

Abstract
RAS proteins require membrane association for their biological activity, making this association a logical target for anti-RAS therapeutics. Lipid modification of RAS proteins by a farnesyl isoprenoid is an obligate step in that association, and is an enzymatic process. Accordingly, farnesyltransferase inhibitors (FTIs) were developed as potential anti-RAS drugs. The lack of efficacy of FTIs as anti-cancer drugs was widely seen as indicating that blocking RAS membrane association was a flawed approach to cancer treatment. However, a deeper understanding of RAS modification and trafficking has revealed that this was an erroneous conclusion. In the presence of FTIs, KRAS and NRAS, which are the RAS isoforms most frequently mutated in cancer, become substrates for alternative modification, can still associate with membranes, and can still function. Thus, FTIs failed not because blocking RAS membrane association is an ineffective approach, but because FTIs failed to accomplish that task. Recent findings regarding RAS isoform trafficking and the regulation of RAS subcellular localization have rekindled interest in efforts to target these processes. In particular, improved understanding of the palmitoylation/depalmitoylation cycle that regulates RAS interaction with the plasma membrane, endomembranes and cytosol, and of the potential importance of RAS chaperones, have led to new approaches. Efforts to validate and target other enzymatically regulated post-translational modifications are also ongoing. In this review, we revisit lessons learned, describe the current state of the art, and highlight challenging but promising directions to achieve the goal of disrupting RAS membrane association and subcellular localization for anti-RAS drug development.
Introduction
The three RAS genes (HRAS, NRAS, KRAS) are the most commonly mutated oncogenes in human cancers (1,2). The role of oncogenic RAS proteins as key drivers in both common and uncommon cancers has led to intensive efforts over more than three decades to develop therapeutics that target RAS, encompassing both direct and indirect approaches. Oncogenically mutated RAS proteins fail to cycle “off” from the active, GTP-bound state to the resting GDP-bound state, and thereby accumulate in the “on” configuration. Early direct approaches sought to attack this impaired molecular switch. Attempts to identify antagonists of GTP-binding or to identify drug-like mimics of the negative regulatory GAP proteins have been unsuccessful, although new strategies of stabilizing conformational states may yet bear fruit, as discussed elsewhere in this CCR Focus section (3). More recent efforts to target specific RAS mutations (e.g., KRAS G12C), to interfere with RAS binding to its activator SOS1, and to block association with effectors such as RAF1 have been reviewed recently (1,4). The consensus at present is that the most fruitful direction for anti-RAS therapeutics in the near future is indirect targeting of RAS signaling via inhibiting its downstream effectors, particularly the RAF-MEK-ERK and PI3K-AKT-MTOR kinase cascades that have been shown to be critical for RAS driver functions in specific cancers. These efforts are discussed elsewhere (1,4,5). Other approaches, such as attempts to identify additional targets for co-inhibition with RAS, through synthetic lethality screens or metabolic dependencies, are also discussed elsewhere in this CCR Focus section (6,7). Here, our focus is on direct targeting of RAS by interfering with its membrane association and trafficking. We argue that this approach, while challenging, remains both logical and potentially tractable, given information that has emerged over the past few years. Because the association of RAS proteins with membranes is absolutely required for their function, targeting this requirement can be viewed as the functional equivalent not of turning off the defective switch that is oncogenic RAS, but of removing it from the circuit.
CAAX Processing and RAS Membrane Association
The critical need for RAS protein association with cellular membranes has been appreciated for decades (8,9). RAS association with the plasma membrane (PM) and with other membrane compartments upon which signaling occurs (10,11) is promoted by a well-described series of post-translational modifications at RAS C-terminal CAAX motifs (Fig. 1), where C = cysteine, A = (usually) aliphatic amino acids and X = a variable amino acid; in RAS, X = S or M (12,13). In the initial and obligate step, a 15-carbon farnesyl polyisoprene lipid is added by farnesyltransferase (FTase) to the cysteine of the CAAX motif through a stable thioether linkage. Subsequently the AAX amino acids are cleaved off by the farnesylcysteine-directed endoprotease, RAS converting CAAX endopeptidase 1, also known as RAS converting enzyme 1 (RCE1). The carboxyl group of the now C-terminal farnesylcysteine is next methylesterified by isoprenylcysteine carboxylmethyltransferase (Icmt) to produce RAS proteins with hydrophobic tails that have affinity for membranes. Both RCE1 and ICMT are restricted to the endoplasmic reticulum (14,15), indicating that RAS must traffic to the PM through this compartment, and suggesting multiple layers of location-based regulation (Fig.2). Each of the enzymes involved in these CAAX processing steps has been a target for drug discovery.
Figure 1.
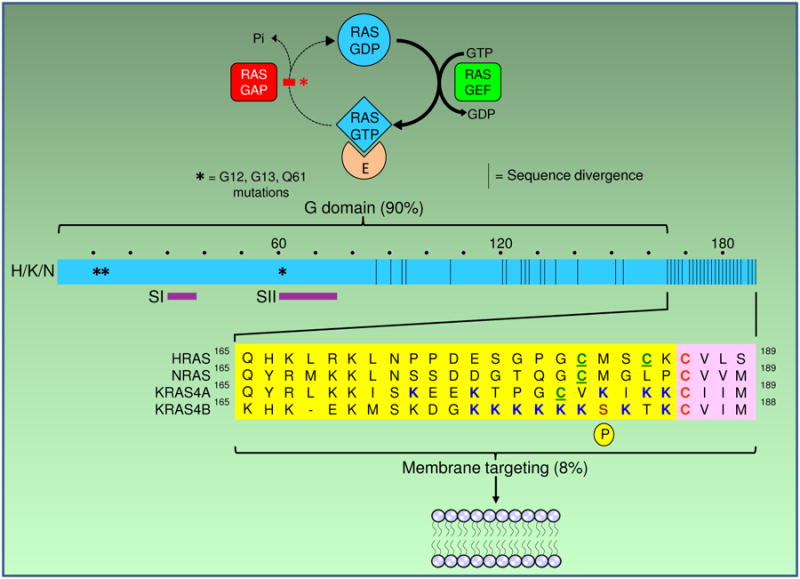
Membrane targeting sequences of RAS proteins. Top: the RAS on/off switch that is broken in oncogenically mutated RAS and fails to turn off from the active, GTP-bound state that interacts with effectors (E) to transmit downstream signals. Since membrane association is required for proper effector interaction, interfering with membrane targeting can impair signal transmission, like unwiring an electrical switch to prevent it from carrying current. Bottom: ribbon diagram of the four RAS proteins, which are 90% similar throughout their G domains that bind the guanine nucleotides, regulators and effectors (including switch regions SI, SII), but differ greatly at their C-terminal membrane targeting domains. The latter consist of a carboxyterminal CAAX tetrapeptide motif (pink boxes) with an invariant cysteine that is the site of farnesylation, and an upstream hypervariable region (yellow boxes) that include the “second signals” of one (NRAS) or two (HRAS, KRAS4A) palmitoylatable cysteines or clusters of positively charged (polybasic) residues (PBR), as well as “third signals” comprised of the surrounding residues. KRAS4B has a stretch of six contiguous lysines and no palmitoylatable cysteine, whereas KRAS4A has a hybrid motif of both a bipartite PBR and a palmitoylatable cysteine. Numbers refer to amino acid residues. Asterisks indicate sites of mutational hotspots at G12, G13 and Q61. Dots above the ribbon mark each 10 amino acid stretch. Brown bars in the ribbon mark sites of sequence variation. P, phosphorylation of KRAS4B at Serine181.
Figure 2.
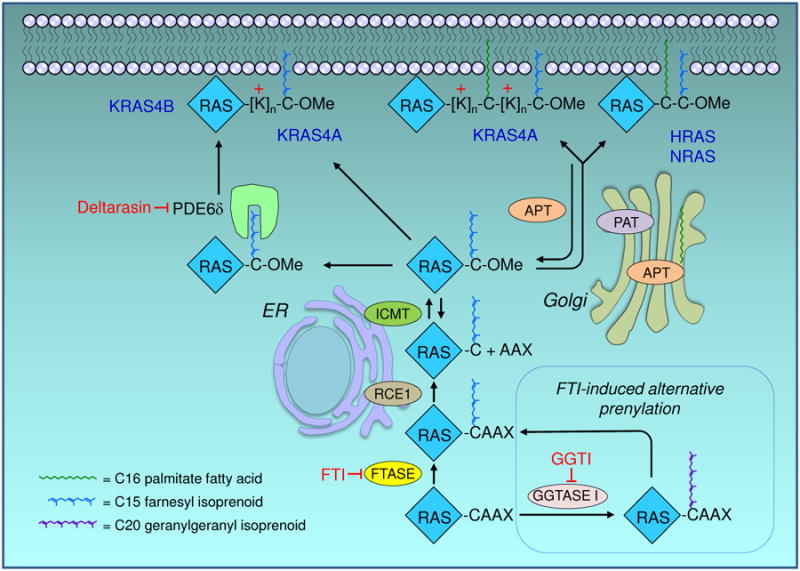
RAS trafficking pathway. Nascent RAS proteins leaving the polysome are rapidly modified by farnesyltransferase (FTase), which attaches a C15 farnesyl isoprenoid lipid to the cysteine of the CAAX motif. This provides them sufficient affinity for the endoplasmic reticulum (ER), where they are further modified by RAS-converting CAAX endopeptidase 1 (RCE1)-catalyzed proteolytic removal of the AAX residues, and by reversible isoprenylcysteine carboxylmethyltransferase (ICMT)-catalyzed carboxylmethylation of the now terminal farnesylated cysteine residue. By preventing the first and obligate step, FTase inhibitors (FTIs) prevent all three of these modifications. In the presence of FTIs, KRAS and NRAS are alternatively prenylated by geranylgeranyltransferase I (GGTase I), which attaches a C20 geranylgeranyl isoprenoid that allows the same subsequent processing steps. RAS trafficking to the inner leaflet of the plasma membrane (PM) requires a second membrane-targeting element that dictates the pathway it will take to the PM. NRAS and HRAS have one or two cysteine residues, respectively, that undergo reversible acylation by a Golgi-resident protein acyltransferase (PAT) to promote their trafficking to the PM. Rapid deacylation by an acyl protein thioesterase (APT1/2) frees them up to be re-acylated and trafficked back to the PM. The nonpalmitoylated pool of APT in the cytosol is the active form, and is in dynamic equilibrium with a palmitoylated pool on the Golgi. KRAS4B, which has no palmitoylatable cysteine but a stretch of 6 lysines (polybasic region) does not go to the Golgi but trafficks to the more directly to the PM, where it binds by virtue of its electrostatic charge. KRAS4A, which has a hybrid motif of a palmitoylated cysteine and a bifurcated polybasic region, undergoes an intermediate form of trafficking. Phosphodiesterase-6δ (PDE6δ) recognizes the farnesyl isoprenoid and solubilizes nonpalmitoylated RAS proteins from any compartment, thereby promoting their availability for restoration to the PM; deltarasin blocks this interaction. Not pictured: other chaperone proteins that guide the lipidated RAS proteins between and within membrane regions. Each enzyme depicted has been a target for drug discovery.
Targeting CAAX Prenylation: FTase and GGTase, Statins
RAS farnesylation by FTase is the first, irreversible and rate-limiting step of CAAX processing. As it was quickly determined to be both an obligate modification for oncogenic RAS biological activity and a process governed by an enzyme that recognized a simple short tetrapeptide CAAX motif, this step was rapidly exploited for inhibition. Both rational drug design and library screening were employed in numerous intensive and successful efforts to identify FTase inhibitors (FTIs) (16,17). Two of these, lonafarnib and tipifarnib, progressed to advanced clinical trials but failed to show efficacy against KRAS-driven cancers (16,17). The failure of FTIs, once anticipated to bemagic bullets for RAS-driven cancers, to serve as broadly effective anti-RAS drugs led to a widespread misperception that even RAS itself is not a good target. However, it is critical to recognize that, in the presence of FTIs, NRAS and KRAS, but not HRAS, become substrates for geranylgeranyltransferase I (GGTase I) through a process known as alternative prenylation (18,19). This phenomenon was revealed only when FTIs became available. Since geranylgeranylated RAS proteins still associate with membranes and are still biologically active, FTIs were ineffective despite hitting their FTase target. Thus, FTIs failed as anti-RAS drugs not because blocking RAS association with membranes is a flawed approach, but rather because FTIs failed to achieve this goal. Accordingly, we and others have continued to pursue the RAS modification and trafficking pathways as logical targets for potential therapeutics. One potential solution to the problem of alternative prenylation might be dual inhibition of FTase and GGTase I (20), either by combining individual inhibitors of each enzyme, or by dual specificity inhibitors. Such inhibitors have been developed and some have reached the clinic (16,17), but have been limited by toxicity (21-23). There are still hopes that a therapeutic window can be found, possibly by targeting their delivery to RAS-driven cancer cells (17), where oncogenic RAS rendered cytosolic by these agents could sequester effectors and act as dominant negatives (24,25).
Because farnesyl pyrophosphate (FPP) is both an intermediate in cholesterol biosynthetic pathway and is also used for modification of CAAX proteins, it has long been speculated that statins may limit cancer by inhibiting prenylation of RAS or related small GTPases. However, due to the differential KMs for squalene synthase versus FTase, cholesterol synthesis is 1,000-fold more easily inhibited by loss of FPP than is FTase-mediated modification of RAS (26). Consistent with this, only suprapharmacological levels of statins block protein prenylation in cell culture (27) and cause mislocalization of RAS (28), whereas therapeutic levels do not (28). Moreover, the effects of statins on cell growth are RAS-independent (27); nor is there evidence for these effects on RAS in animals administered pharmacologically relevant doses. Thus, statins show no promise for use as anti-RAS drugs.
Targeting Post-Prenylation CAAX Processing: RCE1 and ICMT
Although it is clear that the post-prenylation CAAX processing enzymes also contribute to RAS membrane association, making them potentially attractive targets for drug development, there are numerous challenges to overcome before inhibition of either RCE1 or ICMT can be translated to the clinic. These include uncertainty regarding the requirements for RCE1 and ICMT in oncogenic RAS functions, perplexing results from genetic validation studies, lack of understanding of the mechanism of RCE1 CAAX protease activity, and difficulties in developing inhibitors of these enzymes that can achieve sufficient potency and selectivity to become clinically efficacious drugs. Nevertheless, resolving these issues is likely to reveal important facets of RAS biology that may in turn reveal additional targets, warranting some discussion here.
RCE1 generates the substrate for ICMT: a prenylcysteine with anα carboxyl group. Thus, ICMT can act only after RCE1. The sequential nature of these modifications implies that RCE1 deficiency should produce cell biological consequences at least as strong as ICMT deficiency, if one assumes that the effects of partial processing are similar for each substrate. At present, due to the lack of drug-like potent and selective inhibitors, this can be tested genetically but not pharmacologically. These studies reveal significant context-dependence of deficiencies in these enzymes. Contrary to initial suppositions, Icmt null mice die earlier in gestation than do Rce1 null mice (29,30), and Icmt deficiency completely blocked the transformed growth of RAS-transformed rodent fibroblasts whereas Rce1 deficiency had modest effects (31). Further, the consequences of genetic loss of Rce1 and Icmt can be both opposing and context-dependent. In a mouse model, the same myeloproliferative disease driven by oncogenic Kras G12D that was ameliorated by Icmt deficiency (32) was enhanced by Rce1 deficiency (33). However, Icmt deficiency accelerated the disease in a mouse model of pancreatic cancer driven by Kras G12D (34), due to inhibition of signaling from Notch-1, which acts as a tumor suppressor in this model (35). Importantly, Icmt deficiency ameliorated Kras-driven disease in other mouse tumor models (M.G. Dalin and M.O. Bergo;unpublished results), supporting the context-dependency of ICMT impairment. These results suggest that better understanding the biology of CAAX processing will be critical to further validation of RCE1 and ICMT as potential drug targets, particularly with respect to the indications and populations in which they may be best applied. The complexity inherent in the context-dependent roles of RCE1 and ICMT is likely explained by the myriad substrates of these enzymes other than RAS, many of which are signaling molecules.
Many drugs fail in development due to cardiac toxicity, which is a demonstrated consequence of Rce1 depletion in the heart (36). Similarly, targeted depletion of Rce1 in the retina resulted in rapid degeneration of specific photoreceptor cells (37). On the other hand, the cleavage of RHOA by the bacterial toxin and cysteine protease YopT requires RCE1 but not ICMT (38). Thus, even if they can be developed, RCE1 inhibitors may be more toxic overall than ICMT inhibitors. Nevertheless, attempts have been made to develop inhibitors of both RCE1 (39) and ICMT (40-42), although each will necessarily also affect alternate substrates in addition to RAS. Disappointingly, even the most potent and selective of the RCE1 inhibitors have recently been shown to lack mechanism-based activity (43). The anti-proliferative effects of ICMT inhibitors on RAS-transformed cells may be more compelling (41,42); whether they can be converted into pharmacological leads is currently unclear.
RAS Trafficking: “Second Signals,” the Acylation/Deacylation Cycle, and RAS Chaperones
RAS membrane association and trafficking are regulated in a complex manner that has yet to be fully unraveled (Fig. 2). This complexity has revealed both additional challenges and additional targets for drug discovery. First, in addition to modification of the CAAX sequence, RAS PM association requires a proximal “second signal” that is either palmitoylation of one (NRAS) or two cysteines (HRAS) or a polybasic stretch of lysine residues (KRAS4B) (44,45) to confer additional hydrophobicity or an electrostatic interaction with the negatively charged headgroups of the phospholipids at the inner leaflet of the PM, respectively (Fig. 1). KRAS4A is unique among the four RAS proteins in possessing a dual membrane targeting motif that consists of both a palmitoylated cysteine and two short polybasic regions flanking that acylated cysteine (46) (Fig. 1) Second, RAS association with membranes is not a one-way street. Instead, RAS proteins undergo a cycle of delivery to the PM followed by return to endomembranes for recycling, where they can interact with a Golgi-resident palmitoylacyl transferase (47). Depalmitoylation of NRAS and HRAS at the PM initiates the recycling process(48,49), whereas KRAS4B dissociation from the PM is mediated in part by PKC-catalyzed phosphorylation of serine 181 within the polybasic region (50). Third, RAS protein trafficking is also modulated by interactions with GDI-like chaperones (51,52) that help to shepherd the lapidated RAS proteins among hydrophobic membranes through aqueous cytosol, and within specific PM regions (53). Among these chaperones are galectin-1 and-3 that escort HRAS and KRAS4B, respectively (54-56), and nucleolin that shepherds NRAS (57,58). Phosphodiesterase-6δ (PDE6δ) has been variously proposed to chaperone NRAS (46,59) or nonpalmitoylated HRAS (59), and KRAS4B (46,60-62) but not KRAS4A (46). RAS proteins are released from PDE6δ upon binding of ARL2/3 to an allosteric site on PDE6δ (63). Each of these interactions is potentially a source of targets for anti-RAS drugs, and each has been pursued to varying degrees.
Targeting RAS Trafficking by Disrupting Palmitoylation: Protein Acyl (Palmitoyl) Transferases
Aside from CAAX-signaled modifications, enzymatic targets in the RAS processing pathway include those that regulate the palmitoylation/depalmitoylation cycle. KRAS, the predominant RAS gene mutated in human cancers, is expressed in two splice variants. The nonpalmitoylated KRAS4B has been generally accepted to be the major driver of cancer. However, recent work implicating the palmitoylated splice variant KRAS4A in colorectal adenocarcinoma (46) may force a re-evaluation of the notion that palmitoylation is not a good target in RAS-driven cancers. Moreover, palmitoylation inhibitors may help to limit the activity of oncogenic NRAS (64,65), a key driver of melanoma and hematopoietic malignancies. As inhibitors of palmitoylation and depalmitoylation are developed, it may also be important to better understand the roles of palmitoylated wild type RAS isoforms in supporting or impairing oncogenic RAS-driven cancers (1). Nonspecific inhibitors of protein palmitoylation such as 2-bromopalmitate (66) have been useful as tool compounds, but are unlikely to be developed into drugs given the vast number of palmitoylated proteins in the human genome (67). The recent identification of 23 DHHC (aspartic acid-histidine-histidine-cysteine tetrapeptide motif) proteins in the mammalian repertoire of protein acyltransferases (PATs) supports the feasibility of developing palmitoylation inhibitors that are specific for a subset of substrates such as RAS proteins (68). Consensus motifs to link specific palmitoylated substrates with their respective PATs have not yet been determined, but identification of the DHHC9/GPC16 complex as the PAT that modifies NRAS and HRAS (47) suggests the possibility of targeting this process selectively. Although progress in this direction has been limited to date (66,68,69), the recent advent of metabolic labeling with the bioorthogonal fatty acid 17-octadecynoic acid (17-ODYA) followed by click chemistry-mediated retrieval of labeled substrates promises to simplify transferase assays taht can be applied to compound screens. Additionally, because sequences around the palmitoylation sites stabilize membrane association and signaling of palmitoylated RAS proteins without affecting palmitoylation status itself (70,71), these may also need to be taken into account for successful PAT inhibitor development.
Targeting RAS Trafficking by Disrupting Depalmitoylation: APT1/LYPLA1 Acyl Protein Thioesterases
There has been more progress in the area of inhibiting depalmitoylation, although translating the present tool compounds to drugs is also likely to be challenging. Counterintuitive although it may seem at first glance, the rationale for inhibiting depalmitoylation of palmitoylated RAS proteins is that depalmitoylation on all cellular membranes seems to be required for dynamic cycling of NRAS and HRAS among their membrane locations (72). This process is facilitated by acyl protein thioesterase 1 (APT1, also known as lysophospholipase 1, LYPLA1) and possibly also APT2, which are themselves reversibly S-acylated (73). The existence of APT in both palmitoylated Golgi-bound and nonpalmitoylated cytosolic pools (74) due to autodepalmitoylating activity of APTs (74) may explain how cytosolic (nonpalmitoylated) APT can promote cycling of palmitoylatable isoforms of RAS proteins to and from membranes, ultimately resulting in enrichment of the palmitoylated forms at the PM and Golgi (72,74). These features provide the rationale for development of APT1/2 inhibitors as potential anti-RAS agents. The ability of a series of beta-lactone-based inhibitors such as palmostatins B and M (derived from the over-the-counter weight loss drug tetrahydrolipstatin (75,76)) to mislocalize HRAS and NRAS proteins to internal membranes (75,76) and to inhibit the proliferation of myeloid progenitor cells expressing oncogenic NRAS in treated mice (25) suggests that such inhibitors have the potential to disrupt oncogenic NRAS function, although questions remain regarding kinetics and target specificity (77). Additional tool compounds such as boron-based APT1/2 inhibitors have been identified (78). Other recent screens have identified LYPLA1/- and LYPLA2-selective inhibitors based on a triazole urea scaffold (79). Interestingly, APTs are not the only regulators of RAS depalmitoylation; for example, HRAS becomes depalmitoylated following the peptidyl-prolyl isomerase activity of FKBP12 on a proline near the palmitoylated cysteines (80). Understanding and optimizing the possible effects of such inhibitors may be complicated by the differential dynamics of wild type and oncogenic RAS palmitoylation (81), and by the numerous non-RAS substrates of APT1/2/LYPLA1/2 that also undergo dynamic palmitoylation, including proteins as varied as heterotrimeric G protein alpha subunits, MAGUK scaffolds, Src family tyrosine kinases, nucleoporin and BK ion channels (69). Nevertheless, the advent of new labeling technologies and new probes are making it possible to better study dynamic palmitoylation (82), which in turn is expected to reveal novel paths toward improved inhibitors of this key modification.
Targeting RAS Trafficking by Disrupting Other Modifications: PKC-mediated Phosphorylation
In addition to CAAX processing and palmitoylation, other post-translational modifications of RAS are potentially targetable (1,83). Of these, phosphorylation of KRAS4B on serine 181, a process that is mutually exclusive with calmodul in binding (84), can alter subcellular localization dramatically, displacing the modified GTPase from the PM (50,85), and converting KRAS4B from a growth-promoting to a growth-suppressing protein (86). Consistent with this, a PKC agonist, bryostatin, slowed the growth of mouse tumors driven by oncogenic KRAS4B but not those driven by a phosphorylation-deficient KRAS4B mutant (50). It is unclear why, in another study, rodent fibroblasts transformed with phosphorylation-deficient KRAS4B12V, 181A failed to produce tumors in nude mice (87), leading these investigators to conclude that phosphorylation of KRAS4B is required for oncogenesis rather than leading to growth suppression. We have found that crossing p48-Cre mice with animals with a phosphorylation-deficient LSL-Kras4B-G12D/S181A double knock-in allele produced pancreatic tumors with equal frequency as crossing to mice with a phosphomimetic LSL-Kras4B-G12D/S181S allele (unpublished results), arguing strongly against a requirement for KRAS4B phosphorylation. Importantly, a recent study (88) showed that numerous cancer-associated mutations of PKC isoforms are loss-of-function mutations. This finding suggests that these PKCs act as tumor suppressors, consistent with the effect of phosphorylating KRAS4B on S181. Regardless of the explanation, although bryostatin and analogs have been under preclinical and clinical investigation as anti-cancer treatments, are reasonably well tolerated and have anti-tumor activity (89), enthusiasm for this approach to anti-KRAS therapy is diminished by low probability of finding a drug that stimulates KRAS4B phosphorylation without affecting PKC-mediated activation of other signaling molecules that promote tumor growth and/or lead to toxicities.
Targeting RAS Trafficking by Disrupting RAS-Chaperone Interactions
In addition to blocking enzymatic activities regulating RAS membrane interactions, several distinct approaches have been taken to disrupting farnesylated RAS binding to chaperones, largely with the intent to block oncogenic KRAS4B specifically.
Inhibitors of the Ras-PDE6δ interaction have recently been identified. Deltarasin, at low micromolar levels, impaired the accumulation of KRAS4B on the PM and slowed the growth in vitro and in vivo of a tumor cell line harboring mutant KRAS (62,90). However, the degree of dependence of KRAS on PDE6δ is not yet clear. For example, PDE6d knockout mice are viable and fertile (91), whereas knockout of Kras in mice is embryonic lethal (92), indicating that KRAS can still function in the absence of PDE6δ. On the other hand, the crystal structure of PDE6δ indicates little to no specificity for one farnesylated protein over another (63), which may be reflected in the discordant observations of RAS interactions mentioned above. It may also be that, similarly to effective multi-kinase inhibitors that were once disparaged as “dirty” drugs, inhibition of multiple farnesylated proteins contributes to the salutary effects of deltarasin. Future translation of current PDE6δ inhibitors to clinical leads will likely require a better understanding of its chaperone specificity (93) and trafficking patterns.
Salirasib, also known as farnesylthiosalicylic acid (FTS), resembles the S-farnesylated cysteine on Ras (94,95) and is proposed to compete with it for binding to chaperones such as galectins (52,56,96). A small clinical trial showed that salirasib was well tolerated in pancreatic cancer patients (97), yet definitive answers as to whether it effectively perturbed KRAS function in patients, and can provide clinical benefit, remain to be determined. Like the other inhibitors of RAS membrane association, salirasib is not specific for RAS, but also inhibits other farnesylated proteins including RHEB and MTOR (1). Larger trials are merited, but await a better understanding of salirasib mechanism of action and elucidation of tractable biomarkers.
Meanwhile, galectins are also known to modulate RAS nanoclustering and localization within defined membrane microdomains (53-56,98), that in turn are regulated by phosphatidylserine (98). Thus, perhaps it was not surprising that an unbiased high-content screen to observe mislocalization of KRAS4B from PM to endomembranes revealed that staurosporine and analogs blocked endosomal recycling of phosphatidylserine and displaced KRAS4B from the PM to endosomes where it was degraded (99). Interestingly, fendiline, an L-type calcium channel blocker, was also identified as a compound that induced KRAS4B mislocalization (100); however, this activity was channel-independent. The third class of compound identified in this visual screen was metformin, that also displaced KRAS4B from the PM (101). These findings are very exciting because they suggest that unbiased screens have the potential to uncover existing drugs that can be repurposed to block KRAS4B membrane association. To further identify new regulators of KRAS4B membrane association, the Philips lab has designed a genome-wide siRNA screen employing a dual luciferase assay that quantitatively reports displacement of KRAS4B from cellular membranes (unpublished). We expect that additional roles for still other proteins in membrane targeting and/or stabilization have yet to be revealed. As this new information adds to the surge in understanding how nascent RAS proteins are delivered to the PM, we expect the RAS trafficking pathway to continue to provide a target-rich environment for drug discovery.
Acknowledgments
Grant Support: A.D. Cox and C.J. Der were supported by the NIH under award numbers R01CA042978, R21CA179193, and R01CA175747; a grant from the Lustgarten Foundation for Pancreatic Cancer Research; and the 2012 Pancreatic Cancer Action Network-AACR Innovative Grant, supported by Tempur-Pedic Retailers, grant number 12–60-25-DER. M.R. Philips was supported by the NIH under award numbers R01GM055279 and R01CA116034.
Footnotes
Note: A.D. Cox, C.J. Der, and M.R. Philips share senior authorship.
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission Possible? Nat Rev Drug Discov. 2014;13:828–51. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcus K, Mattos C. Direct attack on RAS: intramolecular communication and mutation-specific effects. Clin Cancer Res. 2015;21:xxx–xxx. doi: 10.1158/1078-0432.CCR-14-2148. [DOI] [PubMed] [Google Scholar]
- 4.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–81. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 5.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Downward J. RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin Cancer Res. 2015;21:xxx–xxx. doi: 10.1158/1078-0432.CCR-14-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimmelman AC. Metabolic dependencies in RAS-driven cancers. Clin Cancer Res. 2015;21:xxx–xxx. doi: 10.1158/1078-0432.CCR-14-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson JH, Cochrane CG, Bourne JR, Solski PA, Buss JE, Der CJ. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc Natl Acad Sci U S A. 1990;87:3042–6. doi: 10.1073/pnas.87.8.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310:583–6. doi: 10.1038/310583a0. [DOI] [PubMed] [Google Scholar]
- 10.Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ, et al. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 2003;424:694–8. doi: 10.1038/nature01806. [DOI] [PubMed] [Google Scholar]
- 11.Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4:343–50. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- 12.Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47:883–91. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, et al. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem. 1998;273:15030–4. doi: 10.1074/jbc.273.24.15030. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc Natl Acad Sci U S A. 1998;95:11175–80. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775–91. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamanoi F, Lu J. Recent progress in developing small molecule inhibitors designed to interfere with ras membrane association: toward inhibiting K-Ras and N-Ras functions. Enzymes. 2013;34 Pt. B:181–200. doi: 10.1016/B978-0-12-420146-0.00008-1. [DOI] [PubMed] [Google Scholar]
- 18.Rowell CA, Kowalczyk JJ, Lewis MD, Garcia AM. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem. 1997;272:14093–7. doi: 10.1074/jbc.272.22.14093. [DOI] [PubMed] [Google Scholar]
- 19.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–64. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 20.Liu M, Sjogren AK, Karlsson C, Ibrahim MX, Andersson KM, Olofsson FJ, et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc Natl Acad Sci USA. 2010;107:6471–6. doi: 10.1073/pnas.0908396107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.deSolms SJ, Ciccarone TM, MacTough SC, Shaw AW, Buser CA, Ellis-Hutchings M, et al. Dual protein farnesyltransferase-geranylgeranyltransferase-I inhibitors as potential cancer chemotherapeutic agents. J Med Chem. 2003;46:2973–84. doi: 10.1021/jm020587n. [DOI] [PubMed] [Google Scholar]
- 22.Lobell RB, Liu D, Buser CA, Davide JP, DePuy E, Hamilton K, et al. Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther. 2002;1:747–58. [PubMed] [Google Scholar]
- 23.Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61:8758–68. [PubMed] [Google Scholar]
- 24.Fiordalisi JJ, Holly SP, Johnson RL, 2nd, Parise LV, Cox AD. A distinct class of dominant negative Ras mutants: cytosolic GTP-bound Ras effector domain mutants that inhibit Ras signaling and transformation and enhance cell adhesion. J Biol Chem. 2002;277:10813–23. doi: 10.1074/jbc.M107684200. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Hedberg C, Dekker FJ, Li Q, Haigis KM, Hwang E, et al. Inhibiting the palmitoylation/depalmitoylation cycle selectively reduces the growth of hematopoietic cells expressing oncogenic Nras. Blood. 2012;119:1032–5. doi: 10.1182/blood-2011-06-358960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinensky M, Beck LA, Leonard S, Evans R. Differential inhibitory effects of lovastatin on protein isoprenylation and sterol synthesis. J Biol Chem. 1990;265:19937–41. [PubMed] [Google Scholar]
- 27.DeClue JE, Vass WC, Papageorge AG, Lowy DR, Willumsen BM. Inhibition of cell growth by lovastatin is independent of ras function. Cancer Res. 1991;51:712–7. [PubMed] [Google Scholar]
- 28.Cho KJ, Hill MM, Chigurupati S, Du G, Parton RG, Hancock JF. Therapeutic levels of the hydroxmethylglutaryl-coenzyme A reductase inhibitor lovastatin activate ras signaling via phospholipase D2. Mol Cell Biol. 2011;31:1110–20. doi: 10.1128/MCB.00989-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Gomes AQ, et al. Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J Biol Chem. 2001;276:5841–5. doi: 10.1074/jbc.C000831200. [DOI] [PubMed] [Google Scholar]
- 30.Lin X, Jung J, Kang D, Xu B, Zaret KS, Zoghbi H. Prenylcysteine carboxylmethyltransferase is essential for the earliest stages of liver development in mice. Gastroenterology. 2002;123:345–51. doi: 10.1053/gast.2002.34279. [DOI] [PubMed] [Google Scholar]
- 31.Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, et al. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 2002;22:171–81. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wahlstrom AM, Cutts BA, Liu M, Lindskog A, Karlsson C, Sjogren AK, et al. Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood. 2008;112:1357–65. doi: 10.1182/blood-2007-06-094060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wahlstrom AM, Cutts BA, Karlsson C, Andersson KM, Liu M, Sjogren AK, et al. Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood. 2007;109:763–8. doi: 10.1182/blood-2006-05-024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Court H, Amoyel M, Hackman M, Lee KE, Xu R, Miller G, et al. Isoprenylcysteine carboxylmethyltransferase deficiency exacerbates KRAS-driven pancreatic neoplasia via Notch suppression. J Clin Invest. 2013 doi: 10.1172/JCI65764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanlon L, Avila JL, Demarest RM, Troutman S, Allen M, Ratti F, et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010;70:4280–6. doi: 10.1158/0008-5472.CAN-09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergo MO, Lieu HD, Gavino BJ, Ambroziak P, Otto JC, Casey PJ, et al. On the physiological importance of endoproteolysis of CAAX proteins: heart-specific RCE1 knockout mice develop a lethal cardiomyopathy. J Biol Chem. 2004;279:4729–36. doi: 10.1074/jbc.M310081200. [DOI] [PubMed] [Google Scholar]
- 37.Christiansen JR, Kolandaivelu S, Bergo MO, Ramamurthy V. RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2011;108:8862–6. doi: 10.1073/pnas.1103627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fueller F, Bergo MO, Young SG, Aktories K, Schmidt G. Endoproteolytic processing of RhoA by Rce1 is required for the cleavage of RhoA by Yersinia enterocolitica outer protein T. Infect Immun. 2006;74:1712–7. doi: 10.1128/IAI.74.3.1712-1717.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manandhar SP, Hildebrandt ER, Schmidt WK. Small-molecule inhibitors of the Rce1p CaaX protease. J Biomol Screen. 2007;12:983–93. doi: 10.1177/1087057107307226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Judd WR, Slattum PM, Hoang KC, Bhoite L, Valppu L, Alberts G, et al. Discovery and SAR of methylated tetrahydropyranyl derivatives as inhibitors of isoprenylcysteine carboxyl methyltransferase (ICMT) J Med Chem. 2011;54:5031–47. doi: 10.1021/jm200249a. [DOI] [PubMed] [Google Scholar]
- 41.Lau HY, Ramanujulu PM, Guo D, Yang T, Wirawan M, Casey PJ, et al. An improved isoprenylcysteine carboxylmethyltransferase inhibitor induces cancer cell death and attenuates tumor growth in vivo. Cancer Biol Ther. 2014;15:1280–91. doi: 10.4161/cbt.29692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winter-Vann AM, Baron RA, Wong W, dela Cruz J, York JD, Gooden DM, et al. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc Natl Acad Sci USA. 2005;102:4336–41. doi: 10.1073/pnas.0408107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dechert AM, MacNamara JP, Breevoort SR, Hildebrandt ER, Hembree NW, Rea AC, et al. Modulation of the inhibitor properties of dipeptidyl (acyloxy)methyl ketones toward the CaaX proteases. Bioorg Med Chem. 2010;18:6230–7. doi: 10.1016/j.bmc.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991;10:4033–9. doi: 10.1002/j.1460-2075.1991.tb04979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133–9. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- 46.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112:779–94. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, et al. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem. 2005;280:31141–8. doi: 10.1074/jbc.M504113200. [DOI] [PubMed] [Google Scholar]
- 48.Goodwin JS, Drake KR, Rogers C, Wright L, Lippincott-Schwartz J, Philips MR, et al. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J Cell Biol. 2005;170:261–72. doi: 10.1083/jcb.200502063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–52. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 50.Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–93. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 51.Nancy V, Callebaut I, El Marjou A, de Gunzburg J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J Biol Chem. 2002;277:15076–84. doi: 10.1074/jbc.M109983200. [DOI] [PubMed] [Google Scholar]
- 52.Rotblat B, Niv H, Andre S, Kaltner H, Gabius HJ, Kloog Y. Galectin-1(L11A) predicted from a computed galectin-1 farnesyl-binding pocket selectively inhibits Ras-GTP. Cancer Res. 2004;64:3112–8. doi: 10.1158/0008-5472.can-04-0026. [DOI] [PubMed] [Google Scholar]
- 53.Abankwa D, Gorfe AA, Inder K, Hancock JF. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc Natl Acad Sci U S A. 2010;107:1130–5. doi: 10.1073/pnas.0903907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ashery U, Yizhar O, Rotblat B, Elad-Sfadia G, Barkan B, Haklai R, et al. Spatiotemporal organization of Ras signaling: rasosomes and the galectin switch. Cell Mol Neurobiol. 2006;26:471–95. doi: 10.1007/s10571-006-9059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belanis L, Plowman SJ, Rotblat B, Hancock JF, Kloog Y. Galectin-1 is a novel structural component and a major regulator of h-ras nanoclusters. Mol Biol Cell. 2008;19:1404–14. doi: 10.1091/mbc.E07-10-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shalom-Feuerstein R, Plowman SJ, Rotblat B, Ariotti N, Tian T, Hancock JF, et al. K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008;68:6608–16. doi: 10.1158/0008-5472.CAN-08-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farin K, Schokoroy S, Haklai R, Cohen-Or I, Elad-Sfadia G, Reyes-Reyes ME, et al. Oncogenic synergism between ErbB1, nucleolin, and mutant Ras. Cancer Res. 2011;71:2140–51. doi: 10.1158/0008-5472.CAN-10-2887. [DOI] [PubMed] [Google Scholar]
- 58.Inder KL, Lau C, Loo D, Chaudhary N, Goodall A, Martin S, et al. Nucleophosmin and nucleolin regulate K-Ras plasma membrane interactions and MAPK signal transduction. J Biol Chem. 2009;284:28410–9. doi: 10.1074/jbc.M109.001537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2012;14:148–58. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- 60.Bhagatji P, Leventis R, Rich R, Lin CJ, Silvius JR. Multiple cellular proteins modulate the dynamics of K-ras association with the plasma membrane. Biophys J. 2010;99:3327–35. doi: 10.1016/j.bpj.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmick M, Vartak N, Papke B, Kovacevic M, Truxius DC, Rossmannek L, et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell. 2014;157:459–71. doi: 10.1016/j.cell.2014.02.051. [DOI] [PubMed] [Google Scholar]
- 62.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–42. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 63.Ismail SA, Chen YX, Rusinova A, Chandra A, Bierbaum M, Gremer L, et al. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat Chem Biol. 2011;7:942–9. doi: 10.1038/nchembio.686. [DOI] [PubMed] [Google Scholar]
- 64.Song SP, Hennig A, Schubert K, Markwart R, Schmidt P, Prior IA, et al. Ras palmitoylation is necessary for N-Ras activation and signal propagation in growth factor signalling. Biochem J. 2013;454:323–32. doi: 10.1042/BJ20121799. [DOI] [PubMed] [Google Scholar]
- 65.Cuiffo B, Ren R. Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood. 2010;115:3598–605. doi: 10.1182/blood-2009-03-213876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Resh MD. Targeting protein lipidation in disease. Trends Mol Med. 2012;18:206–14. doi: 10.1016/j.molmed.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang W, Di Vizio D, Kirchner M, Steen H, Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 2010;9:54–70. doi: 10.1074/mcp.M800448-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chavda B, Arnott JA, Planey SL. Targeting protein palmitoylation: selective inhibitors and implications in disease. Expert Opin Drug Discov. 2014;9:1005–19. doi: 10.1517/17460441.2014.933802. [DOI] [PubMed] [Google Scholar]
- 69.Hernandez JL, Majmudar JD, Martin BR. Profiling and inhibiting reversible palmitoylation. Curr Opin Chem Biol. 2013;17:20–6. doi: 10.1016/j.cbpa.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Willumsen BM, Cox AD, Solski PA, Der CJ, Buss JE. Novel determinants of H-Ras plasma membrane localization and transformation. Oncogene. 1996;13:1901–9. [PubMed] [Google Scholar]
- 71.Laude AJ, Prior IA. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Sci. 2008;121:421–7. doi: 10.1242/jcs.020107. [DOI] [PubMed] [Google Scholar]
- 72.Rocks O, Gerauer M, Vartak N, Koch S, Huang ZP, Pechlivanis M, et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010;141:458–71. doi: 10.1016/j.cell.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 73.Kong E, Peng S, Chandra G, Sarkar C, Zhang Z, Bagh MB, et al. Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-ras product and growth-associated protein-43. J Biol Chem. 2013;288:9112–25. doi: 10.1074/jbc.M112.421073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vartak N, Papke B, Grecco HE, Rossmannek L, Waldmann H, Hedberg C, et al. The autodepalmitoylating activity of APT maintains the spatial organization of palmitoylated membrane proteins. Biophys J. 2014;106:93–105. doi: 10.1016/j.bpj.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010;6:449–56. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 76.Hedberg C, Dekker FJ, Rusch M, Renner S, Wetzel S, Vartak N, et al. Development of highly potent inhibitors of the Ras-targeting human acyl protein thioesterases based on substrate similarity design. Angew Chem Int Ed Engl. 2011;50:9832–7. doi: 10.1002/anie.201102965. [DOI] [PubMed] [Google Scholar]
- 77.Cox AD. Protein localization: Can too much lipid glue stop Ras? Nat Chem Biol. 2010;6:483–5. doi: 10.1038/nchembio.399. [DOI] [PubMed] [Google Scholar]
- 78.Zimmermann TJ, Burger M, Tashiro E, Kondoh Y, Martinez NE, Gormer K, et al. Boron-based inhibitors of acyl protein thioesterases 1 and 2. Chembiochem. 2013;14:115–22. doi: 10.1002/cbic.201200571. [DOI] [PubMed] [Google Scholar]
- 79.Adibekian A, Martin BR, Chang JW, Hsu KL, Tsuboi K, Bachovchin DA, et al. Probe Reports from the NIH Molecular Libraries Program [monograph on the Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2010. [cited 2015 Jan 26]. Characterization of a selective, reversible inhibitor of lysophospholipase 1 (LYPLA1) Available from http://www.ncbi.nlm.nih.gov/books/NBK189924/ [PubMed] [Google Scholar]
- 80.Ahearn IM, Tsai FD, Court H, Zhou M, Jennings BC, Ahmed M, et al. FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol Cell. 2011;41:173–85. doi: 10.1016/j.molcel.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baker TL, Zheng H, Walker J, Coloff JL, Buss JE. Distinct rates of palmitate turnover on membrane-bound cellular and oncogenic H-ras. J Biol Chem. 2003;278:19292–300. doi: 10.1074/jbc.M206956200. [DOI] [PubMed] [Google Scholar]
- 82.Martin BR. Chemical approaches for profiling dynamic palmitoylation. Biochem Soc Trans. 2013;41:43–9. doi: 10.1042/BST20120271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Villalonga P, Lopez-Alcala C, Chiloeches A, Gil J, Marais R, Bachs O, et al. Calmodulin prevents activation of Ras by PKC in 3T3 fibroblasts. J Biol Chem. 2002;277:37929–35. doi: 10.1074/jbc.M202245200. [DOI] [PubMed] [Google Scholar]
- 85.Plowman SJ, Ariotti N, Goodall A, Parton RG, Hancock JF. Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol Cell Biol. 2008;28:4377–85. doi: 10.1128/MCB.00050-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sung PJ, Tsai FD, Vais H, Court H, Yang J, Fehrenbacher N, et al. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci USA. 2013;110:20593–8. doi: 10.1073/pnas.1306431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barcelo C, Paco N, Morell M, Alvarez-Moya B, Bota-Rabassedas N, Jaumot M, et al. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014;74:1190–9. doi: 10.1158/0008-5472.CAN-13-1750. [DOI] [PubMed] [Google Scholar]
- 88.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, et al. Cancer-associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kollar P, Rajchard J, Balounova Z, Pazourek J. Marine natural products: bryostatins in preclinical and clinical studies. Pharm Biol. 2014;52:237–42. doi: 10.3109/13880209.2013.804100. [DOI] [PubMed] [Google Scholar]
- 90.Zimmermann G, Schultz-Fademrecht C, Kuchler P, Murarka S, Ismail S, Triola G, et al. Structure guided design and kinetic analysis of highly potent benzimidazole inhibitors targeting the PDEdelta prenyl binding site. J Med Chem. 2014;57:5435–48. doi: 10.1021/jm500632s. [DOI] [PubMed] [Google Scholar]
- 91.Zhang H, Li S, Doan T, Rieke F, Detwiler PB, Frederick JM, et al. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2007;104:8857–62. doi: 10.1073/pnas.0701681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–81. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Azoulay-Alfaguter I, Strazza M, Mor A. Chaperone-mediated specificity in Ras and Rap signaling. Crit Rev Biochem Mol Biol. 2014:1–9. doi: 10.3109/10409238.2014.989308. [DOI] [PubMed] [Google Scholar]
- 94.Elad G, Paz A, Haklai R, Marciano D, Cox A, Kloog Y. Targeting of K-Ras 4B by S-trans, trans-farnesyl thiosalicylic acid. Biochim Biophys Acta. 1999;1452:228–42. doi: 10.1016/s0167-4889(99)00144-5. [DOI] [PubMed] [Google Scholar]
- 95.Haklai R, Weisz MG, Elad G, Paz A, Marciano D, Egozi Y, et al. Dislodgment and accelerated degradation of Ras. Biochemistry. 1998;37:1306–14. doi: 10.1021/bi972032d. [DOI] [PubMed] [Google Scholar]
- 96.Kloog Y, Elad-Sfadia G, Haklai R, Mor A. Ras chaperones: new targets for cancer and immunotherapy. Enzymes. 2013;33 Pt A:267–89. doi: 10.1016/B978-0-12-416749-0.00012-9. [DOI] [PubMed] [Google Scholar]
- 97.Laheru D, Shah P, Rajeshkumar NV, McAllister F, Taylor G, Goldsweig H, et al. Integrated preclinical and clinical development of S-trans, trans-Farnesylthiosalicylic Acid (FTS, Salirasib) in pancreatic cancer. Invest New Drugs. 2012;30:2391–9. doi: 10.1007/s10637-012-9818-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou Y, Liang H, Rodkey T, Ariotti N, Parton RG, Hancock JF. Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Mol Cell Biol. 2014;34:862–76. doi: 10.1128/MCB.01227-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cho KJ, Park JH, Piggott AM, Salim AA, Gorfe AA, Parton RG, et al. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J Biol Chem. 2012;287:43573–84. doi: 10.1074/jbc.M112.424457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van der Hoeven D, Cho KJ, Ma X, Chigurupati S, Parton RG, Hancock JF. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol. 2013;33:237–51. doi: 10.1128/MCB.00884-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cho KJ, van der Hoeven D, Hancock JF. Inhibitors of K-Ras plasma membrane localization. Enzymes. 2013;33 Pt A:249–65. doi: 10.1016/B978-0-12-416749-0.00011-7. [DOI] [PubMed] [Google Scholar]