maturation, while all aforementioned transcription factors that have been identified as participating in this process are positive regulators. Foxos are transcription factors whose expression is associated with the generation of common lymphoid progenitors and the regulation of T cell and B cell development and function (Chow et al., 2013; Hedrick et al., 2012; Hess Michelini et al., 2013; Kim et al., 2013; Ouyang et al., 2012; Staron et al., 2014; Togher et al., 2015). Some of these elegant studies also demonstrate that Foxo1 and Foxo3 regulate their target genes in a highly cell- and context-specific mechanism. This underscores the need for exploring Foxo’s unique role in NK cell development and function. Here we show that Foxo1, and/or to a lesser extent Foxo3, control NK cell homing, maturation and anti-tumor activity. In addition, we demonstrate that the inhibitory role of Foxo1 on NK cell maturation depends on its repressive activity on Tbx21 expression. These findings highlight the importance of negative regulatory checkpoints on NK cell development and activity, and reveal novel opportunities for manipulating NK cell activity.
RESULTS
Foxo transcription factors control NK cell homing
Intrinsic negative regulators of NK cell development have generally not been well described. Phosphorylated Akt was reported to inactivate Foxo transcription factors by inducing their exit from the nucleus (Calnan and Brunet, 2008). While the Foxo family of transcription factors include four members – Foxo1, 3, 4 and 6 – extensive comparative analysis of gene expression databases revealed that NK cells express Foxo1, and to a lesser extent Foxo3, but have no apparent expression of Foxo4 or Foxo6 (data not shown). To determine their role in NK cell biology, we crossed Ncr1iCre mice (Narni-Mancinelli et al., 2011) with mice carrying floxed Foxo1 alleles (Foxo1fl/fl) as well as floxed Foxo3 alleles (Foxo3fl/fl) to generate three NK cell-specific conditional knock-out models, referred as to Foxo1ΔNK, Foxo3ΔNK and Foxo1,3ΔNK respectively. Whereas the expression of a single Foxo1 or Foxo3 allele was sufficient to maintain expression of the proteins, intracellular flow cytometry confirmed the efficient and specific depletion of Foxo1 and/or Foxo3 in CD3−CD19−NKp46+ cells when both floxed alleles were present (Figure 1A). Initial flow cytometric analyses showed that Foxo1 and/or Foxo3 deficiency does not alter the quantity of Lin−CD122+ cells, nor the proportions of NK1.1+NKp46+ cells in the bone marrow, thus excluding a role of Foxo1 and 3 in early steps of NK cell development (Figure 1B). However, looking in the periphery, we found that while NK cell proportions and cell numbers were almost unchanged in spleen and liver, they were selectively decreased in the lymph nodes of Foxo1ΔNK and Foxo1,3ΔNK mice, suggesting impaired homing properties (Figure 1C). Foxo1 has been shown to control T cell trafficking by regulating the expression of several homing receptors including CD62L (Kerdiles et al., 2009). Consistent with these data, we observed an increased proportion of CD62L− NK cells in lymphoid organs of Foxo1ΔNK but not Foxo3ΔNK mice, which was mostly attributed to defective expression of CD62L on immature CD11b−CD27+ and, to a lesser extent, on CD11b+CD27+ cells (Figure 1D). Importantly, we also found that this phenotype was enhanced in Foxo1,3ΔNK when compared to Foxo1ΔNK NK cells. These data demonstrated that Foxo1 transcription factor regulates NK cell homing and further suggested that both Foxo1 and Foxo3 control CD62L expression in developing NK cells, although Foxo1 is able to compensate for the absence of Foxo3 (Figure 1D). Altogether, these data revealed that Foxo transcription factors are dispensable for the early steps of NK cell development but redundantly control NK cell homing in the periphery, by regulating CD62L expression.
Figure 1. Foxo transcription factors deficiency impairs NK cell homing.

(A) Intracellular flow cytometric analysis of Foxo1 and Foxo3 expression in splenic B cells (CD3−CD19+), T cells (CD3+NK1.1−) and NK cells (CD19−CD3−NK1.1+NKp46+) from mice of the indicated genotypes. (B) Flow cytometric analysis and enumeration of Lin−CD122+ bone marrow (BM) cells. (C) Frequency and enumeration of NK cells in the indicated organs (pLN, periphery lymph node). (D) Flow cytometry analysis of CD62L expression on CD19−CD3− NK1.1+NKp46+ cells from the indicated organs, as well as on the indicated NK cell subpopulations in the spleen. Each dot indicates one mouse (*p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001; unpaired two-tailed Student’s t test with Welsh’s correction).
Foxo transcription factors control NK cell maturation
We then examined the role of Foxo1 and Foxo3 in NK cell maturation. In the spleen, we noticed an overall increase in the frequency of the most mature CD11b+CD27− population in Foxo1ΔNK, Foxo3ΔNK, Foxo1,3ΔNK mice, which was associated with a decrease in the CD11b+CD27+ population in Foxo1ΔNK and Foxo1,3ΔNK mice (Figure 2A). Despite the reduced representation of mature NK cells in these organs, we observed a similar phenotype in the bone marrow and lymph nodes of Foxo1ΔNK and Foxo1,3ΔNK mice. During the final stages of maturation, NK cells sequentially acquire the expression of CD11b, downregulate CD27 gene expression, and finally upregulate the expression of CD43 (Chiossone et al., 2009; Hayakawa and Smyth, 2006; Kim et al., 2002; Yokoyama et al., 2004). The above results support the idea that Foxo transcription factors inhibit the progression of CD11b+ NK cells across the latest maturation stages. Accordingly, a specific analysis of CD11b+ NK cells revealed a strong bias towards an overrepresentation of CD27− cells over CD27+ cells in the spleen and lymph nodes of Foxo1ΔNK and Foxo1,3ΔNK mice (Figure 2B). In further support of these results, CD43lo NK cells from Foxo1ΔNK and Foxo1,3ΔNK mice displayed a robust downregulation of CD27 expression, which was also apparent – though less important – in Foxo3ΔNK mice (Figure 2C). Finally, we also observed that Foxo1 deficiency, Foxo3 deficiency, and their double knock-out were associated with increased proportions of KLRG1+ cells, whose expression is generally associated with NK cell terminal differentiation (Huntington et al., 2007; Narni-Mancinelli et al., 2011) (Figure 2D).
Figure 2. Foxo transcription factors inhibit NK cell maturation.

(A) Flow cytometric analysis and cumulative frequencies of NK cell subpopulations in the indicated organs, based on CD11b and CD27 expression (pLN, periphery lymph nodes; BM, bone marrow). (B) Calculated ratio between CD27− versus CD27+ cells among CD11b+ NK cells, based on data displayed in (A). (C) Flow cytometric analysis and cumulative results of CD27 expression on CD43lo gated NK cells. (D) Cumulative frequencies of KLRG1-expressing NK cells (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001; unpaired two-tailed Student’s t test with Welsh’s correction). See also Figure S1.
To investigate whether Foxo1 deficiency affects the kinetics of NK cell maturation, we separately transplanted an equal number of bone marrow cells from CD45.2 Foxo1ΔNK mice or Foxo1fl/fl control mice into lethally irradiated CD45.1 congenic mice. At one, two, and three weeks after transplantation, significantly more CD11b+CD27− (mature) but fewer CD11+CD27+ (immature) NK cells were observed in the CD45.1 mice that received bone marrow cells from Foxo1ΔNK donors, as compared to those infused with bone marrow cells from Foxo1fl/fl control donors (Figure S1A). To determine whether this increase in mature NK cells was stem cell intrinsic or microenvironment-dependent, we created chimeras in Rag2−/−Il2rg−/− mice (lacking T cells, B cells and NK cells) by injecting bone marrow cells from CD45.1 WT and CD45.2 Foxo1ΔNK mice, mixed at a 1:1 ratio. As shown by flow cytometry at 6 weeks post-transplantation, a greater proportion of mature but fewer immature NK cells were derived from CD45.2 Foxo1ΔNK bone marrow cells than from CD45.1 WT control cells (Figure S1B), suggesting that NK cell maturation controlled by Foxo1 is cell intrinsic.
Our above data thus revealed that, in addition to their role in regulating NK cell homing, Foxo transcription factors also repress maturation of CD11b+CD27+ to CD11b+CD27− NK cells. Moreover, although the data indicate that both Foxo1 and Foxo3 act redundantly, they strongly suggest that Foxo1 plays a prominent role, possibly owing to its higher expression relative to Foxo3 in NK cells as we aforementioned.
Foxo protein deficiency enhances NK cell effector functions
Our finding that Foxo deficiency enhances NK cell maturation motivated us to determine if Foxo transcription factors also repress effector functions. We first tested whether the absence of Foxo1 and/or Foxo3 affect NK cell response to cytokine stimulation. When stimulated with the highly potent IL-12 and IL-18 cytokine combination or PMA and ionomycin, we observed that the overall proportion of IFN-γ-secreting cells was not affected regardless of genotype. However, the absence of Foxo factors was associated with an increased ‘per-cell’ response to an IL-12 and IL-18 co-stimulation, as shown by the increased IFN-γ geometric (g) MFI of secreting cells (Figure 3A). In order to validate and gain further insight into this effect, we turned to an in vitro system using the human NK cell line, NKL, and a PINCO-Foxo1-GFP plasmid allowing us to overexpress Foxo1 (Figure S2A). Cytokine induction of IFN-γ production was diminished in NKL cells overexpressing Foxo1 compared to those infected with an empty control vector (Figure 3B). The inhibitory effect of Foxo1 on cytokine activated NK cells was discernible even in the presence of inhibitory TGF-β signaling (Figure 3B). Conversely, we found that IFNG mRNA transcript was increased when Foxo1 expression was knocked down by short hairpin (sh) RNA in NKL cells (Figure 3C and Figure S2B).
Figure 3. Enhanced IFN-γ secretion following cytokine stimulation of Foxo-deficient NK cells.

(A) Intracellular flow cytometric analysis of IFN-γ production by splenic NK cells co-stimulated with IL-12 and IL-18, or PMA and ionomycin (Iono) for 4 hours in the presence of Brefeldin A (Error bars, S.D of experimental duplicates. One out of four experiments with a total of 6 to 10 mice per group). (B) NKL cells transduced with PINCO-Foxo1 or PINCO empty vector were stimulated with the cytokines indicated for 18 hours for ELISA analysis of IFN-γ. (C) NKL cells expressing Foxo1 shRNA (shFoxo1) or empty vector control (EV) were treated with IL-12 for 12 hours and harvested for real-time RT-PCR to assess IFNG mRNA. (D) NKL cells expressing Foxo1-ER were treated with 4-OH-tamoxifen or ethanol carrier for indicated time points, followed by real-time RT-PCR analysis of IFNG mRNA. Data shown in (B)-(D) are representative of at least two independent experiments, and error bars represent S.D. * p < 0.05 and ** p < 0.01 (unpaired two-tailed Student’s t test). See also Figure S2.
To further examine the negative regulatory effects of Foxo1 on IFN-γ gene expression, an inducible gene regulation approach was undertaken. We infected NKL cells with a retrovirus encoding a fusion protein consisting of Foxo1 and the estrogen receptor ligand-binding domain (Foxo1-ER) (Amin and Schlissel, 2008), which becomes activated with the addition of the estrogen analog 4-hydroxytamoxifen (4-OH-tamoxifen). After confirming expression of Foxo1-ER in NKL cells with intracellular staining (Figure S2C), we found that IFNG mRNA transcript in cytokine-activated NKL cells was decreased at 3 and 6 hours after the addition of 4-OH-tamoxifen (Figure 3D). Collectively, these data indicate that Foxo1 is a negative regulator of NK cell effector function, in that its deficiency enhances NK cell IFN-γ production and its overexpression diminishes it.
We then tested the cytotoxic potential of Foxo-deficient NK cells. Directly ex vivo, Foxo1-deficient NK cells had increased cytotoxic potential against the prototypic Yac-1 target cells (Figure 4A). Further, overexpression of Foxo1 in NKL cells inhibits killing of K562 cells (Figure 4B). In this context, we postulated that loss of Foxo1 in NK cells might enhance their ability to prevent in vivo tumor progression. We utilized the B16F10 melanoma model of tumor metastasis (Werneck et al., 2008) to investigate NK cell-mediated tumor surveillance in vivo by enumerating the pulmonary tumor nodules two weeks after intravenous injection of B16F10 melanoma cells. We observed a marked decrease in the number of melanoma nodules in the lungs of Foxo1ΔNK mice compared with WT littermate controls (Figure 4C). Altogether, these data demonstrate that Foxo transcription factors restrain NK cell functions and anti-metastatic activity in vivo.
Figure 4. Loss of Foxo1 enhances cytotoxic and anti-metastatic potential of NK cells.
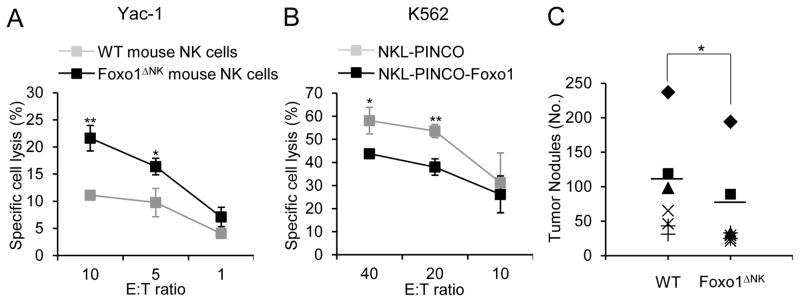
(A) Assessment of natural cytotoxicity of FACS-sorted splenic NK cells against Yac-1 target cells using standard 51Cr-release assay (Error bars, S.D; n = 6; * p < 0.05 and ** p < 0.01; unpaired two-tailed Student’s t test). (B) NKL cells transduced with PINCO-Foxo1 or PINCO empty vector were used for a standard 51Cr-release assay to determine cytotoxicity against target K562 cells (representative of at least two independent experiments, and error bars represent S.D. * p < 0.05 and ** p < 0.01; unpaired two-tailed Student’s t test). (C) Representative and summarized data of lung nodules from 8- to 12-week-old WT and Foxo1ΔNK mice 2 weeks after I.V. injection of 0.25×106 B16F10 melanoma cells. Each symbol represents an individual mouse, and the same symbols from the two groups indicate littermate pairs (* p < 0.05; generalized linear models; n = 7 to 10 for each genotype). See also Figure S2.
Pro-inflammatory cytokines and in vivo tumor challenge inactivate Foxo1
We previously showed that pro-inflammatory cytokines can inhibit negative regulators such as SHIP1 and SMAD in NK cells (Trotta et al., 2005; Yu et al., 2006). IL-12 is an important cytokine for regulating the NK cell effector functions, such as IFN-γ production, and we and others also previously showed that pro-inflammatory cytokines such as IL-15 and IL-2 are critical factors for controlling NK cell development and function (Freud et al., 2005; Yu et al., 2013). Phosphorylation of Foxo transcription factors may interfere with their DNA binding activity and trigger their sequestration in the cytoplasm, thereby leading to their inactivation (Calnan and Brunet, 2008). Thus, if Foxo transcription factors are negative regulators of NK cell development and function, it is possible that their phosphorylation status is impacted by these pro-inflammatory cytokines. To assess this, the human NK cell line NK-92 was incubated with IL-15, IL-2, or IL-12, separately, for 5–120 min, and Foxo1 phosphorylation was measured. Compared to untreated samples, IL-15 induced phosphorylation of Foxo1 as early as 5 min, and the phosphorylation remained elevated at subsequent time points. IL-2 and IL-12 also induced phosphorylation of Foxo1 at all time-points, while the phosphorylation was highest at 120 min after treatment (Figure 5A). We further confirmed these human data in mouse spleen NK cells by intracellular staining for phospho-Foxo1Ser256 (Figure 5B). These observations therefore suggest that the pro-inflammatory cytokines that activate NK cell effector function and/or promote NK cell development can antagonize inhibitory signaling through the phosphorylation and subsequent inactivation of Foxo1. These data are consistent with our previous findings that pro-inflammatory cytokine signaling has a capacity to antagonize inhibitory TGF-β signaling (Yu et al., 2006). We also found that the ratio of phospho-Foxo1Ser256/Foxo1 was higher in murine NK cells belonging to CD11b+CD27+ population than CD11b− CD27+ population, and was highest in CD11b+CD27− cells (Figure S3). We next determined the amount of Foxo1 phosphorylation after NK cells had been activated by tumor cells in vivo. After challenged with B16F10 tumor cells, lung tissue NK cells also showed increased Foxo1 phosphorylation, while the amount of total Foxo1 remained unchanged (Figure 5C). Together with their inhibitory role in effector functions, our data therefore suggest that Foxo1 transcription factor inactivation upon NK cell stimulation is required for their ‘full-blown’ activation. Furthermore, Foxo1’s regulation of activity might act as an important negative checkpoint controlling NK activation.
Figure 5. Pro-inflammatory cytokines and tumor challenges induce Foxo1 phosphorylation.

(A) NK-92 cells were stimulated with IL-15 (100 ng/ml), IL-2 (200 IU) or IL-12 (10 ng/ml). At indicated time points, cells were harvested and lysed, followed by immunoblot using a phosphorylated (p)-Foxo1Ser256 antibody and a β-actin antibody (internal control). Numbers under each lane represent densitometric quantification of p-Foxo1Ser256, after normalizing to β-actin. Data shown are representative of two independent experiments with similar results. (B) Intracellular staining of p-Foxo1 in splenic NK cells (gated on CD3−NK1.1+) after being stimulated with or without IL-15 (100 ng/ml), IL-2 (200 IU) or IL-12 (10 ng/ml) for 60 minutes. (C) Intracellular staining of p-Foxo1 and total Foxo1 in isolated lung tissue NK cells seven days after I.V. injection with B16F10 melanoma cells. Data in (B) and (C) are representative of three different experiments with three mice per group for each experiment. See also Figure S3
Differential mechanisms of Foxo1 suppressing Tbx21 expression in human and mouse NK cells
Foxo1 regulates NK cell development and effector function in a manner which is opposite to that reported for Tbx21 (Gordon et al., 2012; Townsend et al., 2004), but both control NK cell maturation at the late-stage checkpoint. To investigate whether Foxo1 may regulates Tbx21 expression or vice versa during NK cell maturation, the temporal and spatial expression of Foxo1 and Tbx21 was assessed during NK cell maturation across the developmental stages defined by CD11b and CD27 surface expression. Both intracellular flow cytometry and real-time reverse transcriptase (RT)-PCR demonstrated higher Foxo1 expression in CD11b−CD27+ NK cell subpopulation compared with CD11b+CD27+ and CD11b+CD27− subpopulations (Figure 6A), whereas Tbx21 had the opposite expression pattern (Figure 6B).
Figure 6. Foxo1 inhibits Tbx21 expression in human NK cells through direct promoter binding and in murine NK cells through recruitment by Sp1 to the proximal promoter region.

(A, B) Intracellular staining and mRNA content of Foxo1 (A) and Tbx21 (B) in CD11b−CD27+, CD11b+CD27+ and CD11b+CD27− splenic NK cells from 8-week-old WT C57BL/6 mice. The relative mean fluorescence intensity (MFI) and mRNA of Foxo1 or Tbx21 in each NK cell subset, normalized to that detected in the CD11b−CD27+ subset, is shown in the middle and right panels, respectively. (C) Tbx21 protein expression assessed by intracellular staining of splenic NK cells from WT or Foxo1ΔNK mice. (D) Tbx21 mRNA expression in FACS-sorted splenic NK cells from WT or Foxo1ΔNK mice measured by real-time RT-PCR. (E) Relative MFI of Tbx21 protein in CD11b−CD27+, CD11b+CD27+ and CD11b+CD27− splenic NK cells of WT and Foxo1ΔNK mice. (F) TBX21 transcript quantification by real-time RT-PCR in NKL cells expressing Foxo1-ER after addition of 4-OH-tamoxifen for the indicated time in the presence of IL-12. (G) TBX21 transcript quantification by real-time RT-PCR in NKL cells expressing shRNA (shFoxo1) or the empty vector control (EV) following stimulation with IL-12 for 12 hours. (H) Schematic structure of the human TBX21 promoter with putative Foxo1 binding sites (top panel) and ChIP assay of Foxo1 binding to the TBX21 promoter in human NKL cells (bottom panel). The DNA precipitated by Foxo1 antibody was detected by PCR. (I, J) Immunoblotting of endogenous Sp1 and Foxo1 in Foxo1 immunoprecipitates from murine splenocytes (I) or Foxo1 and Sp1 immunoprecipitates from murine NK cells (J). (K) Recruitment of Foxo1 to the mouse Tbx21 promoter region by Sp1. Chromatin from wild-type murine NK cells was precipitated with anti-Foxo1 antibody, anti-Sp1 antibody, or non-immune IgG. The precipitated DNA fragments were subjected to PCR amplification using specific primers directed against the proximal Tbx21 promoter region spanning from −160 to +110 bp relative to the transcription start site. *p < 0.05, and **p < 0.01 (unpaired two-tailed Student’s t test, A-G). Data shown are representative of two (F, H, J, K) or three (C, D, E, G, I) independent experiments. Error bars, S.D. [n = 4 to 6 mice per group (A, B, E), n = 6 to 7 mice per group (C, D), n = 2 cultures per group (F, H), n = 3 cultures per group (G), and NK cells combined from 6 wild type mice for one experiment (J, K)].
Building on this inverse correlation between Foxo1 and Tbx21 expression, we next asked whether Tbx21 regulated Foxo1 gene expression. Real-time RT-PCR indicated that Foxo1 mRNA expression was unchanged between NK cells isolated from Tbx21−/− mice and those isolated from their WT littermate control mice (data not shown), arguing against a role of Tbx21 in the regulation of Foxo1 expression at the transcriptional level. We then explored whether Foxo1 regulated Tbx21 expression. In NK cells isolated from Foxo1ΔNK and control mice, we assessed both Tbx21 mRNA and protein expression by real-time RT-PCR and intracellular flow cytometry, respectively. Tbx21 mRNA and protein expression were both increased in total NK cells isolated from Foxo1ΔNK mice compared to those isolated from control mice (Figure 6C, D). Considering that loss of Foxo1 enhanced late-stage NK cell maturation, we then used intracellular flow cytometry to compare Tbx21 protein expression in NK cell developmental stages isolated from Foxo1ΔNK and control mice. We found that Tbx21 expression was elevated in CD11b+CD27+ and CD11b+CD27− NK cells from Foxo1ΔNK mice compared to those from control mice (Figure 6E). These data suggest that the downregulation of Foxo1 triggers the elevated Tbx21 expression during late stage maturation of NK cell development.
We next tested these data in human NK cells. Addition of 4-OH-tamoxifen to induce Foxo1 activity in NKL cells expressing Foxo1-ER, suppressed TBX21 mRNA expression at the 6 hour time point (Figure 6F), while knockdown of Foxo1 augmented TBX21 mRNA expression (Figure 6G).
Since we found that Foxo1 also inhibited TBX21 expression in human NK cells (Figure 6F, G), we first asked if Foxo1 binds to the TBX21 promoter in the human NKL cell line, which possesses a potential forkhead-binding site of TTGTTTT at −1187 to −1181 bp relative to the transcription start site. Our chromatin immunoprecipitation (ChIP) assay revealed that the chromatin fragments containing the potential forkhead-binding site from the human TBX21 promoter were indeed specifically immunoprecipitated by anti-Foxo1 antibody in NKL cells (Figure 6H). We next performed ChIP assay in purified murine NK cells to determine whether Foxo1 binds to the three putative forkhead-binding sites previously described (around −4635 bp, −1541 bp and −744 bp regions) (Rao et al., 2012) at the proximal region as well as another one at the remote region (around −16.6 kb) on the Tbx21 promoter. Consistent with the previous observation (Rao et al., 2012), we did not find any direct binding activity of Foxo1 at all these putative forkhead-binding sites on the Tbx21 promoter in mouse NK cells (data not shown). To further determine the mechanism, we also performed a microarray analysis on highly purified primary mouse NK cells from Foxo1ΔNK and control mice. We aimed to find whether Foxo1 regulated other factors, such as previously identified Tbx21 regulators, Tox and Ezh2 (Tong et al., 2014; Yun et al., 2011), and thereby indirectly regulated Tbx21 expression. We failed to identify such a mechanism (data not shown).
A previous report indicates that about 83% of genes regulated by Foxo1 do not contain any consensus forkhead-binding sites in the promoter regions (Dong et al., 2008). Together with the above negative data that we collected, we hypothesized that Foxo1 is recruited to the Tbx21 promoter through a protein-protein interaction and thereby regulated Tbx21 expression. After working on several potential binding partners of Foxo1, we started to test this hypothesis in murine cells by investigating the interaction between Foxo1 and Sp1, a Foxo1 protein binding partner previously identified in other cell types (Li et al., 2007). Indeed we demonstrated such an interaction in murine splenocytes (Figure 6I), which was further validated in murine NK cells (Figure 6J). We previously identified six putative SP1 binding sites on the proximal TBX21 promoter spanning from −160 to +110 bp relative to transcription start site, which were highly homologous among different mammalian species, including humans and mice (Yu et al., 2007). Previously we demonstrated the binding of SP1 to the TBX21 promoter in human cells (Yu et al., 2007), and recently this was independently validated by a ChIP-seq from another group (Araya et al., 2014). Based on these current and previous findings, we postulated that, if our above hypothesis was correct, Foxo1 would be associated with the Tbx21 proximal promoter at the Sp1 binding region. Our ChIP assay demonstrated that in purified primary murine NK cells, Foxo1 was indeed associated with the Tbx21 proximal promoter at the Sp1 binding region (Figure 6K). Consistent with our previous findings in humans (Yu et al., 2007), Sp1 also showed binding activity at this region in mouse NK cells (Figure 6K). All the above evidences indicate that Foxo1 suppresses Tbx21 expression through recruitment by Sp1 to the Tbx21 proximal promoter region, leading to impaired transactivation of Tbx21 by Sp1 in murine NK cells.
Foxo1 requires Tbx21 to exert its effect on NK cell maturation
We undertook a genetic approach to further confirm our above finding, showing that Foxo1 acts upstream of Tbx21 to negatively regulate its expression. A previous study demonstrated that Tbx21−/− mice almost completely lose late-stage NK cell maturation (Townsend et al., 2004). We therefore hypothesized that Foxo1’s role inregulating NK cell maturation required Tbx21 (Figure S4). To test this, we generated Tbx21−/− and Tbx21+/− Foxo1-deficient mice, i.e. Foxo1ΔNKTbx21−/− and Foxo1ΔNKTbx21+/−. Parallel analysis of NK cells from these mice by flow cytometry indeed indicated that the function of Foxo1 in NK cell maturation relied on the existence of Tbx21. Specifically, when both alleles of Tbx21 are WT, consistent with the data shown in Figure 2, compared to control mice, loss of Foxo1 in Foxo1ΔNK mice was associated with an increased proportion of CD11b+CD27− cells [Figures 7A (left two columns) and 7C]. However, when Tbx21 was absent in Foxo1ΔNKTbx21−/− mice, compared to control Tbx21−/− mice, the loss of Foxo1 in Foxo1ΔNK Tbx21−/− mice had no further effect on late-stage NK cell maturation; i.e. genetic deletion of Foxo1 failed to rescue the absence of late-stage NK cell maturation observed in Tbx21−/− mice [Figures 7 A (right two columns) and 7C]. When only a single allele of Tbx21 was present (i.e. Tbx21+/−), the loss of Foxo1 in Foxo1ΔNK Tbx21+/− mice rescued the defect in NK cell maturation that occurred in Tbx21+/− mice so that it was similar to that observed in WT control mice, but reduced compared to maturation in Foxo1ΔNK mice (Figures 7B and 7D). These data indicate that Foxo1 requires Tbx21 to exert its negative effect on NK cell maturation.
Figure 7. Promotion of late-stage NK cell maturation by loss of Foxo1 requires Tbx21 expression.

(A, B) Analysis of CD11b versus CD27 expression in NK cells (gated on CD3−NK1.1+ cells) in the bone marrow (BM), spleen, periphery lymph node (pLN), and peripheral blood of 8-week-old Foxo1fl/fl (WT), Foxo1ΔNK, Foxo1ΔNK Tbx21−/− and Tbx21−/− [Tbx21 homozygotes (HO)] mice (A) and WT, Foxo1ΔNK, Foxo1ΔNK Tbx21+/−, and Tbx21+/− [Tbx21 heterozygotes (HE)] mice (B). Cell percentages of each NK cell developmental stage defined by CD11b and CD27 surface expression are presented in the quadrants of dot plots, and the ratios (red) of stage 4 (CD11b+CD27−) to stage 3 (CD11b+CD27+) NK cells are displayed at the right side of each plot. Flow cytometry plots are representative of at least three independent experiments (n ≥ 3 for each). (C, D) Statistical analyses of the percentages of NK cells from each population defined by CD11b and CD27 expression in the spleens of the mice described in (A) and (B), respectively. NS, not significant (p > 0.05); * p < 0.05; ** p < 0.01; ***p < 0.001 (one-way ANOVA, C, D). Error bars, S.D. (n = 6 to 7 for each genotype). See also Figure S4.
DISCUSSION
The generation and maturation of NK cells are the result of an elaborate series of cellular and molecular events that are often associated with developmental regulators. Previously, a study using systemic Tbx21−/− mice identified Tbx21 as an intrinsic positive regulator during a late stage of NK cell maturation from relatively immature NK cells (Townsend et al., 2004). Numerous other studies using similar approaches of systemic knockout mice have identified other positive regulators of NK cell development and effector functions (Hesslein and Lanier, 2011; Ramirez and Kee, 2010). Our focus has been on negative regulators, or checkpoints, as they are undoubtedly involved in this process yet are largely unknown. Understanding negative regulatory factors in cell development is critical to define and ultimately modulate these processes. We previously found that TGF-β plays a role in negatively regulating NK cell function by antagonizing positive signaling pathways (Yu et al., 2006) and, subsequently, other groups found that TGF-β also acts as an extrinsic negative regulator in NK cell development (Marcoe et al., 2012). Of note, inhibition of TGF-β signaling with neutralizing antibodies and other approaches has been used to treat cancer (Bouquet et al., 2011; Liu et al., 2012; Zwaagstra et al., 2012).
In the current study, we utilized Ncr1iCre mice (Narni-Mancinelli et al., 2011) to generate NK cell conditional deletion of Foxo1 and/or Foxo3. Using this cell-specific approach, we found that Foxo transcription factors act in multiple ways to regulate NK cells homeostasis and function. Indeed, during the earliest steps of NK cell maturation, Foxo transcription factors are involved in the control of cell homing by regulating the expression of CD62L (Martin-Fontecha et al., 2004), whereas they act as negative regulators in the checkpoint controlling CD27 downregulation and final maturation in CD11b+ NK cells. We also found that Foxo1 negatively regulates NK cell effector functions. Mechanistically, our data showed that Foxo1 inhibits Tbx21 expression in NK cells. In humans this appears to occur via direct binding to the TBX21promoter, while in mice, Foxo1 is recruited by Sp1 to associate with the proximal promoter region of Tbx21 where Sp1 binds. Consistent with this, Foxo1 and Tbx21 expression inversely correlate with each other from immature NK cells to mature NK cells. Our findings support a model in which the dynamic and inverse changes in Foxo1 and Tbx21 expression control NK cell maturation and homeostasis (Figure S5). During this process, Foxo1 is highly expressed while Tbx21 is present in low amounts in immature NK cells. Along with NK maturation, Foxo1 expression gradually decreases and releases its control as a negative regulator of Tbx21, leading to late-stage NK cell maturation. We believe that this negative regulation of Tbx21 is important for tight control of NK cell homeostasis. Loss of this balance may be associated with disease, as too many NK cells could promote autoimmune disease and too few NK cells may result in earlier or more probable malignant transformation.
In support of this proposed mechanism, our ChIP results demonstrate that Foxo1 associates with a binding site (−1187 to −1181) upstream of the transcription start site of TBX21 in human NK cells. Consistent with previous work in T cells (Rao et al., 2012), we were unable to show a direct binding of Foxo1 to the Tbx21 promoter in murine NK cells. A previous report shows that about 83% of genes regulated by Foxo1 do not contain any consensus forkhead-binding sites in the promoter regions (Dong et al., 2008). This suggests that protein-protein interactions may be responsible for Tbx21 suppression by Foxo1 in murine cells. Consistent with this, our co-immunoprecipitation experiment using both murine splenocytes and NK cells demonstrates that Foxo1 interacts with Sp1, and the ChIP experiment shows that both Foxo1 and Sp1 are associated with the proximal promoter region of Tbx21 at the Sp1 binding region. We believed that we have unraveled one important mechanism underlying inhibition of Tbx21 by Foxo1 in murine NK cells although some other mechanisms may also exist. Our current study is in accordance with previous data from another group showing that Foxo1 interacts with Sp1 in another cell type (Li et al., 2007), and as well corroborated by the data from other groups and ours demonstrating that Sp1 binds to the proximal promoter region of TBX21 and regulates its expression (Araya et al., 2014; Yu et al., 2007). Regulation of Tbx21 by Foxo1 seems to be important for controlling NK cell effector functions and late-stage maturation, as our preliminary data (not shown) suggested that in murine NK cells, Foxo1 seems not to regulate other transcription factors known to be required for NK cell development, such as Ikaros, Pu.1, VDUP-1, E4BP4, MEF, GATA-3, and IRF2.
Given the solid evidence that Tbx21 provides a positive deterministic signal for late-stage NK cell maturation (Gordon et al., 2012; Soderquest et al., 2011; Townsend et al., 2004), identification of a new molecule acting as an upstream negative regulator is of great significance. Our data support a notion that Tbx21 is a downstream target of Foxo1, which we found negatively regulates late-stage NK cell maturation and function. In addition to the inverse expression between Foxo1 and Tbx21 in developing and mature NK cells, two more findings support this regulation of Tbx21 by Foxo1. First, Tbx21 transcription and protein expression were found to be increased in NK cells with a specific depletion of Foxo1, whereas overexpression of Foxo1 was found to inhibit Tbx21 expression. Second, Foxo1 deficiency could not rescue the late-stage maturation of NK cells in Tbx21−/− mice; however, Foxo1 deficiency promoted NK maturation when one or both alleles of Tbx21 were present (Figure 7B, D). Together, these results demonstrate that promotion of late-stage NK cell maturation by reducing Foxo1 expression requires Tbx21.
Dynamic changes in transcription factor regulation or expression may depend on extrinsic soluble factors secreted by other cells, such as dendritic cells and/or monocytes, and external stimuli. Thus, it would be intriguing to know whether the dynamic changes occurring between Foxo1 and Tbx21 are regulated by extrinsic factors and tumor challenges. In fact, we found that stimulation of both NK-92 cells and murine NK cells with IL-15, IL-2 and IL-12, even for a short time, led to Foxo1 phosphorylation. In vivo tumor challenges with B16F10 melanoma cells also induce Foxo1 phosphorylation in mouse NK cells. Foxo1 phosphorylation has been shown to be associated with its cytoplasmic translocation, leading to reduced nuclear activity of Foxo1, which may in turn evoke an increase in Tbx21 expression. Consistent with this, we and others have previously shown that the above pro-inflammatory cytokines can upregulate Tbx21 expression (Townsend et al., 2004; Yu et al., 2006).
In summary, we have demonstrated that in NK cells, Foxo transcription factors regulate homing, late-stage maturation and effector functions. Mechanistically, we provide evidence that Foxo1 inhibits Tbx21 expression through direct association with the Tbx21 promoter at a consensus forkhead-binding site in human NK cells, and through interference with Sp1-mediated transactivation of Tbx21 after recruitment by Sp1 in murine NK cells. Our study indicates that Foxo factors represent a novel negative regulatory element or a checkpoint in NK cells, and provides a new avenue to target NK cells for viral clearance and tumor surveillance based on Foxo1 activity.
EXPERIMENTAL PROCEDURES
Mice
All mice were bred and housed in specific pathogen-free conditions in accordance with Ohio State University Animal Care and Use Guidelines as well as French and European guidelines for animal care. All animal work was approved by The Ohio State University Animal Care and Use Committee, as well as Marseille’s ethical committee for animal use.
C57BL/6 Foxo1fl/fl and Foxo3f/f mice were a kind gift from Dr. Ming Li at the Memorial Sloan-Kettering Cancer Center as well as Dr. Ronald A. DePinho at the Dana Farber Cancer Institute. Congenic C57BL/6 CD45.1 and Tbx21−/− were purchased from The Jackson Laboratory. Rag2−/−Il2rg−/− mice were purchased from Taconic Biosciences. All mice used were 8- to 12-week-old.
Intravenous lung metastasis assay
8- to 12-week-old Foxo1fl/fl (WT) or Ncr1iCre/+Foxo1fl/fl (Foxo1ΔNK) mice were used for lung experimental-metastasis assays following intravenous injection of 0.25 ×106 B16F10 melanoma cells. Two weeks after the injection, lung tumor nodules were counted. To determine the amount of Foxo1 phosphorylation in NK cells in lung tissue, wild-type C57BL/6 mice were challenged with I.V. injections of 0.25 ×106 B16F10 melanoma cells for 7 days. Lung tissue lymphocytes were isolated to detect the Foxo1 phosphorylation by flow cytometry.
Immunoprecipitation (IP) assay
Splenocytes were pooled from 6 mice, and 5 × 106 primary NK cells were purified from the splenocytes by using a mouse NK negative selection kit (Miltenyi Biotec), followed by FACS-sorting. Cell lysates were prepared using a NP40 cell lysis buffer, supplemented with 10 mM phenylmethanesulfonyl fluoride and 1× proteinase inhibitor cocktail (Sigma). An equal amount of protein was used in each immunoprecipitation, and was pre-cleared with 1 μg rabbit (for Foxo1 IP) or mouse (for Sp1 IP) IgG control antibodies plus 50 μl Dynabeads Protein A or Dynabeads Protein G, respectively, for 2 hours at 4 °C. The precleaned lysates were then incubated with anti-Foxo1 (C29H4) (1:100, Cell Signal Technology) or anti-Sp1 (E3) (5 μg, Santa Cruz Technology) antibodies at 4 °C overnight. An equal volume of cell lysate, incubated with the same species normal IgG, was taken as a control (Cell Signaling Technology or Abcam). The immunoprecipitates were subsequently incubated with 50 μl Dynabeads Protein A or Dynabeads Protein G for additional 4 hours at 4 °C, washed with lysis buffer, and subjected to immunoblotting against Foxo1 (C29H4) (Cell Signaling Technology) or Sp1 (Millipore). A Clean-Blot IP Detection Kit (HRP) (Pierce) was used as a secondary antibody.
Chromatin immunoprecipitation (ChIP)
ChIP assay was carried out using an EZ-ChIP assay kit (Upstate Biotechnology, Lake Placid, NY) as described previously (Deng et al., 2014). Briefly, an equal amount (10 μg) of rabbit anti-Foxo1 (Abcam) antibody or normal rabbit IgG antibody (Santa cruz) was used to precipitate the cross-linked DNA/protein complexes of 10 × 106 human NKL cells. Similarly, an equal amount (7.5 μg) of anti-Foxo1, anti-Sp1 antibody (Abcam), or normal rabbit IgG (Cell Signaling Technology) was used to precipitate the cross-linked DNA/protein complexes from 5 × 106 mouse primary NK cells. Following reversal of cross-linking, the amount of chromatin precipitated by the indicated antibody was detected by PCR. The PCR primers were designed specially against the target regions and were listed in online supplemental procedures.
Flow cytometric analysis, in vitro assessment of NK function, retroviral transduction and infection, and quantitative real-time RT-PCR
Standard protocols were followed for these procedures, which were presented in our previous publication (Yu et al., 2010) and were also detailed in online supplemental procedures.
Statistical analysis
Unpaired Student’s t test was utilized to compare two independent groups for continuous endpoints if they are normally distributed, or non-parametric method (e.g. Wilcoxon Rank Sum test) was used if they are non-normally distributed. One-way ANOVA was performed when three or more independent groups were compared and the assumptions of normality and equal variance hold. Linear mixed models were used to account for the correlation due to the repeated measures from the same mouse. Generalized linear models were used in the randomized block design with litters as the block factor. P values were adjusted for multiple comparisons using Holm’s procedure. All tests were two-sided. A P value less than 0.05 is considered statistically significant. Data are presented as means ± S.D.
Supplementary Material
Acknowledgments
The authors would like to thank Ming O. Li (Memorial Sloan-Kettering Cancer Center, US) and Ronald A. DePinho (Dana Farber Cancer Institute, Harvard Medical School, US) for providing Foxo1fl/fl and Foxo3f/f mice. This project was supported in part by grants from US National Institutes of Health (CA155521 and OD018403 to J.Y., CA095426, CA163205 and CA068458 to M.A.C., and CA185301 to M.A.C and J.Y.). J.Y. was supported by an American Cancer Society Research Scholar Grant (RSG-14-243-01-LIB) as well as by Gabrielle’s Angel Foundation for Cancer Research. This project was also supported by a grant from the Natural Science Foundation of China (81273507 to X.L. and 81370631 to S.Y.). E.V. lab is supported by the European Research Council (THINK Advanced Grant), the Ligue Nationale contre le Cancer (Equipe Labellisée) and by institutional grants from INSERM, CNRS and Aix-Marseille University to CIML. E.V. is a scholar of the Institut Universitaire de France. The authors would like to sincerely thank Dr. Xian C. Li at Houston Methodist Research Institute for supporting the project and Dr. David M. Lucas at The Ohio State University for his critical reading of this manuscript.
Footnotes
Supplemental Information including five gures, supplemental procedures, and supplemental references can be found online.
AUTHOR CONTRIBUTIONS
Y-C.D., Y.K., J.C., S.Y., Y.W., X.C., H.M., L.Z., Y-F.D., Q.Z., and F.W. performed experiments; J.Z. analyzed the data; T.H. performed experiments and reviewed the manuscripts; X.Z., C-G.L., and A.G.F. designed the research; X.L., M.A.C., E.V, and J.Y. equally contributed to this project, designed the research, and edited the manuscript; J.Y., Y-C.D., and Y.K. wrote the manuscript;
COMPETING FINANCIAL INTERESTS
E.V. is the cofounder and a shareholder of Innate Pharma. The other authors have no conflicting financial interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan DS, Rybalov B, Awong G, Zuniga-Pflucker JC, Kopcow HD, Carlyle JR, Strominger JL. TGF-beta affects development and differentiation of human natural killer cell subsets. Eur J Immunol. 2010;40:2289–2295. doi: 10.1002/eji.200939910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol. 2008;9:613–622. doi: 10.1038/ni.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya N, Sato T, Ando H, Tomaru U, Yoshida M, Coler-Reilly A, Yagishita N, Yamauchi J, Hasegawa A, Kannagi M, et al. HTLV-1 induces a Th1-like state in CD4+CCR4+ T cells. J Clin Invest. 2014;124:3431–3442. doi: 10.1172/JCI75250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, Babb JS, Lonning SM, DeWyngaert JK, Formenti SC, Barcellos-Hoff MH. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. 2011;17:6754–6765. doi: 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113:5488–5496. doi: 10.1182/blood-2008-10-187179. [DOI] [PubMed] [Google Scholar]
- Chow KT, Timblin GA, McWhirter SM, Schlissel MS. MK5 activates Rag transcription via Foxo1 in developing B cells. J Exp Med. 2013;210:1621–1634. doi: 10.1084/jem.20130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci F, Caligiuri MA, Di Santo JP. What does it take to make a natural killer? Nat Rev Immunol. 2003;3:413–425. doi: 10.1038/nri1088. [DOI] [PubMed] [Google Scholar]
- Deng Y, Chu J, Ren Y, Fan Z, Ji X, Mundy-Bosse B, Yuan S, Hughes T, Zhang J, Cheema B, et al. The Natural Product Phyllanthusmin C Enhances IFN-gamma Production by Human NK Cells through Upregulation of TLR-Mediated NF-kappaB Signaling. J Immunol. 2014;193:2994–3002. doi: 10.4049/jimmunol.1302600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Santo JP. Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol. 2006;24:257–286. doi: 10.1146/annurev.immunol.24.021605.090700. [DOI] [PubMed] [Google Scholar]
- Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freud AG, Becknell B, Roychowdhury S, Mao HC, Ferketich AK, Nuovo GJ, Hughes TL, Marburger TB, Sung J, Baiocchi RA, et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity. 2005;22:295–304. doi: 10.1016/j.immuni.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. doi: 10.1016/j.immuni.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006;176:1517–1524. doi: 10.4049/jimmunol.176.3.1517. [DOI] [PubMed] [Google Scholar]
- Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. 2012;12:649–661. doi: 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. 2013;210:1189–1200. doi: 10.1084/jem.20130392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslein DG, Lanier LL. Transcriptional control of natural killer cell development and function. Adv Immunol. 2011;109:45–85. doi: 10.1016/B978-0-12-387664-5.00002-9. [DOI] [PubMed] [Google Scholar]
- Huntington ND, Nutt SL, Carotta S. Regulation of murine natural killer cell commitment. Front Immunol. 2013;4:14. doi: 10.3389/fimmu.2013.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington ND, Tabarias H, Fairfax K, Brady J, Hayakawa Y, Degli-Esposti MA, Smyth MJ, Tarlinton DM, Nutt SL. NK cell maturation and peripheral homeostasis is associated with KLRG1 up-regulation. J Immunol. 2007;178:4764–4770. doi: 10.4049/jimmunol.178.8.4764. [DOI] [PubMed] [Google Scholar]
- Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor foxo1 controls central-memory CD8(+) T cell responses to infection. Immunity. 2013;39:286–297. doi: 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, Yokoyama WM. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol. 2002;3:523–528. doi: 10.1038/ni796. [DOI] [PubMed] [Google Scholar]
- Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- Li H, Liang J, Castrillon DH, DePinho RA, Olson EN, Liu ZP. FoxO4 regulates tumor necrosis factor alpha-directed smooth muscle cell migration by activating matrix metalloproteinase 9 gene transcription. Mol Cell Biol. 2007;27:2676–2686. doi: 10.1128/MCB.01748-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, Ancukiewicz M, Boucher Y, Jain RK, Xu L. TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A. 2012;109:16618–16623. doi: 10.1073/pnas.1117610109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luevano M, Madrigal A, Saudemont A. Transcription factors involved in the regulation of natural killer cell development and function: an update. Front Immunol. 2012;3:319. doi: 10.3389/fimmu.2012.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoe JP, Lim JR, Schaubert KL, Fodil-Cornu N, Matka M, McCubbrey AL, Farr AR, Vidal SM, Laouar Y. TGF-beta is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat Immunol. 2012;13:843–850. doi: 10.1038/ni.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci U S A. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. 2012;491:554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez K, Kee BL. Transcriptional regulation of natural killer cell development. Curr Opin Immunol. 2010;22:193–198. doi: 10.1016/j.coi.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity. 2012;36:374–387. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderquest K, Powell N, Luci C, van Rooijen N, Hidalgo A, Geissmann F, Walzer T, Lord GM, Martin-Fontecha A. Monocytes control natural killer cell differentiation to effector phenotypes. Blood. 2011;117:4511–4518. doi: 10.1182/blood-2010-10-312264. [DOI] [PubMed] [Google Scholar]
- Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO, Kaech SM. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802–814. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togher S, Larange A, Schoenberger SP, Feau S. FoxO3 is a negative regulator of primary CD8(+) T-cell expansion but not of memory formation. Immunol Cell Biol. 2015;93:120–125. doi: 10.1038/icb.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q, He S, Xie F, Mochizuki K, Liu Y, Mochizuki I, Meng L, Sun H, Zhang Y, Guo Y, Hexner E. Ezh2 regulates transcriptional and posttranslational expression of T-bet and promotes Th1 cell responses mediating aplastic anemia in mice. J Immunol. 2014;192:5012–5022. doi: 10.4049/jimmunol.1302943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, Gapin L, Glimcher LH. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20:477–494. doi: 10.1016/s1074-7613(04)00076-7. [DOI] [PubMed] [Google Scholar]
- Trotta R, Parihar R, Yu J, Becknell B, Allard J, 2nd, Wen J, Ding W, Mao H, Tridandapani S, Carson WE, Caligiuri MA. Differential expression of SHIP1 in CD56bright and CD56dim NK cells provides a molecular basis for distinct functional responses to monokine costimulation. Blood. 2005;105:3011–3018. doi: 10.1182/blood-2004-10-4072. [DOI] [PubMed] [Google Scholar]
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werneck MB, Lugo-Villarino G, Hwang ES, Cantor H, Glimcher LH. T-bet plays a key role in NK-mediated control of melanoma metastatic disease. J Immunol. 2008;180:8004–8010. doi: 10.4049/jimmunol.180.12.8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol. 2004;22:405–429. doi: 10.1146/annurev.immunol.22.012703.104711. [DOI] [PubMed] [Google Scholar]
- Yu J, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol. 2013;34:573–582. doi: 10.1016/j.it.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Mao HC, Wei M, Hughes T, Zhang J, Park IK, Liu S, McClory S, Marcucci G, Trotta R, Caligiuri MA. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood. 2010;115:274–281. doi: 10.1182/blood-2009-04-215491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, Jaung MS, Blaser BW, Sun J, Benson DM, Jr, et al. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24:575–590. doi: 10.1016/j.immuni.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Yu J, Wei M, Boyd Z, Lehmann EB, Trotta R, Mao H, Liu S, Becknell B, Jaung MS, Jarjoura D, et al. Transcriptional control of human T-BET expression: the role of Sp1. Eur J Immunol. 2007;37:2549–2561. doi: 10.1002/eji.200737088. [DOI] [PubMed] [Google Scholar]
- Yun S, Lee SH, Yoon SR, Kim MS, Piao ZH, Myung PK, Kim TD, Jung H, Choi I. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol Lett. 2011;136:29–36. doi: 10.1016/j.imlet.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Zwaagstra JC, Sulea T, Baardsnes J, Lenferink AE, Collins C, Cantin C, Paul-Roc B, Grothe S, Hossain S, Richer LP, et al. Engineering and therapeutic application of single-chain bivalent TGF-beta family traps. Mol Cancer Ther. 2012;11:1477–1487. doi: 10.1158/1535-7163.MCT-12-0060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.