

Abstract
Objective
Hyperhomocysteinemia (HHcy) is a risk factor for cardiovascular disease and has been reported to inhibit endothelial cell (EC) growth. Notwithstanding, precisely how HHcy regulates EC growth in vivo remains unknown. In this study, we established a mouse model of endothelial injury and reendothelialization and examined the role and mechanism of HHcy in endothelial repair.
Methods and results
A mouse model of carotid artery air-dry endothelium denudation and reendothelialization was established and used to evaluate post-injury endothelial repair in mice with the gene deletion of cystathionine-β-synthase (CBS). Moderate and severe HHcy were induced in CBS+/+ and CBS−/+ mice through a high-methionine diet. Post-injury reendothelialization, which correlated with increased post-injury neointima formation, was impaired in severe HHcy mice. To elucidate the underlying mechanism, we examined circulating endothelial progenitor cells (EPC) in HHcy mice and studied the effect of homocysteine (Hcy) on proliferation, migration, and adhesion of human umbilical vein endothelial cells (HUVEC). The peripheral EPC population was not significantly altered in HHcy mice. Hcy had a profound inhibitory effect on EC proliferation and migration at physiologically relevant concentrations and inhibited EC adhesion at concentrations of 200 μM and higher.
Conclusion
We have established a convenient and accurate mouse model of carotid injury in which the reendothelialization process can be precisely quantified. In addition, we have observed impaired reendothelialization and increased neointimal formation in severe HHcy mice. The capacity of Hcy to inhibit proliferation and migration of EC may be responsible for impaired reendothelialization and contribute to arteriosclerosis in HHcy.
Keywords: Homocysteine, Carotid injury, Reendothelialization
1. Introduction
Hyperhomocysteinemia (HHcy) is an established independent risk factor for myocardial infarction and stroke [1,2]. Yet, the mechanisms by which homocysteine (Hcy) promotes cardiovascular disease (CVD) remain unclear Several biological mechanisms have been suggested to explain cardiovascular pathological changes associated with HHcy [3]. These include endothelial cell (EC) damage [4], impaired endothelial function [5], dysregulation of cholesterol and triglyceride biosynthesis [6], thrombosis [7], and stimulation of vascular smooth muscle cell (VSMC) proliferation [8,9].
We have proposed that Hcy promotes atherosclerosis by inhibiting endothelial regeneration and inducing endothelium dysfunction [10]. It has been shown that supraphysiological Hcy levels have a direct toxic effect on the endothelium [11]. We provided initial evidence demonstrating that, in the presence of adenosine, pathophysiologically relevant concentrations of Hcy inhibit EC growth-but not that of other cell types-through a hypomethylation related mechanism [4]. Recently, we found that Hcy specifically inhibits cyclin A transcription, and that adenovirus-transduced cyclin A expression rescued EC DNA synthesis and growth from the inhibitory effect of Hcy [12]. Although these studies have documented that Hcy has profound biological effects on EC in vitro, much less is known regarding the actions and mechanisms by which HHcy regulates EC growth in vivo.
Vascular disease is initiated by endothelial injury resulting from chemical, microbiological, immunological, or mechanical insults to the vessel wall. Endothelial injury is a key feature of arteriosclerosis, leading to platelet aggregation, activation of coagulation, VSMC proliferation, and atherosclerosis. In response to injury, the endothelium undergoes a process of rapid wound repair–which involves EC adhesion, migration, and proliferation–to reestablish the integrity of the endothelium. Recent studies have shown that increased endothelial recovery, or reendothelialization, correlates with diminished neointimal hyperplasia [13,14]. EC regrowth plays a pivotal role in regulating neointima formation after injury. The rate of luminal endothelial repair is a critical modulator of arterial lesion formation after injury [13,14]. Strategies to improve endothelial repair/revascularization should alter the pathobiology of vascular injury, thereby improving the prognosis of patients with myocardial/skeletal muscle ischemia, peripheral arterial disease, and organ transplantation, where endothelial damage is a major characteristic. Therapeutic angiogenesis, which has become an emerging field of cardiovascular medicine, involves inducing EC growth for revascularization in ischemic tissue via use of growth factors or gene therapy [15–17].
Endothelial injury is an early event in vascular disease and, since endothelial regeneration determines the onset of atherosclerosis, it is important to characterize the effect of Hcy on endothelial regeneration and determine the underlying biological mechanisms. In the present study, we established a mouse model of carotid air-dry endothelial denudation and regeneration, and evaluated the effect of HHcy on post-injury reendothelialization and neointima formation in mice with targeted deletion of the gene for cystathionine-β-synthase (CBS), which converts Hcy to cystathionine. We also investigated the effect of HHcy on the proliferation, migration, and adhesion of human umbilical vein endothelial cells (HUVEC).
2. Methods
2.1. CBS mice and Hcy measurement
CBS mice were purchased from Jackson Laboratory (Bar Harbor, Maine) and further backcrossed five generations, for a total twelve-generation backcross, to achieve approximately 99.9% purity in a C57BL/B6 genetic background. Mice were genotyped by PCR, as previously described [18]. CBS littermates with evenly distributed sex were selected for study. Mice were fed a control (CT) rodent diet (0% cholesterol, 5.23% fat, 0.37% methionine, 2.39 mg/g choline, 3.19 mg/kg folate, 54.6 Ag/kg B12, 14.5 mg/kg B6, [TD91354, Harlan Teklad, Madison, WI]), or a high methionine (HM) diet (TD02143, TD91354 added methionine to 2% [Harlan Teklad]) at 8 weeks of age. After 2 months on the respective diets, mouse plasma was collected for EPC analysis and Hcy measurements using liquid chromatography-electrospray tandem mass spectrometry methods, as we have previously described [19]. All animal care and procedures conform to NIH guidelines.
2.2. Flow cytometry analysis of mouse EPC
To investigate the effect of HHcy on the EPC population in vivo, mouse circulating EPC were isolated from venous blood and analyzed by flow cytometry. EPC were defined by double-positive staining for CD34, a stem cell marker, and FLK-1, an EC marker. 100 μl peripheral mouse blood was incubated with 1 μg FITC-conjugated rat anti-mouse CD34 monoclonal antibody and 0.5 μg PE-conjugated rat anti-mouse FLK-1 monoclonal antibody for 30 min. Five isotype-identical antibodies, which served as background controls, were incubated with 100 μl mouse blood: FITC-rat IgG2a, FITC-rat anti-mouse CD34, PE-rat IgG2a, PE-rat anti-mouse FLK-1, and FITC-rat anti-mouse CD34 plus PE-Rat anti-mouse FIK1. All antibodies were purchased from BD Biosciences/Pharmingen (San Diego, CA). After incubation, red blood cells were lysed with ACK buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA, pH7.4). The remaining cells were washed in PBS, fixed in 2% formaldehyde, and analyzed on an EPICS XL-MCL flow cytometer (Beckman Coulter, Miami, FL). Fluorospheres (1000 beads) (Beckman Coulter) were added to each sample in order to normalize cell number calculation. Double-stained cells were identified as EPC [20].
2.3. Carotid artery air-dry endothelial denudation and reendothelialization
After 4 weeks on their respective diets, mice were anesthetized with intraperitoneal sodium pentobarbital (50 mg/kg) and subjected to carotid artery dilation and air-dry endothelial denudation surgery. This procedure was accomplished by modifying and combining the mouse air-drying model [21] with in vivo Evans blue staining [22]. As indicated in Fig. 1A, we occluded the mouse left common carotid (LCC) and dilated it with 2 atmospheres of pressure for 1 min using an angioplasty inflation device. The inflation device was replaced with an air-filled 60 ml syringe controlled by a digital pump to deliver high-speed air for 15 min (30 ml/min) in order to dry and kill the endothelium. We placed a suture at the air-exit hole to mark the injury board (Fig. 1A). Then, the injured carotid was refilled with saline, the external carotid was ligated, and blood flow was restored. At the end of the experiment, anesthetized mice were perfused with 5% Evans blue, washed with phosphate-buffered saline (PBS), and fixed with 10% neutral buffered formalin. Both the LCC and right common carotid (RCC) were dissected, cut longitudinally, pinned on a silicon dissecting dish, and photographed under a dissecting microscope (Fig. 1B). Deendothelialized areas were defined as those areas that stained blue between the branch point of the external carotid at the proximal end and the suture placed at the air-exit hole at the distal end, using the updated Image-Pro Plus program (Media Cybernetics, Silver Spring, MD). The reendothelialization rate was calculated by dividing the deendothelialization area with the remaining blue stained area and subtracting that figure from 100%. To verify whether the Evans blue staining accurately depicted the presence or absence of endothelium, acetone-fixed frozen longitudinal sections of completely or partially deendothelialized carotid arteries were immunostained with rat anti-mouse platelet-endothelial cell adhesion molecule-1 (PECAM-1) at 1:200 dilutions (BD Biosciences/Pharmingen) (Fig. 1C).
Fig. 1.
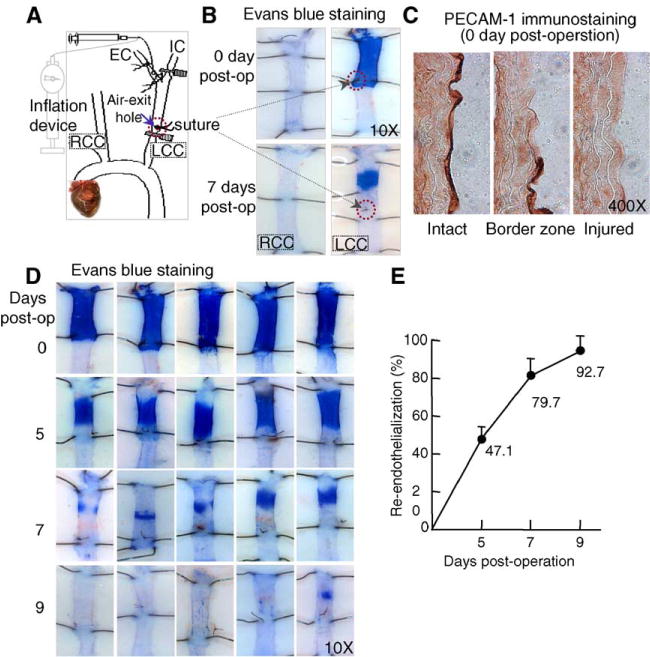
Mouse model of carotid artery air-dry endothelial denudation and reendothelialization. A. Schematic illustration of air-dry endothelial denudation. The LCC was dilated with 2 atmospheres of pressure, followed by endothelial denudation via injecting high-speed air through the carotid. A suture was placed to mark the air-exit hole. B. Evans blue staining. Animals were perfused with Evans blue at the indicated time. Excised carotids were pinned on dissecting plates. At the site of endothelial denudation, the vessel takes up dye and appears blue. C. PECAM-1 immunostaining. Acetone-fixed frozen longitudinal sections of completely or partially deendothelialized carotid arteries were immunostained with PECAM-1 antibody. The intact endothelium stained dark brown. The border zone of the injury site reveals the transition from absence to presence of endothelium. The injured section shows an absence of endothelium. D and E. Time course of post-injury reendothelialization. Images are representative of Evans blue stained mouse carotids harvested at the indicated time points. The area of endothelial denudation is defined as the branching point of EC, from IC to the suture marker at the air-exit hole. The graph shows the percentage of reendothelialization over time in the injured carotid. LCC, left common carotid; RCC, right common carotid; EC, external carotid; IC, internal carotid; PECAM-1, Platelet-Endothelial Cell Adhesion Molecule-1.
2.4. Cross-sectional neointima analysis
Four weeks after carotid artery dilation and air-dry endothelial denudation surgery, mice were perfused with PBS. The injured LCC segment was excised from the suture line at the air-exit hole of the carotid bifurcation, fixed in 2% paraformaldehyde for 40 min, and embedded in paraffin for cross-sectional analysis. Every other section (5 μM each) was collected–about 160 sections in toto –and these sections were divided into over 20 slides, so that every slide contained eight sections at 1400-μM intervals. Vessel sections were stained with an Elastic stain kit (Sigma-Aldrich Corporation, St. Louis, MO) for morphological analysis. The neointima (NI) was defined as the region between the lumen and internal elastic lamina. The medial wall (MW) was defined as the region between the internal and external elastic laminas. NI and MW areas were measured using the Image Plus program. The percentage of lumen narrowing was calculated as 100 × (area of the NI÷area inside the internal elastic lamina).
2.5. HUVEC culture and [3H]thymidine incorporation
HUVEC (Clonetics) were cultured, as previously described, and used from passages 6 to 8 [4]. DL-Hcy was directly added to the cell culture media in all the experiments. For the [3H]thymidine incorporation assay, cells were plated onto 24-well plates (4×104 cells/well) and grown to 70–80% confluence (about 24 h). The cells were then incubated with fresh medium, with or without 25 μM adenosine, plus 10 μM erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA) for 24 h and exposed to DL-Hcy for 24 h. Cells were metabolically labeled with 1 μCi/ml [methyl-3H]thymidine (DuPont/NEN, Boston MA) for the last 3 h. Incorporated [methyl-3H]thymidine was measured in a liquid scintillation counter, as previously described [4].
2.6. Adhesion assay
Confluent HUVEC were incubated with DL-Hcy in the presence or absence of adenosine, as described above, for 2 days. Pretreated cells were gently detached with 0.5 mmol/L EDTA in PBS and replated on a 35 mm culture dish (1 × 105 cells/dish) coated with human fibronectin (20 μg/ml) (Sigma-Aldrich), and incubated with consistent treatment for 30 min. Adherent cells were washed twice with PBS, and fixed with 4% formaldehyde. Using a digital camera (AxioCam color, Zeiss), images from 7 fields were randomly taken. Cell numbers were counted by blinded independent investigators using the Image Plus program [23,24].
2.7. Scratch wound assay
The migration of HUVEC was assessed by scratch wound assays [25]. Confluent HUVEC were cultured on 35 mm dishes and preincubated with DL-Hcy for 2 days. Cells were wounded by scratching with a micropipette tip, rinsed with PBS, and then incubated with consistent treatment for 20 h. Wound closure was monitored through the use of digital photography and measured using the Image Plus program. Cell migration was expressed as the percentage of distance migrated divided by the length of the initial wound.
2.8. Statistical analysis
All in vitro assays were repeated 3 times. Results are expressed as the mean ± SD. Statistical comparison of single parameters between two groups were performed by paired Student’s t test. Kruskal–Wallis One-way ANOVA was used to compare the means of multiple groups, followed by Dunn’s test. A p value ≥ 0.05 was considered significant.
3. Results
CBS mice and plasma Hcy concentration. Consistent with previous observations, plasma Hcy levels were increased about 2-fold in CBS−/+ mice compared with CBS+/+ mice, which corresponds to mild human HHcy The HM diet induced moderate HHcy (plasma Hcy 40.6 μM) in the CBS+/+ mice and severe HHcy (plasma Hcy 140 μM) in the CBS−/+ mice, which is similar to what is observed in human HHcy (Table 1). CBS genotype was confirmed with PCR amplification of a wild-type fragment (550 bp) and a knockout DNA fragment (450 bp) of CBS mouse genomic DNA (Fig. 2B). Hepatic CBS protein levels and enzymatic activities were reduced by greater than 50% in CBS−/+ mice, compared to their CBS+/+ littermates [26].
Table 1.
Plasma levels of Hcy and EPC population in CBS mice
| Genotype | Diet | Hcy (μM) | EPC (cell/ml blood) |
|---|---|---|---|
| CBS+/+ | CT | 3.7±1.2 | 558±322 |
| CBS−/+ | CT | 6.8±1.6* | 612±326 |
| CBS+/+ | HM | 40.7±18.9*†‡ | 512±223 |
| CBS−/+ | HM | 140.3±51.5*†‡ | 404±249 |
Mice were fed either a CT or an HM diet, starting at 8 weeks of age, for 2 months. Mouse plasma was collected at 16 weeks of age. Hcy levels were measured by mass spectrometry. EPC population was determined by flow cytometric analysis. Values are mean ± SEM, n = 10;
P<0.01 versus CBS+/+;
P<0.01 versus CBS−/+;
P<0.01 versus CBS+/+ on HM diet;
CT, control; Hcy, homocysteine; EPC, endothelial progenitor cells; HM, high methionine.
Fig. 2.

Post-injury reendothelialization and vascular remodeling in HHcyc mice. CBS mice were fed a CT or an HM diet at 8 weeks of age. Air-dry endothelial denudation surgery was performed after 4 weeks. Mouse plasma was collected at the end of the experiment. Hcy levels were measured by mass spectrometry A. Evans blue staining. In vivo Evans staining was performed 7 days post-injury. B. CBS genotype and reendothelialization quantification. An agarose gel image shows CBS genotype by PCR. C. Cross-sectional analysis using Verhoeff-van Gieson staining. Carotids were harvested 4 weeks post-injury for a cross-sectional analysis. D. Quantitative analysis of cross-section. The values represent the means ± SD of data generated on 10 animals; *, p<05 versus CBS+ + mice; †, P<0.05 versus CBS−/+; ‡, P<0.05 versus CBS+/+ on HM diet; CT, control; HM, high methionine; LCC, left common carotid; RCC, right common carotid; WT, wild-type; KO, knockout; NI, neointima; MW, medial wall.
3.1. Peripheral EPC population in CBS mice
We evaluated the effect of HHcy on circulating EPC population by flow cytometry analysis. CD34 and Flk-1 double-stained cells were judged as EPC. The EPC population was decreased by HHcy, but this reduction did not achieve statistical significance. These data indicate that moderate and severe HHcy mouse models, which correspond to moderate and severe HHcy in humans, can be conveniently created in CBS mice and that the peripheral EPC population is not significantly altered in HHcy mice.
3.2. Characterization of a mouse model of endothelial injury and reendothelialization
To quantitatively monitor in vivo endothelial repair, we modified and combined the mouse carotid air-drying injury with the in vivo Evans blue staining procedures. The air-dry model was initially established in the rat by Fishman et al. [27], and adopted in mice by Simon et al. [21] for the purpose of restenosis analysis. We found that denudation of endothelium was clearly achieved, as reflected by in vivo Evans blue staining (Fig. 1B). Evans blue dye binds only to nonviable cells; thus, nonviable endothelium appears blue. Complete endothelial denudation was confirmed by EC-specific PECAM-1 immunostaining on longitudinal sections after injury on day 0, post-operation (Fig. 1C). The in vivo Evans stained nonviable endothelium blue zone was stable during the course of recovery (Fig. 1D). Reendothelialization appeared to spread from the margins of the initially deendothelialized area to the central portion. Increased reendothelialization was observed from day 0 to day 9 postendothelial injury. A small area of denudation was still observed at day 7, and endothelial regeneration was almost complete on day 9 (Fig. 1D). With the easily identified initial denudation margin labeled by the suture, reendothelialization was calculated as 0% immediately after endothelial injury on day 0; however, reendothelialization reached 47.06±6.76% on day 5, 79.66±11.43% on day 7, and 92.66±8.12% on day 9, after surgery in the control mice (Fig. 1E).
3.3. Severe HHcy suppresses reendothelialization and accelerates neointimal hyperplasia in CBS mice
We evaluated the effect of HHcy on post-injury reendothelialization in CBS mice 7 days after endothelial denudation. Neither mild HHcy in CBS−/+ mice nor moderate HHcy in CBS+/+ on HM diet affected endothelial repair after surgical injury. The post-injury reendothelialization rate was 79.66±11.43% in CBS+/+ mice, 77.43 ± 9.39% in CBS−/+ mice, and 75.43 ± 5.39% in CBS+/+ on HM diet. However, severe HHcy in CBS−/+ mice fed the HM diet resulted in a significant decrease in reendothelialization to 64.86±9.09% (Fig. 2A and B). To determine whether impaired endothelial regeneration leads to enhanced neointima (NI) formation, we examined post-injury vascular remodeling in CBS mice. Four weeks after endothelial denudation, the injured carotids developed a neointima and appeared to be rigid, with stretched-out elastic laminas (Fig. 2C). Severe HHcy CBS−/+ mice on the HM diet had significantly greater neointimal hyperplasia, with an average NI area of 0.0171 ± 0.0112 mm2, compared with 0.0070 ± 0.0043 mm in CBS+/+ mice, 0.0074 ± 0.0020 mm2 in CBS−/+ mice, and 0.0070T0.0052 mm in CBS+/+ mice on the HM diet (Fig. 2D). There were no changes in the MW area. The ratio of NI/MW of the injured carotid was significantly increased in CBS−/+ mice on the HM diet (0.45 ±0.19), compared with that of CBS+ + mice (0.22T0.12), CBS−/+ mice (0.28±0.10), and CBS+/+ mice on the HM diet (0.21 ± 0.09) (Fig. 2D), mainly due to an increase in the NI area. As with endothelial repair, severe HHcy in CBS led to a noticeable dose-sensitive acceleration in the percentage of lumen narrowing. However, significant lumen narrowing was only achieved in severe HHcy CBS−/+ mice on the HM diet (21.20±9.17%), compared to CBS−/+ mice (9.73±4.20%), CBS−/+ mice (13.28±4.45%), and CBS−/+ mice on the HM diet (15.01 ± 3.55%) (Fig. 2E). These data indicate that severe HHcy inhibits post-injury endothelium repair, leading to neointimal hyperplasia.
3.4. Hcy inhibits proliferation of HUVEC
Since EC proliferation plays a key role in endothelial repair, we examined the effect of Hcy on EC proliferation by measuring [3H]thymidine uptake in HUVEC 24 h after Hcy treatment. In contrast to our previous studies, we examined the effect of Hcy, both in the absence of adenosine and under adenosine conditions that sensitize the hypomethylation mechanism [4]. Hcy concentrations comparable to those observed in severe HHcy in humans (100 μM or higher) resulted in a dose-dependent inhibition in DNA synthesis in HUVEC (Fig. 3). However, in the presence of 50 μM adenosine, as little as 20 μM Hcy-a concentration seen in moderate human HHcy-decreased DNA synthesis to 58%.
Fig. 3.

Effect of Hcy on proliferation of HUVEC. Subconfluent HUVEC were incubated with DL-Hcy in the absence or presence of 50 μM adenosine plus 10 μM EHNA for 24 h. [3H]thymidine incorporation was measured during the last 4 h. The values represent the means ± SEM of three independent experiments (n =9); *, P <0.001 versus blank control; #, P <0.001 versus adenosine control.
3.5. Hcy inhibits migration of HUVEC
Because endothelial migration plays an important role in wound healing, we examined the effect of Hcy on the migratory properties of HUVEC using a scratch-wound assay, a well-established in vitro wound healing model [25]. Confluent monolayers of HUVEC were wounded, and wound closure was monitored by still photography 20 h after the scratch assay (Fig. 4A). Control HUVEC migrated out about 0.43 mm from the scratched edge and wound closure was almost complete (98.5%) by 20 h. However, a significant area of the wound remained cell-free in HUVEC treated with Hcy. The inhibitory effect of Hcy was dose-dependent and potentiated in the presence of adenosine (Fig. 4B and D).
Fig. 4.
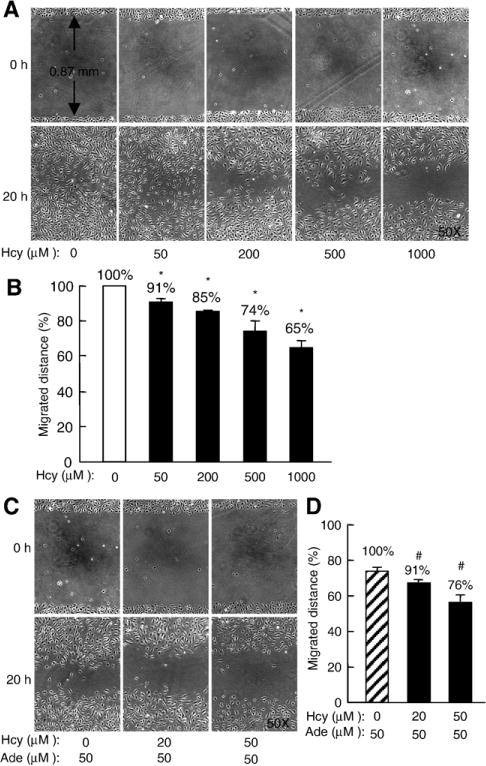
Effect of Hcy on migration of HUVEC. Confluent HUVEC were incubated with DL-Hcy in the absence or presence of 25 μM adenosine plus 10 μM EHNA for 48 h; cells were wounded by scratching with a micropipette tip. Cell migration was monitored 20 h after wounding. A and C. Representative images of wound area (0 h) and wound closure (20 h). B and D. Quantitative analysis of cell migration. The values represent the means ± SEM of three independent experiments (n =9); *, P <0.001 versus blank control; #, P <0.001 versus adenosine control.
3.6. Hcy inhibits adhesion of HUVEC
It is established that both the migration and adhesion of EC adjacent to the injury margin play a critical role in endothelial repair. We found that 200 μM Hcy reduced HUVEC adhesion to 77% (Fig. 5). Interestingly, HUVEC adhesion was not changed by Hcy in the presence of adenosine, suggesting that a hypomethylation-related mechanism is not involved in Hcy-reduced EC adhesion.
Fig. 5.
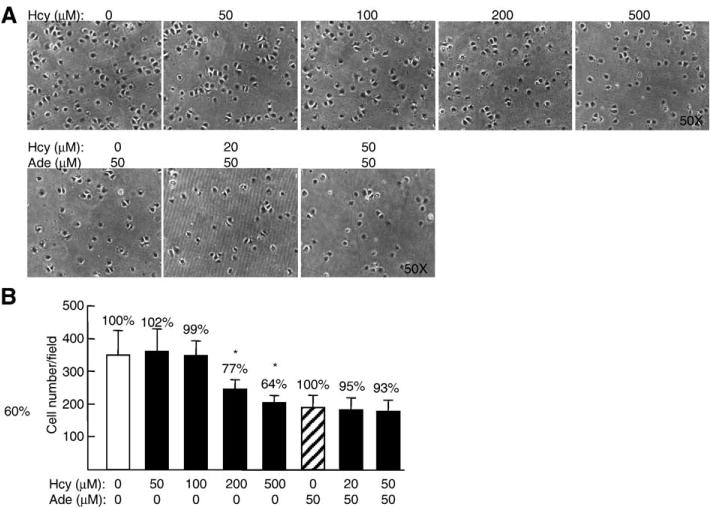
Effects of Hcy on adhesion of HUVEC. Confluent HUVEC were incubated with DL-Hcy in the absence or presence of 25 μM adenosine plus 10 μM EHNA for 48 h, replated on fibronectin-coated dishes, and incubated for 30 min. Adherent cells were fixed, photographed, and counted. The values represent the means ± SEM of three independent experiments (n =9); *, P <0.001 versus blank control; #, P <0.001 versus adenosine control.
4. Discussion
In this study, we investigated the role and mechanism of HHcy in regulating endothelial regeneration. In order to evaluate the effect of Hcy on endothelial repair in vivo, we established a convenient and accurate mouse model of endothelium injury/regeneration. Moderate and severe HHcy that are pathophysiologically relevant to humans were created in CBS+/+ and CBS−/+ mice after two months on the HM diet. Utilizing this injury model, we found that severe HHcy significantly impairs post-injury reendothelialization and accelerates neointimal hyperplasia. HHcy did not significantly decrease the circulating EPC population. Moreover, we observed that Hcy has a profound inhibitory effect on EC proliferation and migration at pathophysiologically relevant concentrations and that it inhibits EC adhesion at concentrations of 200 μM and higher-concentrations observed in human severe HHcy.
Several arterial injury models have been developed in the mouse to study endothelium regeneration. Mouse carotid or femoral arteries injured by passing a flexible wire, or by applying a laser or an electrical current can achieve both endothelial denudation and medial damage, which results in progressive intimal thickening [28]. However, the latter two approaches severely damage the medial layer of smooth muscle cells, which can impact endothelial repair; therefore, they may not be relevant to clinical circumstances. Although the wire injury model is often employed, in our experience, the extent of vascular injury is highly inconsistent. This is likely due to variations in arterial size. In addition, there is a high incidence of thrombosis in this model and there are recovery complications related to the severe injury of the medial layer. A major limitation in monitoring endothelial repair with any of these models is the difficulty in identifying the original injury border. Therefore, precise quantification is not readily achieved. In light of this, we established a model of carotid air-dry endothelial denudation and regeneration by modifying the air-drying mouse model [21], which was initially developed to study neointimal hyperplasia. This procedure produces endothelial denudation similar to that caused by coronary angioplasty, which is commonly used in the clinical treatment of acute myocardial infarction. One of the key modifications we made was placing a suture on the air-exit hole; this precisely defines the injury border, which can then be easily identified following reendothelialization. Thus, we have developed a reliable, reproducible model of mouse carotid endothelial injury-without medial wall damage-in which the reendothelialization process can be accurately quantified.
By adding 2% methionine to the chow, an HM diet induces moderate HHcy (plasma Hcy 40 μM) in CBS+/+ mice and severe HHcy (plasma Hcy 140 μM) in CBS−/+ mice, which are comparable to similar conditions observed in human HHcy. Previously, moderate HHcy (plasma Hcy 20–30 μM) was induced in mice by adding methionine to the drinking water (0.5%); this was associated with vascular dysfunction [29]. Recently, severe HHcy (plasma Hcy 244 μM) was created in apolipoprotein E null mice using a B vitamin-deficient diet [30], which contains only 18%, 22%, and 15%, respectively, of the B12, folate, and B6 essential requirements recommended by the National Laboratory Animal Nutrition Committee [31]. Nonetheless, such severe B vitamin depletion may perturb metabolic processes and complicate data interpretation. Our approach using an HM diet in CBS−/+ animals is the first to easily produce severe HHcy, while not interfering with basic nutritional requirements. This strategy also permits large-scale in vivo functional assessments that were not previously possible with CBS−/− mice, due to their low survival rate.
Acute removal of the vascular endothelium results in a well-characterized response-to-injury reaction. It has been suggested that the rate of reendothelialization determines post-injury neointima formation. Damage and removal of endothelium results in arterial lesion formation with a severity that is proportional to the duration of denudation [32,33]. Studies in animal models have demonstrated that cardiovascular risk factors delay reendothelialization and that improved endothelial repair correlates with inhibited intimal hyperplasia after vascular injury [13,14]. Thus, there is an important link between the integrity of luminal endothelium and the extent of neointima formation. We found that 7 days after endothelial denudation injury, reendothelialization was attenuated by 19% in severe HHcy mice, compared with control mice. At 28 days, severe HHcy mice presented with significantly increased intimal thickening and a decreased luminal area. Moderate HHcy did not result in a significant difference in post-injury reendothelialization and intimal thickening, which is consistent with in vitro data where modest Hcy concentrations did not affect EC growth and attachment. Our data suggest that impaired endothelial repair is a major determinant of increased atherosclerosis in HHcy, and this supports our initial hypothesis: that the endothelium is a primary target of Hcy injury [10]. A previous study using a rat dietary-HHcy model observed decreased reendothelialization with a less-defined quantification method after carotid balloon injury, which also induces medial wall injury [34]. Thus, we have clearly established a role for Hcy in inhibiting EC growth [4,12] and endothelial regeneration in a physiologically relevant model. We conclude that Hcy induces atherosclerosis, at least in part, by inhibiting endothelial repair. Considering that endothelial damage is a key pathological characteristic in angioplasty and organ transplantation, preclinical studies should evaluate the beneficial effect of a combined Hcy-lowering and angiogenic therapy in preventing the consequences of vascular injury events in HHcy.
Increasing evidence supports a positive role for bone marrow-derived EPC in endothelialization and vascular repair [35]. Circulating EPC can be recruited to denuded areas and incorporated into nascent endothelium [36,37]. We found that the EPC population was slightly decreased in moderate and severe HHcy; however, this reduction did not achieve statistical significance. These data suggest that a decrease in the circulating EPC population may not play a significant role in HHcy-impaired reendothelialization.
Our Evans blue staining images suggest that EC moves from the border zone to fill the wound region, because the blue stain disappears from both ends of the injury margins. It is known that, in response to injury, endothelial cells undergo an active repair process involving proliferation, migration, and adhesion in order to reestablish endothelial integrity. We found that Hcy significantly inhibits the proliferation and migration of HUVEC in a dose-dependent manner. These inhibitory effects are observed at Hcy concentrations that are found in human severe HHcy (>100μM), which induce oxidative stress, [38,39] or in mild HHcy (>20 μM) in the presence of adenosine, a condition that sensitizes hypomethylation. Rather than the inhibition of proliferation and migration, Hcy inhibited the adhesion of HUVEC at Hcy concentrations of 200 μM or higher, but did not further inhibit EC adhesion in the presence of adenosine, a normal constituent of all body fluids. The intracellular level of adenosine is approximately 15 μM in most cells, and can rise to 100 μM during hypoxia and ischemia [40]. Hcy can utilize adenosine, a process termed Hcy adenosylation, to form S-adenosylhomocysteine (SAH), a potent inhibitor of methyltransferase. We proposed that hypomethelation, resulting from Hcy-mediated SAH accumulation, is a key chemical event in Hcy-induced EC injury [10]. This hypothesis is supported by clinical studies showing that elevated Hcy levels in patients are linked to increased SAH and impaired erythrocyte membrane protein methylation; [41]. it is also supported by animal studies showing that CBS KO mice have increased SAH levels and decreased DNA methylation [42,43]. Therefore, our data suggest that HHcy impairs reendothelialization by inhibiting EC proliferation and migration, potentially through two different biochemical mechanisms: oxidative stress and methylation inhibition.
In conclusion, our findings indicate that severe HHcy promotes post-injury neointima formation, at least in part, by impairing reendothelialization via inhibition of EC proliferation and migration. Because endothelial injury and repair play key roles in the development of arteriosclerosis, identification of the molecular basis by which Hcy inhibits endothelial function may provide important insights into the role of Hcy in CVD, and enable the identification of new therapeutic strategies to facilitate reendothelialization.
Acknowledgments
This work was supported in part by NIH grants HL67033, HL74925, and HL77288 (HW); HL36045 (AIS); and HL59976 (WD). The authors have no competing financial interests to disclose regarding this manuscript.
References
- 1.Selhub J, Jacques PF, Bostom AG, D’Agostino RB, Wilson PW, Belanger AJ, et al. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N Engl J Med. 1995;332:286–91. doi: 10.1056/NEJM199502023320502. [DOI] [PubMed] [Google Scholar]
- 2.Stampfer MJ, Malinow MR, Willett WC, Newcomer LM, Upson B, Ullmann D, et al. A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA. 1992;268:877–81. [PubMed] [Google Scholar]
- 3.Wang H, Tan H, Yang F. Mechanisms in homocysteine-induced vascular disease. Drug Discov Today (Disease mechanisms) 2005;2:25–31. [Google Scholar]
- 4.Wang H, Yoshizumi M, Lai K, Tsai JC, Perrella MA, Haber E, et al. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J Biol Chem. 1997;272:25380–5. doi: 10.1074/jbc.272.40.25380. [DOI] [PubMed] [Google Scholar]
- 5.Lentz SR, Sobey CG, Piegors DJ, Bhopatkar MY, Faraci FM, Malinow MR, et al. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia [see comments] J Clin Invest. 1996;98:24–9. doi: 10.1172/JCI118771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107:1263–73. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loscalzo J. The oxidant stress of hyperhomocyst(e)inemia [editorial; comment] J Clin Invest. 1996;98:5–7. doi: 10.1172/JCI118776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai JC, Perrella MA, Yoshizumi M, Hsieh CM, Haber E, Schlegel R, et al. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc Natl Acad Sci U S A. 1994;91:6369–73. doi: 10.1073/pnas.91.14.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai JC, Wang H, Perrella MA, Yoshizumi M, Sibinga NE, Tan LC, et al. Induction of cyclin A gene expression by homocysteine in vascular smooth muscle cells. J Clin Invest. 1996;97:146–53. doi: 10.1172/JCI118383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee ME, Wang H. Homocysteine and hypomethylation. A novel link to vascular disease. Trends Cardiovasc Med. 1999;9:49–54. doi: 10.1016/s1050-1738(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 11.Harker LA, Harlan JM, Ross R. Effect of sulfinpyrazone on homocysteine-induced endothelial injury and arteriosclerosis in baboons. Circ Res. 1983;53:731–9. doi: 10.1161/01.res.53.6.731. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Jiang X, Yang F, Chapman GB, Durante W, Sibinga NE, et al. Cyclin A transcriptional suppression is the major mechanism mediating homocysteine-induced endothelial cell growth inhibition. Blood. 2002;99:939–45. [PMC free article] [PubMed] [Google Scholar]
- 13.Hutter R, Carrick FE, Valdiviezo C, Wolinsky C, Rudge JS, Wiegand SJ, et al. Vascular endothelial growth factor regulates reendothelialization and neointima formation in a mouse model of arterial injury. Circulation. 2004;110:2430–5. doi: 10.1161/01.CIR.0000145120.37891.8A. [DOI] [PubMed] [Google Scholar]
- 14.Asahara T, Bauters C, Pastore C, Kearney M, Rossow S, Bunting S, et al. Local delivery of vascular endothelial growth factor accelerates reendothelialization and attenuates intimal hyperplasia in balloon-injured rat carotid artery. Circulation. 1995;91:2793–801. doi: 10.1161/01.cir.91.11.2793. [DOI] [PubMed] [Google Scholar]
- 15.Hall AV, Jevnikar AM. Significance of endothelial cell survival programs for renal transplantation. Am J Kidney Dis. 2003;41:1140–54. doi: 10.1016/s0272-6386(03)00345-7. [DOI] [PubMed] [Google Scholar]
- 16.Khurana R, Simons M. Insights from angiogenesis trials using fibroblast growth factor for advanced arteriosclerotic disease. Trends Cardiovasc Med. 2003;13:116–22. doi: 10.1016/s1050-1738(02)00259-1. [DOI] [PubMed] [Google Scholar]
- 17.von Degenfeld G, Banfi A, Springer ML, Blau HM. Myoblast-mediated gene transfer for therapeutic angiogenesis and arteriogenesis. Br J Pharmacol. 2003;140:620–6. doi: 10.1038/sj.bjp.0705492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, et al. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A. 1995;92:1585–9. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Jiang X, Yang F, Gaubatz JW, Ma L, Magera MJ, et al. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood. 2003;101:3901–7. doi: 10.1182/blood-2002-08-2606. [DOI] [PubMed] [Google Scholar]
- 20.Heeschen C, Aicher A, Lehmann R, Fichtlscherer S, Vasa M, Urbich C, et al. Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood. 2003;102:1340–6. doi: 10.1182/blood-2003-01-0223. [DOI] [PubMed] [Google Scholar]
- 21.Simon DI, Dhen Z, Seifert P, Edelman ER, Ballantyne CM, Rogers C. Decreased neointimal formation in Mac-1(−/−) mice reveals a role for inflammation in vascular repair after angioplasty. J Clin Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation. 2001;103:423–8. doi: 10.1161/01.cir.103.3.423. [DOI] [PubMed] [Google Scholar]
- 23.Nolte D, Pickelmann S, Swaid S, Holzle F, Wolff KD. Oxygen-carrying solutions improve tissue oxygenation in striated skin muscle subjected to critical ischemia. Mund Kiefer Gesichtschir. 2003;7:31–5. doi: 10.1007/s10006-002-0424-1. [DOI] [PubMed] [Google Scholar]
- 24.Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J, Movafagh S, Ji H, et al. Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ Res. 1998;83:187–95. doi: 10.1161/01.res.83.2.187. [DOI] [PubMed] [Google Scholar]
- 25.Su X, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003;9:171–8. [PubMed] [Google Scholar]
- 26.Vitvitsky V, Dayal S, Stabler S, Zhou Y, Wang H, Lentz SR, et al. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol. 2004;287:11. doi: 10.1152/ajpregu.00036.2004. [DOI] [PubMed] [Google Scholar]
- 27.Fishman JA, Ryan GB, Karnovsky MJ. Endothelial regeneration in the rat carotid artery and the significance of endothelial denudation in the pathogenesis of myointimal thickening. Lab Invest. 1975;32:339–51. [PubMed] [Google Scholar]
- 28.Lindner V, Fingerle J, Reidy MA. Mouse model of arterial injury. Circ Res. 1993;73:792–6. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- 29.Dayal S, Arning E, Bottiglieri T, Boger RH, Sigmund CD, Faraci FM, et al. Cerebral vascular dysfunction mediated by superoxide in hyperhomocysteinemic mice. Stroke. 2004;35:1957–62. doi: 10.1161/01.STR.0000131749.81508.18. [DOI] [PubMed] [Google Scholar]
- 30.Troen AM, Lutgens E, Smith DE, Rosenberg IH, Selhub J. The atherogenic effect of excess methionine intake. Proc Natl Acad Sci U S A. 2003;100:15089–94. doi: 10.1073/pnas.2436385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nutrient requirements of laboratory animals. Fourth revised. Washington DC: National Academy Press; 1995. [PubMed] [Google Scholar]
- 32.Fuster V, Poon M, Willerson JT. Learning from the transgenic mouse: endothelium, adhesive molecules, and neointimal formation. Circulation. 1998;97:16–8. doi: 10.1161/01.cir.97.1.16. [DOI] [PubMed] [Google Scholar]
- 33.Hutter R, Sauter BV, Reis ED, Roque M, Vorchheimer D, Carrick FE, et al. Decreased reendothelialization and increased neointima formation with endostatin overexpression in a mouse model of arterial injury. Circulation. 2003;107:1658–63. doi: 10.1161/01.CIR.0000058169.21850.CE. [DOI] [PubMed] [Google Scholar]
- 34.Morita H, Kurihara H, Yoshida S, Saito Y, Shindo T, Oh-Hashi Y, et al. Diet-induced hyperhomocysteinemia exacerbates neointima formation in rat carotid arteries after balloon injury. Circulation. 2001;103:133–9. doi: 10.1161/01.cir.103.1.133. [DOI] [PubMed] [Google Scholar]
- 35.Sata M, Hristov M, Erl W, Weber PC. Circulating vascular progenitor cells contribute to vascular repair, remodeling, and lesion formation. Endothelial progenitor cells: isolation and characterization. Trends Cardiovasc Med. 2003;13:249–53. doi: 10.1016/s1050-1738(03)00106-3. [DOI] [PubMed] [Google Scholar]
- 36.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 37.Cho HJ, Kim HS, Lee MM, Kim DH, Yang HJ, Hur J, et al. Mobilized endothelial progenitor cells by granulocyte-macrophage colony-stimulating factor accelerate reendothelialization and reduce vascular inflammation after intravascular radiation. Circulation. 2003;108:2918–25. doi: 10.1161/01.CIR.0000097001.79750.78. [DOI] [PubMed] [Google Scholar]
- 38.Welch GN, Upchurch GR, Jr, Loscalzo J. Homocysteine, oxidative stress, and vascular disease. Hosp Pract. 1997;32:81–2. doi: 10.1080/21548331.1997.11443510. [DOI] [PubMed] [Google Scholar]
- 39.Au-Yeung KK, Woo CW, Sung FL, Yip JC, Siow YLOK. Hyper-homocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ Res. 2004;94:28–36. doi: 10.1161/01.RES.0000108264.67601.2C. [DOI] [PubMed] [Google Scholar]
- 40.Frick GP, Lowenstein JM. Studies of 5′-nucleotidase in the perfused rat heart. Including measurements of the enzyme in perfused skeletal muscle and liver. J Biol Chem. 1976;251:6372–8. [PubMed] [Google Scholar]
- 41.Pema AF, Ingrosso D, Zappia V, Galletti P, Capasso G, De Santo NG. Enzymatic methyl esterification of erythrocyte membrane proteins is impaired in chronic renal failure. Evidence for high levels of the natural inhibitor S-adenosylhomocysteine. J Clin Invest. 1993;91:2497–503. doi: 10.1172/JCI116485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caudill MA, Wang JC, Melnyk S, Pogribny IP, Jernigan S, Collins MD, et al. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J Nutr. 2001;131:2811–8. doi: 10.1093/jn/131.11.2811. [DOI] [PubMed] [Google Scholar]
- 43.Dayal S, Bottiglieri T, Arning E, Maeda N, Malinow MR, Sigmund CD, et al. Endothelial dysfunction and elevation of S-adenosylhomocysteine in cystathionine beta-synthase-deficient mice. Circ Res. 2001;88:1203–9. doi: 10.1161/hh1101.092180. [DOI] [PubMed] [Google Scholar]