

Preface
The Aryl hydrocarbon receptor (AHR) is a ligand activated transcription factor that is best known for mediating the toxicity and tumor promoting properties of the carcinogen 2,3,7,8-tetrachlorodibenzo-p-dioxin, commonly referred to as “dioxin”. The AHR influences the major stages of tumorigenesis- initiation, promotion, progression, and metastasis. Physiologically relevant ligands that have been characterized are often formed during disease states or heighten innate and adaptive immune responses. Interestingly, ligand specificity and affinity varies between rodents and humans. Studies of aggressive tumors and tumor cell lines have revealed that AHR levels are elevated and can be found constitutively in the nucleus. This leads to the hypothesis that the AHR is chronically activated in tumors, thus facilitating tumor progression. The potential for therapeutic modulation of AHR activity in tumors will also be discussed.
Introduction
The Aryl hydrocarbon receptor (AHR) is a member of the basic helix–loop–helix–PER– ARNT–SIM (bHLH–PAS) subgroup of the bHLH superfamily of transcription factors and is the only member of this family known to be activated by ligands1. The AHR was discovered as the receptor that binds 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin) with high affinity and is capable of sustained hyperactivation, resulting in a myriad of toxicologic outcomes. The half-life of TCDD in humans is approximately 10 years, due to its inability to be metabolized to a polar derivative that can be excreted. These properties contribute to the potency of TCDD as a promoter of liver and skin carcinogenesis in rodents 2. The AHR has been studied extensively for its ability to induce transcription of several cytochrome P-450s that are important in the metabolism and bioactivation of carcinogens, and in particular, polycyclic aromatic hydrocarbons after heterodimerization with the Ah receptor nuclear translocator (ARNT) (Fig. 1). DNA microarray studies have established that the AHR either directly or indirectly regulates a myriad of genes3. These studies have shown that the AHR regulates genes involved in a wide variety of biochemical pathways, including energy metabolism, lipid and cholesterol synthesis, xenobiotic metabolism and various transporters. AHR knockout mice have provided insight into the physiological role of the AHR and have been useful for exploring the influence of AHR expression on susceptibility to carcinogens. The AHR is now known to be involved in a variety of cellular processes, such as the cell cycle, epithelial barrier function, cell migration, and immune function.
Figure 1. Agonist-mediated activation of the AHR.

The unliganded AHR resides in the cytoplasm of a cell, complexed with a dimer of HSP90 and the co-chaperone protein X-associated protein 2156. The AHR contains both a nuclear localization and a nuclear export signal sequence and undergoes nucleocytoplasmic shuttling. Upon binding an agonist, the AHR complex translocates into the nucleus and ARNT mediates HSP90 displacement, leading to AHR/ARNT heterodimer formation. This heterodimer is capable of binding to a dioxin responsive element (DRE) with the sequence 5′-T/G/TCGTGA/CG/TA/T-3′. Both the AHR and ARNT can recruit coactivators, leading to transcription of a wide variety of genes. The AHR target gene CYP1A1 is almost totally dependent on AHR activity for expression and is highly induced by AHR activation through multiple DREs. CYP1A1 metabolizes a number of pro-carcinogens, such as benzo(a)pyrene, to intermediates that can react with DNA to form adducts, resulting in subsequent mutagenesis.
A high degree of complexity has emerged regarding the role of AHR in cancer, with clear discrepancies between pro- and anti-tumorigenic activities evident when utilizing cell culture versus in vivo models of malignancy. Furthermore, various classes of AHR ligands and indeed ligands within the same class can differentially modulate AHR to influence tumorigenic outcomes. Nevertheless, the AHR is a potentially important drug target that can be effectively modulated with different classes of ligands. Here we will focus on what has been established about the activity of the AHR using animal models and human tissue, with particular emphasis on the role of the AHR in immune surveillance and cancer. An issue that will be covered in this review that has not received sufficient prior attention is the difference between endogenous ligand specificity for the human versus mouse AHR and how this may impact tumor biology. In addition, a wide variety of tumor types exhibit both high levels of AHR expression relative to the parent cell type, and significant constitutive receptor activity, thus AHR antagonists could potentially be employed in cancer treatment.
Mechanisms of AHR activation
AHR agonists
Numerous chemicals exhibit high affinity binding to the AHR, altering its activity in a ligand dependent manner thus rendering this receptor an attractive small molecule target (Table 1). For many years the focus was on the identification of xenobiotics that exhibit strong agonist activity, such as persistent planar halogenated polycyclic hydrocarbons (e.g. TCDD, TCDF, PCBs) and polycyclic aromatic hydrocarbons (e.g. benzo(a)pyrene, benzanthracene). More recently, a wide variety of lower affinity agonists have been identified from diverse sources4. Through the use of various assay systems that detect AHR transcriptional activity, many commercial and consumer products, fruits, vegetables and spices, were determined to have AHR activation potential, which could significantly contribute to our total AHR ligand exposure levels5, 6. However, the identity of specific compounds with AHR activity was not established in these studies. Other research revealed several drugs that exhibit off-target activity through binding to the AHR (Table 1)7. In addition, a number of reports have identified specific dietary constituents that are AHR ligands. For example, certain flavonoids (e.g. quercetin, apigenin, kaempferol) exhibit AHR agonist and antagonist activity in a cell line specific manner8, 9. Moveover, cruciferous vegetables contain significant amounts of indole glucosinolates that, upon consumption, are degraded to indole-3-carbinol. This compound then undergoes condensation reactions in the acidic environment of the stomach, creating several products that are capable of activating the AHR, with the compound of highest affinity for the AHR being indolo[3,2 b]carbazole10.
Compounds that exhibit agonist/antagonist/selective AHR activity
| Activity | Source | Examples |
|---|---|---|
| Agonist | Xenobiotics |
Halogenated aromatic hydrocarbons
2, 3, 7, 8-tetrachlorodibenzo-p-dioxin163, 164 dibenzofurans165 biphenyls166 _ENREF_166 |
|
Polyaromatic hydrocarbons
3-methylchlolanthrene165 benzo(a)pyrene148 benzanthracenes167 benzoflavones168 |
||
|
Pharmaceuticals
tranilast169 leflutamide170 omeprazole169, 171 |
||
| Dietary |
Flavonoids
quercetin148, 172 galangin9 |
|
|
Indoles
indole-3-carbinol10, 168 3, 3-diindoylmethane10, 168 indolo[3, 2b]carbazole10 |
||
| Endogenous |
Tryptophan metabolites
kynurenic acid36 kynurenine32 6-formylindolo[3,2b]carbazole173 indoxyl sulfate38 |
|
|
Others
indirubin30 7-ketocholesterol174 |
||
| Microflora | 3-methylindole11
trypthantrin14 1, 4-dihydroxy-2-naphtoic acid13 malassezin14 |
|
|
| ||
| Antagonist |
Xenobiotic
Dietary |
6, 2, 4,-trimethoxyflavone123
GNF351127 CH-223191125 resveratrol124 |
|
| ||
|
Selective AHR
modulator |
Xenobiotic | SGA360135
3, 4-dimethoxy-a-naphthoflavone136 6-methoxy-1, 3, 8-trichlorodibenzofuran165 |
The presence of AHR ligands at the site of epithelial barriers may indicate that the AHR plays a role in the response to microflora. Metabolism of tryptophan by bacteria in the intestinal tract leads to the formation of AHR ligands such as indole, indole-3-acetate and indole-3-aldehyde11, 12. A chemical library derived from a probiotic bacterium was screened for molecules that activate the AHR and the compound 1,4-dihydroxy-2-naphthoic acid was identified as an AHR agonist13. Specific strains of yeast (e.g. Malassezia species) isolated from patients with certain skin diseases are capable of synthesizing several potent AHR ligands; including indirubin, indolo[3,2b]carbazole and malassezin14. It is possible that the host responds to the presence of these chemicals by mediating AHR activation thus enhancing barrier function, although these effects will likely be tissue and context dependent. Consistent with this concept is the observation that coal tar, a rich source of AHR ligands, is capable of enhancing skin barrier function in an AHR-dependent manner15. Humans have significant exposure to AHR ligands from a variety of sources and whether these exposure levels could lead to enhanced tumorigenesis warrants further investigation. For example, one question that should be addressed is whether AHR ligand concentrations are elevated in cancer patients as the disease progresses.
Ligand independent activation
Another aspect of AHR behavior to consider is the possibility that there is genuine ligand independent activation. In support of this concept is the apparent ability of elevated cAMP levels to activate the AHR16. Furthermore, it is possible that in cells that have relatively high AHR levels, such as many tumor cell lines, the AHR undergoes dynamic nucleocytoplasmic shuttling, which could lead to AHR and ARNT heterodimerization in the absence of ligand17. Support for this concept can be found upon comparing the level of retained nuclear AHR in a human head and neck squamous cell carcinoma cell line, NH30, compared to normal human keratinocytes18. However, the possibility that AHR ligands are present in these cells cannot be excluded. Shear stress in endothelial cells can also lead to AHR activation, although the mechanism of activation is not known 19, 20. Co-expression of a mutant AHR unable to bind ligand (AhR-A375I) and ARNT increased AHR-mediated transcriptional activity suggests that the AHR can potentially heterodimerize with ARNT in the absence of ligand, although heterodimerization potential appear to be quite inefficient21.
TCDD and hepato-carcinogenesis: exemplar of AHR agonist-mediated cancer
TCDD is considered both a complete epigenetic carcinogen and a potent tumor promoter through sustained activation of the AHR and this topic has been extensively reviewed22, 23. High affinity AHR ligands can mediate significant AHR transcriptional activity even in the presence of modest levels of AHR expression, suggesting that hyper-activation of the AHR would affect a wide range of cell types. Many of these studies have focused on the role of the AHR in liver cancer in rodents. Recent progress has been made on the mechanisms of liver tumor promotion with the recognition of inflammatory signaling as a major component of hepatocellular carcinoma24. Using a two-stage carcinogenesis model, it has been shown that the receptors for TNFA, TNFB, IL1A, and Il1B play a major role in the ability of the carcinogen diethylnitrosamine to mediate tumor incidence in mice25. Furthermore, the absence of these receptors greatly attenuated the ability of TCDD to promote the number and size of liver tumors. One possible explanation for these results is the key role that IL6 signaling plays in malignant progression of HCC26. Indeed, the AHR can synergistically induce IL6 in the presence of an inflammatory signal through both dioxin and NFkB response elements in the Il6 promoter27. These observations would support testing whether TCDD can promote carcinogenesis in an Il6 null mouse. Thus, the disruption of excessive AHR activation by the pool of endogenous and exogenous AHR agonist would appear to be a logical goal when considering the AHR as a therapeutic target.
Presence of endogenous AHR ligands that may influence tumorigenesis
Perhaps one of the most fundamental questions concerning the role of the AHR in carcinogenesis is whether endogenous ligands can mediate sufficient receptor activity to influence tumor progression. A number of studies have examined the role of the AHR in tumorigenesis and a complex story has emerged with reports that detail the ability of AHR activation to enhance or repress tumorigenesis. Whether AHR ligands are present in the tumor microenvironment needs to be addressed, as well as how the type of ligand (e.g. endogenous or exogenous) influences AHR activity. Early in the tumorigenesis process a complex multi-cellular inflammatory microenvironment develops. In this situation the level of AHR expression and activity probably resembles that of the tissue of origin and whether AHR ligands present are produced locally or systemically is not known. However, environmental ligands such as halogenated planar polycyclic hydrocarbons, as well as dietary ligands previously described, could facilitate a significant level of activation through systemic circulation. Indeed, AHR ligands have been detected in serum from cancer-free individuals through the use of reporter assay systems28, 29. Several indole-derived potent human AHR ligands, indirubin, indigo and metabolites of 6-formylindolo[3,2-b]carbazole, have been identified in human urine30, 31. Whether these compounds are dietary, generated by the host, or through bacterial metabolism has not been firmly established. Nevertheless, these compounds appear to add to the number of AHR agonists, found in healthy humans (Box 1).
Box 1. The human AHR exhibits differential activity relative to the rodent AHR: An emerging issue.
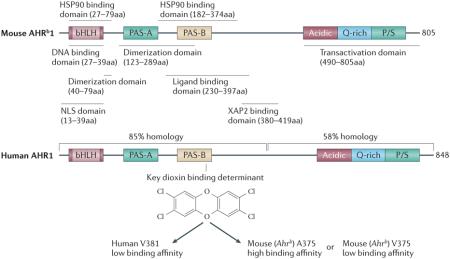
In vivo rodent studies have clearly pointed to a role for the AHR in tumorigenesis. However, there are several significant structural differences between the human and mouse AHR that lead to changes in AHR function. The first difference observed was a decrease in stability of the hAHR/hsp90 interaction compared to the mAHR142. An amino acid sequence comparison between the mouse and human AHR revealed ~85% sequence homology within the N-terminal half of the receptor, while the C-terminal half exhibits much lower sequence homology, the majority of the non-conserved changes occurring in the transactivation domain. Studies using primary hepatocytes from humans, mice, and humanized mice have revealed that the human and mouse AHR differentially mediate gene expression in hepatocytes143-145. One report has shown that the transactivation domain of the hAHR differs relative to the mAHR in terms of its ability to interact with coactivator motifs146. These studies would suggest that there are significant species differences in gene transactivation selectivity and potential. Perhaps the most dramatic difference between the human and mouse AHR is the distinct ligand selectivity. The mAHR binds TCDD with a 10-fold higher affinity compared to the hAHR, due to the difference in a single amino acid residue in the middle of the ligand binding pocket147. Conversely, the hAHR binds endogenous indolic derivatives such as indirubin and indoxyl sulfate with much higher affinity compared to the mAHR38, 148. This in turn may suggest that the human AHR could differ in its “constitutive” activation status in a tumor, which would then imply that studies in mice might underestimate the role of the AHR in human tumorigenesis.
As a tumor grows and a complex inflammatory microenvironment develops, comprised of a number of cell types, endogenous ligands appear to be produced that could set up autocrine/paracrine pathways, leading to sustained AHR activation (Fig. 2). Perhaps the most significant are the tryptophan dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO) pathways that produce kynurenine, a relatively weak AHR ligand, from tryptophan32. In the absence of a disease state, TDO is predominantly expressed in the liver and is the major degradation pathway that controls circulating tryptophan levels, while IDO is expressed in several immune cell types, particularly in an inflammatory environment. TDO and/or IDO, encoded by the TDO2 and IDO1 genes, can be expressed in a variety of tumors, leading to significant production of kynurenine33. Immune cells such as macrophages and dendritic cells within the tumor microenvironment are likely to have elevated levels of IDO1, contributing to the local depletion of tryptophan, along with increased tryptophan metabolites (AHR ligands), which contribute to increased formation of TReg cells and immune tolerance34. There is a positive correlation between,the expression of TDO and the AHR regulated gene CYP1B1 in human glioblastoma tissues. In addition, glioma cell lines can produce high levels of kynurenine capable of activating the AHR 35. These studies also demonstrated that increased CYP1B1 mRNA expression positively correlated with poor survival in patients with glioblastomas. A survey of glioma cell lines revealed that many are capable of producing kynurenine up to a concentration of 60 μM in cell culture35. Whether the level of AHR activity observed in these tumors is predominantly due to kynurenine or the presence of other AHR ligands was not established. Indeed, other products of the IDO degradation pathway, such as kynurenic and xanthurenic acid, are more potent AHR ligands than kynurenine36. In fact, kynurenic acid levels have been observed to positively correlate with the size of pancreatic adenocarcinomas37. Utilizing a glioma line with AHR expression ablated in a xenograph model, results suggested that many of the effects of kynurenine on tumor cell growth appear to be mediated by the AHR. Thus, the high normal circulating levels of tryptophan coupled with increased TDO/IDO expression in late stage tumors, could lead to significant levels of AHR ligand production and subsequent AHR activation.
Figure 2. AHR activity within the tumor micro-environment.

Tumor-associated AHR activity is elevated when compared to surrounding tissue, suggesting that AHR may influence tumor development. The mechanisms that govern enhanced tumor-associated AHR expression are unclear; notwithstanding, increased AHR expression is likely to elevate basal AHR activity within tumors, especially given the systemic omnipresence of AHR agonist ligands derived from xenobiotic, dietary and microbial sources. Indeed, malignant tissue exhibits enhanced nuclear localization of AHR together with increased expression of the prototypical AHR target gene CYP1A1, indicative of higher AHR transcriptional activity. The presence of immune cells (e.g. antigen-presenting cells, APC) within the tumor microenvironment, in conjunction with malignant cells, often exhibit enhanced expression of IDO and TDO 157, 158. These enzymes generate agonistic AHR ligands from tryptophan within the tumor micro-environment, thus adding to the AHR activation potential within tumors. The consequences of enhanced AHR expression and activities arising from systemic and tumor-derived AHR agonists have not been thoroughly investigated; however, such activation is likely to promote tumor growth. AHR activation and inflammatory cytokine signaling, a common feature of tumors, results in synergistic induction of pro-inflammatory factors, including IL6, exacerbating inflammation while simultaneously promoting diffentiation of immune-suppressive Treg cells through increased IL10, TGF, and VEGF expression. Systemic and tumor-localized generation of AHR ligands, heightened AHR expression/activity may establish a pro-inflammatory yet immune-suppressive tumor micro-environment, favoring tumor survival and escape from immune surveillance, which results in tumor progression.
Indole is a weak human AHR agonist that is generated from tryptophan in significant quantities by gut bacteria in the colon11. Indole is then absorbed by the host, circulates to the liver where it is hydroxylated by CYP2E1 followed by sulfation, forming indoxyl sulfate. The kidney then efficiently excretes this metabolite. Indoxyl sulfate is a potent endogenous ligand for the human AHR but exhibits a 40–fold lower activation potential for the mouse AHR38. In healthy humans the serum concentration of indoxyl sulfate is 2 μM, with almost 100% bound to serum proteins. In contrast, the free concentration of indoxyl sulfate in dialysis patients has been estimated to be 12-30 μM, and this level would likely lead to increased AHR activity39, 40. In support of this concept is the observation that in a nephrectomized rat model elevated indoxyl sulfate levels resulted in a significant increase in the AHR target gene Cyp1a2 in kidney and liver 41. Interestingly, the longer a patient is on kidney dialysis the greater the risk of cancer after kidney transplantation 42. Compromised kidney function often occurs in cancer patients. Thus, in the late stage of tumor progression, a wide spectrum of AHR agonists exists that may drive AHR transcriptional activity. This work firmly suggests that AHR activation could play an important role in human tumor progression43.
Consequences of AHR activation in cancer
Genetic manipulation of AHR expression and cancer
One approach to gain insight into the role of the AHR in tumorigenesis would be to utilize Ahr−/− mice. After diethylnitrosamine treatment Ahr−/− mice developed significantly more liver adenomas and exhibited elevated proliferative and inflammatory marker gene expression compared to Ahr+/+ mice44. Cecal tumorigenesis was also enhanced in Ahr−/− mice and heightened inflammatory signaling was observed45. In addition, loss of Ahr expression increased the incidence of prostate tumor formation in a TRAMP mouse model, further supporting the theory that expression of the AHR attenuates tumorigenesis46. In order to examine a model that would exhibit high constitutive AHR transcriptional activity, a transgenic mouse was bred that expresses the AHR with the hsp90-binding domain deleted (a.a. residues 288-421). This mouse line was termed CA-AhR and revealed enhanced levels of liver tumors after diethylnitrosamine exposure47. Elevated levels of gastric tumors were also observed to form spontaneously in CA-AhR mice48. The drawback of using genetic models is that one does not know whether the effects observed are due to developmental issues or are early or late in the carcinogenesis process. In addition, it is difficult to totally assess whether these mouse models truly yield insight as to what effect targeting AHR has on the tumorigenesis process. Nevertheless, these observations support the overall conclusion that AHR expression attenuates carcinogenesis and AHR activation enhances carcinogenesis. However, the lack of AHR expression should not be equated with repression of AHR activity, especially when repression occurs late in the tumorigenesis process, such as the treatment of an existing tumor with an antagonist.
Proliferation
The AHR function with regard to tumor cell proliferation appears to depend on which cell line or tumor model is used and the mechanism of action under investigation. An integrated and comprehensive view of the role of AHR in determining tumor growth is currently lacking due to the fact that most studies have been restricted to cell culture systems using clonal cancer lines with different oncogenic backgrounds. These cloned cell lines are usually derived from different tissues and cultured under optimal conditions, which lack the complex milieu of immune, stromal and tumor-associated signaling that occurs with in vivo malignancy. Nonetheless, it is clear from numerous lines of evidence that AHR does impact tumor cell proliferation through diverse and contradictory mechanisms, thus making AHR a theoretical target for suppression of tumor growth49.
Several growth factors are known to be important in tumor cell proliferation and elevated expression levels are associated with a range of cancers. Importantly, epiregulin and amphiregulin, fibroblast growth factor-9, osteopontin, and vascular endothelial growth factor all exhibit a degree of AHR dependency with regard to their expression50-55. In fact, the promoters of many of these genes (e.g. epiregulin) contain AHR DNA response elements and exhibit enhanced expression following exposure to AHR agonists50. As potent mitogens, enhanced expression of these factors is likely to contribute to tumor cell proliferation 54, 56-58. Indeed, these factors are present at increased levels within tumors of different origins. It has thus far not been established whether the elevated levels can be attributed to heightened AHR activity, although increased AHR expression is often observed in tumors (Box 2), and that, combined with the omnipresence of AHR agonists (environmental, dietary, endogenous and tumor-derived), renders some level of AHR involvement likely. Furthermore, such mitogens have been demonstrated to respond positively to tumor-associated inflammatory stimuli (e.g. Il6 and IL1B) due to the presence of the NFkB response element in their promoters, which in cell culture models provide a synergistic induction in combination with AHR agonist 18, 50. These effects of AHR upon growth factor signaling may account for the tumor promoting activity of TCDD observed in rodents 2. In addition to modulating expression of upstream mitogenic factors, multiple components of the core cell cycle machinery are directly influenced by AHR activity in cell culture (Fig. 3). Several excellent reviews have been published on this subject 59, 60.
BOX 2. Ah receptor expression during tumorigenesis.
The AHR is expressed in most tissues, except skeletal muscle, and expression levels vary widely, with the highest levels observed in liver, lung, spleen, and kidney. Within a given tissue, generally, the greatest level of expression is seen in cells of epithelial origin. Immunohistochemical analysis of breast, prostate, gastric, small cell lung, and liver tumors exhibit increased levels of AHR expression relative to surrounding tissue 64, 149-153. One possible explanation for the enhanced level of expression in human tumors is the ability of activated STAT6 and NFkB to enhance AHR expression, which correlates with inflammatory status154, 155. A specific functional p65/p50 response element was identified in the proximal promoter of the human Ahr gene. Another aspect of receptor expression to address is whether the AHR is actually transcriptionally active. One indirect approach is to examine cytoplasmic versus nuclear localization using immunohistochemical visualization of tissue sections. Grading of prostate tissue sections revealed that increased nuclear localization of the AHR correlated positively with the level of poorly differentiated cells150. Increased levels of nuclear AHR were also associated with poor prognosis for patients with lung squamous cell carcinoma152. In contrast, the levels of AHR in breast cancer were inversely correlated with the histological grade of the tumor149. This may be due to the ability of the AHR to antagonize estrogen receptor activity. Another approach to examine the role of AHR activity in human tumor samples is to determine the level of expression of an AHR target gene (e.g. CYP1B1). For example, a comparison of CYP1B1 expression levels with survival rates of patients with glioma revealed that increased CYP1B1 expression correlated with lower patient survival35. Thus, recent studies clearly support that increased AHR expression or activity correlates with promotion of late stage tumorigenesis in most human tumors that have been examined and may confer a selective advantage.
Figure 3. Proposed mechanisms of cell cycle modulation by the AHR.

Multiple mechanisms are proposed to account for the pro-/anti-proliferative action of AHR agonists observed with tumor cells in vitro. 1) Binding to AHR response elements in promoters of their respective genes, AHR acts as a direct transcriptional activator stimulating expression of growth factors epiregulin and amphiregulin. As mitogens, these contribute to the proliferation of tumor cells and may account for AHR agonist-mediated tumor cell expansion. 2) Agonist-activated AHR binds to the promoter of p27 (CDKN1B) enhancing its expression 159, 160. As an inhibitor of cyclin-dependent kinase activity, increased p27 limits phosphorylation of retinoblastoma protein (Rb) thus restricting E2F-dependent gene expression and progression through the cell cycle. 3) Association of AHR with Rb in an agonist-dependent manner attenuates both phosphorylation of Rb and liberation of E2F, resulting in cell cycle arrest and inhibition of proliferation 161. 4) Agonist activation of AHR promotes association with β-catenin and stimulation of a previously unrecognized ubiquitin ligase function of AHR 45, 67. Ubiquitination of β-catenin in an AHR-dependent fashion promotes proteosomal degradation, restricting cell cycle-dependent gene expression and proliferation. 5) In the absence of ligand, AHR forms a complex with cyclin D and the cyclin-dependent kinases CDK4/6, suppressing phosphorylation of Rb and subsequent E2F-mediated gene expression to promote cell cycle arrest 162. Exposure to AHR agonist favors the dissociation of the AHR/cyclinD/CDK complex to permit cell cycle progression and tumor cell proliferation. The contradictory nature of these mechanisms may reflect cell-type/culture-dependent differences and emphasize the need to investigate the effect of AHR ligands in the context of in vivo proliferation models.
AHR/ER crosstalk and cancer
The endocrine disrupting effects of compounds subsequently identified as AHR ligands, point to the AHR as a modulator of hormone receptor function. Subsequent studies have revealed that both the androgen receptor and the estrogen receptor (ER), in particular, are sensitive targets of AHR inhibition in a ligand-dependent manner61. Consequently, there has been an extensive analysis of the role of AHR in estrogen-dependent breast cancer. A complex picture is emerging with numerous mechanisms proposed to account for the generally anti-estrogenic nature of AHR in breast and other cancers 62-64. For example, AHR agonists stimulate CYP1A1/B1-mediated estrogen depletion 65. Proteosomal degradation of ER is enhanced by AHR acting as a component of the ubiquitin ligase complex. However, its exact role in these complexes has not been established 66, 67. In addition, estrogen-dependent transcription of numerous proliferation/apoptosis target genes is suppressed in an AHR-dependent fashion 68. These mechanisms of AHR activity are largely mediated through direct interaction with ER, resulting in mutual inhibition of DNA binding and disruption of coactivator/repressor recruitment 66, 69, 70. These suppressive mechanisms are believed to account for some of the anti-proliferative/pro-apoptotic effects of AHR agonists upon estrogen-dependent tumor growth models.
Metastasis
The signaling events that initiate metastatic progression of tumors have yet to be fully elucidated, but increasing evidence points to a role for the AHR in the modulation of cell adhesion and migratory potential (Fig. 4)16. Studies have identified increased transient AHR nuclear translocation and activity during loss of cell-cell contact through a mechanism involving Jun N-terminal kinase (JNK) activation 71, 72, as well as augmented migration in multiple cell culture models73, 74. Thus, AHR activation, whether arising from a loss of cell adhesion or agonist stimulation, may promote metastasis. A number of pro-migratory factors appear to be impacted by AHR, including adhesion components 75, proteases74, cytokines 76, signal transduction adaptors 77, 78 and transcription factors 79, 80. In contrast, expression of the chemokine CXCR4 is down regulated by AHR agonist mediated activity in breast cancer cells, suggesting an anti-metastatic effect of AHR activation 81, 82. Clearly, in vivo studies are needed to take into account the effects of AHR activation on metastatic potential within the complex tumor microenvironment.
Figure 4. Proposed role of the AHR in tumor metastasis.

The dual effect of elevated AHR expression and localized AHR agonist generation by tumor and tumor-associated immune cells increases AHR activity, leading to initiation of epithelial-mesenchymal transition (EMT) and facilitating tumor cell migration, invasiveness, and metastasis. Heightened AHR activity promotes the expression of SLUG, which then inhibits E-cadherin expression thus decreasing cell adhesion. AHR-dependent expression of matrix metalloproteases (MMPs) and intra-cellular signaling factors, which promote cytoskeletal rearrangement (e.g. VAV3) render tumor cells increasingly motile 78. The presence of tumor-associated inflammatory cytokine signaling results in a self-sustaining synergistic loop in combination with AHR activation, which enhances cell motility 18. Elevated growth and pro-angiogenic factor gene expression elicited in an AHR-dependent manner provides an escape and proliferative route for motile tumor cells. The generation of AHR agonists by tumor-associated immune cells facilitates the differentiation of immune-suppressive Treg cells, which dampens the immune response to isolated motile tumor cells, thus allowing metastasis and the establishment of distant secondary tumors.
Extrinsic factors that modulate AHR activity in tumors
An important consideration concerning AHR expression in tumor cells is that elevated expression may or may not correlate with increased AHR activity, such that any AHR-mediated response is likely dictated by numerous factors, both intrinsic and extrinsic to AHR signaling. Localized generation and availability of AHR ligands clearly will influence AHR activity at the resolution of a single cell. However, the combination of AHR and its ligands may not be permissive for any given AHR activity in the context of competing signals. For example, estrogen receptor status in breast cancer is likely to influence AHR activity 83, 84. Both receptors exhibit mutual antagonism with regard to the other, indeed this is the basis for the extensive studies purporting the beneficial aspects of AHR agonists in limiting the proliferative effects of the estrogen receptor in models of hormone-dependent breast cancer 70, 85. Estrogen receptor-dependent suppression of AHR activity may attenuate the anti-proliferative action of AHR ligands. Conversely, hormone-dependent inhibition may mitigate AHR-mediated growth factor expression, leading to tumor suppression. Therefore, AHR activity in estrogen receptor positive breast cancer cells likely differs from that in estrogen receptor negative cells. Indeed, the ability of AHR to crosstalk with numerous signaling components introduces a confounding level of complexity when trying to assess the responsiveness of AHR in any given tumor cell. Additional factors may limit AHR responsiveness such as, the metabolic and transporter activities of tumor cells, which may limit exposure to AHR ligands. AHR activity may also vary within a given tumor due to nutrient availability, cell cycle status, redox status, hypoxia, cell-cell contact, or cytokine levels, all of which are reported to influence AHR-mediated signaling 86-89. Thus, it seems likely that the response of AHR to ligands in cancer is probably highly contextual, which may account for the dichotomous observations that AHR exhibits both tumorigenic and suppressor activity. Such contextual complexity is inherently difficult to dissect and cannot be effectively addressed with in vitro cell culture studies. Even in vivo models examining the role of AHR in cancer are highly likely to impart model-dependent contextual bias.
AHR and innate immunity
The central role of innate immune signaling in the development and progression of tumorigenesis is becoming increasingly apparent. Multiple cell types within the tumor contribute to the formation of an anti-apoptotic, increased angiogenic, invasive, and metastatic phenotype90. Within the tumor microenvironment the neoplastic, immune and stromal cells can all participate in the expression of cytokines, chemokines and other inflammatory mediators, leading to positive paracrine feedback loops, which ultimately result in sustained or chronic inflammation. Recent reviews detail the altered expression of a wide variety of inflammatory signaling molecules after AHR ligand exposure63, 91, 92. In Ahr −/− mice, a challenge with lipopolysaccharide (LPS) leads to a heightened induction of a number of cytokines, suggesting that the AHR may play a role in constitutively attenuating inflammation93. Whether AHR activation regulates cytokine/chemokine expression directly or indirectly is important to consider when assessing the role of the AHR in inflammation. Evidence for direct transcriptional regulation of cytokines by the AHR is somewhat limited, but has been established for Il1b, Il6, and Il21 through promoter analysis27, 94, 95. In type 1 regulatory T cells both Il10 and Il21 are directly regulated by c-maf and the AHR in a synergistic manner. Treatment of MCF-7 or ECC-1 cancer cell lines with an AHR ligand in the presence of an inflammatory signal synergistically induces IL696. The mechanism of this synergism is driven by AHR/ARNT occupancy of DRE elements ~3 kB upstream from the transcriptional start site, which in turn mediates displacement of histone deacetylase 1 (HDAC1) from the Il6 promoter and subsequent acetylation of NF-kB (Fig. 4)27. This mode of regulation was also observed in head and neck squamous cell carcinoma lines76. The proximal promoters of a number of cytokine/chemokine genes have DRE elements, raising the possibility that the AHR directly regulates a much larger subset of genes, especially in combination with activation of a transcription factor(s) (e.g. NFkB) associated with inflammation97. Further support for synergistic regulation of cytokine/chemokine expression (e.g. Il6, Il10, Il22, Cxcl3) was observed through AHR activation in the presence of LPS in bone marrow-derived dendritic cells98. Cyclooxygenase 2 gene expression is also directly regulated by activation of both NFkB and AHR in a variety of cell types99, 100. The logic for the AHR mediating inflammation in this manner is most likely based on the role of the AHR in barrier function and as a sensor for ligands generated by flora or the host at the site of an infection. Furthermore, in an inflammatory environment such as a carcinoma line of epithelial origin, the AHR can participate directly in the regulation of several growth factors, consistent with observations in a barrier tissue injury50. Along this line of reasoning, the inflammatory microenvironment within a tumor of epithelial origin likely leads to a broader repertoire of genes altered by the AHR through combinatorial gene regulation.
The AHR appears to regulate innate inflammatory signaling, not only through binding to its cognate response element in association with ARNT, but also through direct binding to RelA and RelB, members of the NFkB family of transcription factors 101, 102. Interaction of the AHR with RelA has been shown to either repress or enhance RelA transcriptional activity in a context specific manner103. The AHR has been shown to bind to RelB and this complex can interact with p52/RelB within the promoter of a number of RelB target genes102, 104. AhR/RelB complexes have also been shown to bind to DREs, further increasing the complexity of target genes influenced by this heterodimer. In addition, interaction of RelB and AHR in the breast cancer cell lines MDA-MB 436 and MCF-7 appears to mediate IL-8 expression105. These studies further support the concept that AHR activation contributes to inflammatory signaling in tumors through multiple mechanisms.
AHR and adaptive immunity
Historically, the importance of adaptive immunity and immune tumor surveillance in tumorigenesis has been controversial. More recent studies have now firmly established that tumors escape immune surveillance through deletion and inactivation of self-reactive lymphocytes and that this is an important early event in tumor development106-108. Locally induced T-regulatory 1 (TR1) and thymus-derived natural TReg cells are believed to play a central role in mediating a suppressed immune environment within a tumor. The AHR has been shown to be an important regulator of T cell differentiation, with AHR levels markedly induced during this process95, 109, 110. The AHR functions in concert with c-Maf to enhance Il10 and Il21 expression, leading to the formation of TR1 cells. TCDD can also induce TReg cell production, suggesting that the presence of endogenous AHR ligands would enhance TReg cell production in the tumor microenvironment111. However, the ability of the AHR to influence chronic disease progression from an adaptive immunity perspective has been examined largely in autoimmune disease models. The AHR was initially shown to favor the formation of TReg cells, following treatment with TCDD112. TCDD has been shown to attenuate autoimmune type1 diabetes in a NOD mouse model and colitis in mice, which correlated with the enhanced production of TReg cells113-115. The AHR can induce both TReg and TH17 cells, with TCDD favoring the production of TReg cells concomitant with a decrease in experimental autoimmune encephalomyelitis (EAE) progression116. In contrast, the AHR agonist FICZ favors TH17 production and a subsequent increase in the severity of EAE. This would clearly suggest that the AHR agonist utilized in a given study could influence the outcome in an autoimmune disease model. A new class of AHR ligands, benzimidazoisoquinolines, has been identified that is capable of mediating enhanced TReg production in a mouse graft-versus-host autoimmune model, leading to suppression of clinical symptoms117. This study supports the concept that an AHR agonist could be developed that would be useful in organ transplants (e.g. bone marrow transplants) if the potential for possible long-term adverse effects of agonist treatment are resolved. One possible contributor to the in vivo production of TReg cells is AHR-mediated induction of IDO and kynurenine 32. In addition, naringenin a dietary flavonoid that is an AHR agonist, is also capable of inducing TReg cell production118. These results suggest that AHR likely participates in the regulation of TReg differentiation due to the presence of endogenous, exogenous, and dietary ligands. Considering the studies examining the effects of AHR ligands on suppression of autoimmunity, it is reasonable to hypothesize that AHR activity within the tumor microenvironment, such as increased production of TReg cells, may in part explain the tumor promoting properties of TCDD.
Potential therapeutic manipulation of AHR activity
Antagonism of AHR activity
AHR antagonists are increasingly being examined as possible therapeutic agents. Perhaps the first AHR antagonist described was a derivative of TCDD, 1-amino-3,7,8-trichlorodibenzo-p-dioxin119. Historically, the most utilized AHR antagonist is α-naphthoflavone, which actually exhibits weak agonist activity at high concentrations120, 121. Other flavone-based antagonists that have been described include 3′methoxy-4′-nitroflavone and 6,2′,4′-trimethoxyflavone, with the latter exhibiting no agonist activity122, 123. Interestingly, some of the beneficial activity of resveratrol has been attributed to its antagonism of the AHR124. More recently, higher affinity AHR antagonists have been identified that appear to have no agonist potential125-127. For example, the compound CH-223191 has been successfully used in rodents128. However, it fails to block agonist activity of β-naphthoflavone, highlighting the possibility that a given AHR antagonist may not block all agonist activity in a given setting129. A high affinity AHR antagonist, StemRegenin 1, was identified by a high throughput screen for compounds that enhance the expansion of hematopoietic stem cells ex vivo126. Interestingly, this compound has much higher affinity for the human versus mouse AHR, illustrating the difference in ligand specificity between species. GNF-351 is an AHR antagonist that was generated during medicinal chemistry optimization of StemReginin 1127. GNF351 does not exhibit any agonist activity and efficiently inhibits both the human and mouse AHR. These recently identified AHR antagonists should serve as useful pharmacologic tools to study the role of the AHR in tumorigenesis.
Development of Selective AHR modulators
Selective nuclear receptor ligands were first described for the estrogen receptor (ER) and were named selective ER modulators (SERMs). These compounds (e.g. tamoxifen, raloxifene) have been shown to alter the spectrum of genes that are activated by the liganded ER complex through differential co-activator/co-repressor recruitment130. This mechanism is based on the concept that the AF-2 transactivation domain in the ER is localized near the ligand-binding domain and SERMs alter the coactivator recruitment interface. The ligand binding and transactivation domains of the AHR appear to be distinct and it is not clear whether different ligands can mediate differential coactivator recruitment. However, selective Ah receptor modulators (SAhRM), such as 6-methyl-1,3,8-trichlorodibenzofuran and 1,1′-dimethyl diindolylmethane, have been identified that exhibit weak AHR agonist activity, yet still fully repress estrogen receptor activity through ER/AHR crosstalk in a manner similar to a full agonist131. Whether or not this anti-nuclear receptor activity is the primary or only therapeutic activity elicited by these compounds warrants further investigation. Another class of selective ER ligands has been generated that dissociates agonist or DNA binding activity from transrepression of NFkB and AP-1 transcriptional activity 132, 133. 134Selective AHR ligands capable of repressing acute phase gene expression, yet exhibiting no AHR agonist activity have been developed (e.g. SGA360 and 3′,4′-dimethoxy-α-naphthoflavone) 134, 135, 136. Results from these studies clearly indicate that the AHR can exhibit robust anti-inflammatory potential in the absence of DRE-mediated activity, and most likely through protein-protein interactions. The identification of this class of ligands also suggests that the AHR can exhibit constitutive anti-inflammatory activity in the apparent absence of DRE-mediated activity.
SAhRM and modulation of tumorigenesis
Numerous studies have been performed to investigate the role of the AHR upon ligand treatment in cultured tumor cells examining specific endpoints. However, there are far fewer studies that have tested the effect of various functionally distinct AHR ligand classes on tumorigenesis, especially from a therapeutic standpoint in vivo. The laboratory of Dr. Stephen Safe has pioneered the development of SAhRMs that show therapeutic potential 64. His work has focused on the development of weak AHR agonists that would not exhibit the toxicity of a full agonist, such as TCDD, using CYP1A1 induction as a measure of transcriptional activity. In the presence of a full agonist these compounds would exhibit AHR antagonistic activity. The SAhRM that was most utilized in in vivo studies is 6-methyl-1,3,8-trichlorodibenzo-p-dioxin (MCDF), which has moderate affinity for the AHR but only exhibits weak agonist activity137. However, this weak agonist is still able to mediate antiestrogenic activity and repress development of rat mammary tumors after exposure to a carcinogen138, 139. MCDF also inhibited growth of ER negative breast cancer and lung metastasis in a xenograph mouse models, suggesting that MCDF exhibited anti-tumorigenic activity through suppresion of tumor cell proliferation and metastasis140, 141. These studies indicate that there is considerable potential for the use of SAhRM in cancer treatments.
Concluding remarks
The study of the effects of AHR agonists together with the development of potent antagonists, and SAhRMs/weak agonists, has highlighted many important, if conflicting and counterintuitive, aspects regarding the role of AHR in tumorigenesis. Evaluation of the therapeutic efficacy of AHR ligands with regard to suppression of tumorigenic outcomes awaits the identification and development of specific high affinity AHR ligands that exhibit improved ADME profiles and their subsequent use in multiple in vivo models of cancer. The differences between mouse and human AHR activities suggest that ‘humanized’ AHR in vivo models would represent the preferred route. Additionally, given the contradictory observations that AHR is both pro-tumorigenic and a tumor suppressor suggests that an efficacious AHR ligand may not exhibit a “one-size-fits-all” role for limiting different types of cancer. Indeed, within a single model any AHR ligand may display temporal effectiveness, being beneficial in a chemopreventative regime but deleterious if a tumor becomes established, or vice versa. AHR agonists appear effective in attenuating cell proliferation/migration in numerous in vitro settings and limited in vivo models. However, their therapeutic application in humans has yet to be examined and is likely to be tainted by the established roles of AHR activation in biotransformation, mutagenesis, tumor promotion and immune suppression. The multitude of activities elicited by agonists evidently cannot be attributed entirely to direct AHR/ARNT heterodimer/DRE-mediated transcription and may reflect the ability of agonists to induce selective AHR activity. Continued examination of these agonist-mediated effects will be informative to the development of potent SAhRMs that can harness and specifically target the beneficial selective component of agonists without the associated toxicities. With the recent development of potent AHR antagonists, it will be important to test their effects on tumorigenesis and, perhaps most importantly, as a treatment for existing tumors. Indeed, the fact that antagonists and agonists are both capable of inhibiting tumor cell phenotype in vitro may suggest that they work through distinctly different mechanisms, yet yield the same end result. Considering the emerging role of the AHR in immune tolerance, testing of AHR antagonists as an adjunct to immunotherapy seems to be warranted. The role of endogenous ligands, in particular those produced in the gut and the tumor microenvironment, needs to be explored in terms of whether they participate in tumor progression in a variety of tumor types. The abundance of evidence demonstrating the importance of AHR in dictating tumorigenic outcomes suggests that therapeutic manipulation of AHR in human cancer is on the horizon. Whether this is through agonist or antagonist/SAhRM will be dictated by data obtained from more extensive in vivo studies using these different classes of AHR ligands as a treatment of existing tumors.
Key Points.
The Ah receptor is a ligand activated transcription factor that is best known for mediating the toxicity and tumor promotional properties of 2,3,7,8-tetrachlorodibenzo-p-dioxin, commonly referred to as “dioxin”.
Three distinct classes of ligands bind to the Ah receptor, agonist, antagonist and selective Ah receptor modulators.
The human Ah receptor compared to the mouse Ah receptor exhibits significant differences in ligand specificity, which may influence the progression of cancer and thus complicates the validity of the mouse model for human carcinogenesis.
Numerous studies demonstrate the ability of the Ah receptor to modulate proliferative and migratory potential.
The Ah receptor is activated by endogenous ligands such as kynurenine, kynurenic acid and indoxyl sulfate.
Physiologically relevant flora can produce potent Ah receptor ligands from tryptophan.
Ah receptor directly modulates inflammatory signaling.
Ah receptor agonist-mediated activity can play a key role in the production of TReg cells and thus could play a role in immune tolerance in cancer.
Ah receptor levels are often increased in tumors, most likely through inflammatory signaling.
Acknowledgements
The authors would like Marcia H. Perdew for critically reviewing the manuscript. The authors apologize to those whose work is not cited due to space limitations. The authors’ research is funded by US National Institutes of Health Grants (ES004869, ES019964 and ES022186).
Glossary
- 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, Dioxin)
A polycyclic halogenated hydrocarbon that is highly toxic to rodents and exhibits high affinity for the AHR
- Full agonist
An AHR ligand that maximally elicits canonical DRE-mediated transcriptional responses
- Weak agonist
An AHR ligand that displays partial agonist activity, eliciting a sub-maximal DRE-mediated transcriptional response. In addition, in the presence of a strong agonist will exhibit antagonist activity
- SAhRM
An AHR ligand that displays functional selectivity, exhibiting negligible DRE-mediated transcriptional responses while maximally stimulating non-DRE mediated AHR activity
- Antagonist
An AHR ligand that inhibits canonical DRE-mediated and non-DRE mediated AHR activity
- Polycyclic aromatic hydrocarbons
A group of over 100 different stable organic molecules comprised of only carbon and hydrogen. They are large, planar molecules assembled from a collection of fused benzene-like rings. They are formed during the incomplete burning of coal, oil and gas, garbage, or other organic substances like tobacco or charbroiled meat
- Barrier function
This term refers to the integrity of a protective epithelial layer that serves as a barrier and allows selective absorption
- Epithelial-mesenchymal transition (EMT)
The process by which cells convert from an epithelial to a mesenchymal phenotype. This process, which is enacted during normal embryonic development, can be abnormally activated in carcinomas, resulting in altered cell morphology, the expression of mesenchymal proteins and increased invasiveness
Biographies
Andrew D. Patterson received his Ph.D. from the Graduate Partnerships Program with George Washington University and the National Institutes of Health with Dr. Albert J. Fornace, Jr. and did his post-doctoral work with Dr. Frank J. Gonzalez. At the Pennsylvania State University, the Patterson lab is focused on understanding the host-microbiome metabolic axis with particular emphasis on the Ah receptor as a major regulator of this complex interaction. The lab employs NMR- and mass spectrometry-based metabolomics, metagenomics, and conventional and gnotobiotic genetically-modified mice to facilitate the study of these pathways and understand their impact on human health and disease.
Iain A. Murray obtained his Ph.D from the University of Manchester in the Endocrine Sciences Research Group under Professors Anne White & Julian Davis. Post-doctoral and subsequent research associate positions with Dr. Gary H. Perdew within the Center for Molecular Toxicology at the Pennsylvania State University have focused on investigating the biology and function of the Aryl hydrocarbon receptor using a range of in vitro and in vivo methodologies. Current research is aimed at the development of novel AHR ligands to dissociate beneficial/therapeutic aspects of AHR activation from established toxicological end-points with their potential application as anti-inflammatory agents.
Gary H. Perdew received his Ph.D from Oregon State University and completed post-doctoral training at McArdle Laboratory for Cancer Research in Dr. Alan Poland’s laboratory. First faculty appointment was in the Department of Foods and Nutrition at Purdue University and after eight years moved to Penn State University where he is currently an endowed professor in the Department of Veterinary and Biomedical Sciences. The Perdew laboratory is interested in the biological function of the Ah receptor and mechanisms of dioxin toxicity mediated by hyperactivation of this receptor. His laboratory also focuses on possible therapeutic potential of selective activation of the Ah receptor.
References
- 1.Bersten DC, Sullivan AE, Peet DJ, Whitelaw ML. bHLH-PAS proteins in cancer. Nat Rev Cancer. 2013;13:827–41. doi: 10.1038/nrc3621. [DOI] [PubMed] [Google Scholar]
- 2.Poland A, Palen D, Glover E. Tumour promotion by TCDD in skin of HRS/J hairless mice. Nature. 1982;300:271–3. doi: 10.1038/300271a0. [DOI] [PubMed] [Google Scholar]
- 3.Sato S, et al. Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol Appl Pharmacol. 2008;229:10–9. doi: 10.1016/j.taap.2007.12.029. [DOI] [PubMed] [Google Scholar]
- 4.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci. 2011;124:1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao B, et al. Common commercial and consumer products contain activators of the aryl hydrocarbon (dioxin) receptor. PLoS One. 2013;8:e56860. doi: 10.1371/journal.pone.0056860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeuken A, et al. Activation of the Ah receptor by extracts of dietary herbal supplements, vegetables, and fruits. J Agric Food Chem. 2003;51:5478–87. doi: 10.1021/jf030252u. [DOI] [PubMed] [Google Scholar]
- 7.Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol Pharmacol. 2007;71:1475–86. doi: 10.1124/mol.106.032748. [DOI] [PubMed] [Google Scholar]
- 8.Van der Heiden E, et al. Food flavonoid aryl hydrocarbon receptor-mediated agonistic/antagonistic/synergic activities in human and rat reporter gene assays. Anal Chim Acta. 2009;637:337–45. doi: 10.1016/j.aca.2008.09.054. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Qin C, Safe SH. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: effects of structure and cell context. Environ Health Perspect. 2003;111:1877–82. doi: 10.1289/ehp.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci U S A. 1991;88:9543–7. doi: 10.1073/pnas.88.21.9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin UH, et al. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol Pharmacol. 2014;85:777–88. doi: 10.1124/mol.113.091165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zelante T, et al. Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity. 2013;39:372–85. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Fukumoto S, et al. Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol Cell Biol. 2014;92:460–5. doi: 10.1038/icb.2014.2. [DOI] [PubMed] [Google Scholar]
- 14.Magiatis P, et al. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J Invest Dermatol. 2013;133:2023–30. doi: 10.1038/jid.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Bogaard EH, et al. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest. 2013;123:917–27. doi: 10.1172/JCI65642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oesch-Bartlomowicz B, et al. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: divergent signaling pathways. Proc Natl Acad Sci U S A. 2005;102:9218–23. doi: 10.1073/pnas.0503488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikuta T, et al. Nucleocytoplasmic shuttling of the aryl hydrocarbon receptor. J Biochem. 2000;127:503–9. doi: 10.1093/oxfordjournals.jbchem.a022633. [DOI] [PubMed] [Google Scholar]
- 18.DiNatale BC, et al. Ah receptor antagonism represses head and neck tumor cell aggressive phenotype. Mol Cancer Res. 2012;10:1369–79. doi: 10.1158/1541-7786.MCR-12-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han Z, et al. Aryl hydrocarbon receptor mediates laminar fluid shear stress-induced CYP1A1 activation and cell cycle arrest in vascular endothelial cells. Cardiovasc Res. 2008;77:809–18. doi: 10.1093/cvr/cvm095. [DOI] [PubMed] [Google Scholar]
- 20.Conway DE, et al. Expression of CYP1A1 and CYP1B1 in human endothelial cells: regulation by fluid shear stress. Cardiovasc Res. 2009;81:669–77. doi: 10.1093/cvr/cvn360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray IA, et al. Evidence that ligand binding is a key determinant of Ah receptor-mediated transcriptional activity. Arch Biochem Biophys. 2005;442:59–71. doi: 10.1016/j.abb.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 22.Bock KW, Kohle C. Ah receptor- and TCDD-mediated liver tumor promotion: clonal selection and expansion of cells evading growth arrest and apoptosis. Biochem Pharmacol. 2005;69:1403–8. doi: 10.1016/j.bcp.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Knerr S, Schrenk D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol Nutr Food Res. 2006;50:897–907. doi: 10.1002/mnfr.200600006. [DOI] [PubMed] [Google Scholar]
- 24.Naugler WE, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 25.Kennedy GD, et al. Liver Tumor Promotion by 2,3,7,8-Tetrachlorodibenzo-p-dioxin is Dependent on the Aryl Hydrocarbon Receptor and TNF/IL-1 Receptors. Toxicol Sci. 2014;140:135–43. doi: 10.1093/toxsci/kfu065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He G, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–96. doi: 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiNatale BC, Schroeder JC, Francey LJ, Kusnadi A, Perdew GH. Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J Biol Chem. 2010;285:24388–97. doi: 10.1074/jbc.M110.118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlezinger JJ, et al. Direct assessment of cumulative aryl hydrocarbon receptor agonist activity in sera from experimentally exposed mice and environmentally exposed humans. Environ Health Perspect. 2010;118:693–8. doi: 10.1289/ehp.0901113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connor KT, et al. AH receptor agonist activity in human blood measured with a cell-based bioassay: evidence for naturally occurring AH receptor ligands in vivo. J Expo Sci Environ Epidemiol. 2008;18:369–80. doi: 10.1038/sj.jes.7500607. [DOI] [PubMed] [Google Scholar]
- 30.Adachi J, et al. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem. 2001;276:31475–8. doi: 10.1074/jbc.C100238200. [DOI] [PubMed] [Google Scholar]
- 31.Wincent E, et al. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J Biol Chem. 2009;284:2690–6. doi: 10.1074/jbc.M808321200. [DOI] [PubMed] [Google Scholar]
- 32.Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pilotte L, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109:2497–502. doi: 10.1073/pnas.1113873109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci. 2013;34:136–43. doi: 10.1016/j.tips.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Opitz CA, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 36.Dinatale BC, et al. Kynurenic Acid is a Potent Endogenous Ah Receptor Ligand that Synergistically Induces Interleukin 6 in the Presence of Inflammatory Signaling. Toxicol Sci. 2010;115:89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Botwinick IC, et al. A biological basis for depression in pancreatic cancer. HPB (Oxford) 2014;16:740–43. doi: 10.1111/hpb.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schroeder JC, et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 2010;49:393–400. doi: 10.1021/bi901786x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niwa T, Takeda N, Tatematsu A, Maeda K. Accumulation of indoxyl sulfate, an inhibitor of drug-binding, in uremic serum as demonstrated by internal-surface reversed-phase liquid chromatography. Clin Chem. 1988;34:2264–7. [PubMed] [Google Scholar]
- 40.Meijers BK, et al. p-Cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:1932–8. doi: 10.2215/CJN.02940509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sindhu RK, Vaziri ND. Upregulation of cytochrome P450 1A2 in chronic renal failure: does oxidized tryptophan play a role. Adv Exp Med Biol. 2003;527:401–7. doi: 10.1007/978-1-4615-0135-0_47. [DOI] [PubMed] [Google Scholar]
- 42.Wong G, et al. Time on dialysis and cancer risk after kidney transplantation. Transplantation. 2013;95:114–21. doi: 10.1097/TP.0b013e31827743b4. [DOI] [PubMed] [Google Scholar]
- 43.Platten M, Litzenburger U, Wick W. The aryl hydrocarbon receptor in tumor immunity. Oncoimmunology. 2012;1:396–397. doi: 10.4161/onci.19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan Y, et al. The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis. Cancer Res. 2010;70:212–20. doi: 10.1158/0008-5472.CAN-09-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ikuta T, et al. ASC-associated inflammation promotes cecal tumorigenesis in aryl hydrocarbon receptor-deficient mice. Carcinogenesis. 2013;34:1620–7. doi: 10.1093/carcin/bgt083. [DOI] [PubMed] [Google Scholar]
- 46.Fritz WA, Lin TM, Cardiff RD, Peterson RE. The aryl hydrocarbon receptor inhibits prostate carcinogenesis in TRAMP mice. Carcinogenesis. 2007;28:497–505. doi: 10.1093/carcin/bgl179. [DOI] [PubMed] [Google Scholar]
- 47.Moennikes O, et al. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004;64:4707–10. doi: 10.1158/0008-5472.CAN-03-0875. [DOI] [PubMed] [Google Scholar]
- 48.Andersson P, et al. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A. 2002;99:9990–5. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Puga A, Xia Y, Elferink C. Role of the aryl hydrocarbon receptor in cell cycle regulation. Chem Biol Interact. 2002;141:117–30. doi: 10.1016/s0009-2797(02)00069-8. [DOI] [PubMed] [Google Scholar]
- 50.John K, Lahoti TS, Wagner K, Hughes JM, Perdew GH. The Ah Receptor Regulates Growth Factor Expression in Head and Neck Squamous Cell Carcinoma Cell Lines. Mol Carcinog. 2013 doi: 10.1002/mc.22032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chuang CY, et al. Up-regulation of osteopontin expression by aryl hydrocarbon receptor via both ligand-dependent and ligand-independent pathways in lung cancer. Gene. 2012;492:262–9. doi: 10.1016/j.gene.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 52.Patel RD, Kim DJ, Peters JM, Perdew GH. The aryl hydrocarbon receptor directly regulates expression of the potent mitogen epiregulin. Toxicol Sci. 2006;89:75–82. doi: 10.1093/toxsci/kfi344. [DOI] [PubMed] [Google Scholar]
- 53.Roman AC, Carvajal-Gonzalez JM, Rico-Leo EM, Fernandez-Salguero PM. Dioxin receptor deficiency impairs angiogenesis by a mechanism involving VEGF-A depletion in the endothelium and transforming growth factor-beta overexpression in the stroma. J Biol Chem. 2009;284:25135–48. doi: 10.1074/jbc.M109.013292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shigeishi H, et al. Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol Rep. 2008;19:1557–64. [PubMed] [Google Scholar]
- 55.Wang CK, et al. Aryl hydrocarbon receptor activation and overexpression upregulated fibroblast growth factor-9 in human lung adenocarcinomas. Int J Cancer. 2009;125:807–15. doi: 10.1002/ijc.24348. [DOI] [PubMed] [Google Scholar]
- 56.Nishimura T, et al. Amphiregulin and epiregulin expression in neoplastic and inflammatory lesions in the colon. Oncol Rep. 2008;19:105–10. [PubMed] [Google Scholar]
- 57.Riese DJ, 2nd, Cullum RL. Epiregulin: roles in normal physiology and cancer. Semin Cell Dev Biol. 2014;28:49–56. doi: 10.1016/j.semcdb.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu Z, et al. Epiregulin is Up-regulated in pancreatic cancer and stimulates pancreatic cancer cell growth. Biochem Biophys Res Commun. 2000;273:1019–24. doi: 10.1006/bbrc.2000.3033. [DOI] [PubMed] [Google Scholar]
- 59.Marlowe JL, Puga A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J Cell Biochem. 2005;96:1174–84. doi: 10.1002/jcb.20656. [DOI] [PubMed] [Google Scholar]
- 60.Elferink CJ. Aryl hydrocarbon receptor-mediated cell cycle control. Prog Cell Cycle Res. 2003;5:261–7. [PubMed] [Google Scholar]
- 61.Vezina CM, Lin TM, Peterson RE. AHR signaling in prostate growth, morphogenesis, and disease. Biochem Pharmacol. 2009;77:566–76. doi: 10.1016/j.bcp.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schlezinger JJ, et al. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol Chem. 2006;387:1175–87. doi: 10.1515/BC.2006.145. [DOI] [PubMed] [Google Scholar]
- 63.Feng S, Cao Z, Wang X. Role of aryl hydrocarbon receptor in cancer. Biochim Biophys Acta. 2013;1836:197–210. doi: 10.1016/j.bbcan.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 64.Safe S, Lee SO, Jin UH. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol Sci. 2013;135:1–16. doi: 10.1093/toxsci/kft128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spink DC, Johnson JA, Connor SP, Aldous KM, Gierthy JF. Stimulation of 17 beta-estradiol metabolism in MCF-7 cells by bromochloro- and chloromethyl-substituted dibenzo-p-dioxins and dibenzofurans: correlations with antiestrogenic activity. J Toxicol Environ Health. 1994;41:451–66. doi: 10.1080/15287399409531856. [DOI] [PubMed] [Google Scholar]
- 66.Wormke M, et al. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol Cell Biol. 2003;23:1843–55. doi: 10.1128/MCB.23.6.1843-1855.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohtake F, Fujii-Kuriyama Y, Kato S. AhR acts as an E3 ubiquitin ligase to modulate steroid receptor functions. Biochem Pharmacol. 2009;77:474–84. doi: 10.1016/j.bcp.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 68.Safe S, Wormke M. Inhibitory aryl hydrocarbon receptor-estrogen receptor alpha cross-talk and mechanisms of action. Chem Res Toxicol. 2003;16:807–16. doi: 10.1021/tx034036r. [DOI] [PubMed] [Google Scholar]
- 69.Madak-Erdogan Z, Katzenellenbogen BS. Aryl hydrocarbon receptor modulation of estrogen receptor alpha-mediated gene regulation by a multimeric chromatin complex involving the two receptors and the coregulator RIP140. Toxicol Sci. 2012;125:401–11. doi: 10.1093/toxsci/kfr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beischlag TV, Perdew GH. ER alpha-AHR-ARNT protein-protein interactions mediate estradiol-dependent transrepression of dioxin-inducible gene transcription. J Biol Chem. 2005;280:21607–11. doi: 10.1074/jbc.C500090200. [DOI] [PubMed] [Google Scholar]
- 71.Cho YC, Zheng W, Jefcoate CR. Disruption of cell-cell contact maximally but transiently activates AhR-mediated transcription in 10T1/2 fibroblasts. Toxicol Appl Pharmacol. 2004;199:220–38. doi: 10.1016/j.taap.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 72.Ikuta T, Kobayashi Y, Kawajiri K. Cell density regulates intracellular localization of aryl hydrocarbon receptor. J Biol Chem. 2004;279:19209–16. doi: 10.1074/jbc.M310492200. [DOI] [PubMed] [Google Scholar]
- 73.Diry M, et al. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene. 2006;25:5570–4. doi: 10.1038/sj.onc.1209553. [DOI] [PubMed] [Google Scholar]
- 74.Peng TL, Chen J, Mao W, Song X, Chen MH. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol. 2009;10:27. doi: 10.1186/1471-2121-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niermann T, Schmutz S, Erne P, Resink T. Aryl hydrocarbon receptor ligands repress T-cadherin expression in vascular smooth muscle cells. Biochem Biophys Res Commun. 2003;300:943–9. doi: 10.1016/s0006-291x(02)02970-4. [DOI] [PubMed] [Google Scholar]
- 76.Dinatale BC, Perdew GH. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Molecular Carcinogenesis. 2011;50:173–183. doi: 10.1002/mc.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bui LC, et al. Nedd9/Hef1/Cas-L mediates the effects of environmental pollutants on cell migration and plasticity. Oncogene. 2009;28:3642–51. doi: 10.1038/onc.2009.224. [DOI] [PubMed] [Google Scholar]
- 78.Fernandez-Salguero PM. A remarkable new target gene for the dioxin receptor: The Vav3 proto-oncogene links AhR to adhesion and migration. Cell Adh Migr. 2010;4:172–5. doi: 10.4161/cam.4.2.10387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ikuta T, Kawajiri K. Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp Cell Res. 2006;312:3585–94. doi: 10.1016/j.yexcr.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 80.Belguise K, et al. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 2007;67:11742–50. doi: 10.1158/0008-5472.CAN-07-2730. [DOI] [PubMed] [Google Scholar]
- 81.Hsu EL, et al. A proposed mechanism for the protective effect of dioxin against breast cancer. Toxicol Sci. 2007;98:436–44. doi: 10.1093/toxsci/kfm125. [DOI] [PubMed] [Google Scholar]
- 82.Jin UH, Lee SO, Pfent C, Safe S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer. 2014;14:498. doi: 10.1186/1471-2407-14-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dohr O, Vogel C, Abel J. Different response of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-sensitive genes in human breast cancer MCF-7 and MDA-MB 231 cells. Arch Biochem Biophys. 1995;321:405–12. doi: 10.1006/abbi.1995.1411. [DOI] [PubMed] [Google Scholar]
- 84.Stark K, et al. Reactivation of estrogen receptor alpha by vorinostat sensitizes mesenchymal-like triple-negative breast cancer to aminoflavone, a ligand of the aryl hydrocarbon receptor. PLoS One. 2013;8:e74525. doi: 10.1371/journal.pone.0074525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wihlen B, Ahmed S, Inzunza J, Matthews J. Estrogen receptor subtype- and promoter-specific modulation of aryl hydrocarbon receptor-dependent transcription. Mol Cancer Res. 2009;7:977–86. doi: 10.1158/1541-7786.MCR-08-0396. [DOI] [PubMed] [Google Scholar]
- 86.Terashima J, Habano W, Gamou T, Ozawa S. Induction of CYP1 family members under low-glucose conditions requires AhR expression and occurs through the nuclear translocation of AhR. Drug Metab Pharmacokinet. 2011;26:577–83. doi: 10.2133/dmpk.DMPK-11-RG-054. [DOI] [PubMed] [Google Scholar]
- 87.Vorrink SU, Domann FE. Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1alpha signaling node. Chem Biol Interact. 2014;218:82–8. doi: 10.1016/j.cbi.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hamouchene H, Arlt VM, Giddings I, Phillips DH. Influence of cell cycle on responses of MCF-7 cells to benzo[a]pyrene. BMC Genomics. 2011;12:333. doi: 10.1186/1471-2164-12-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shin S, et al. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol. 2007;27:7188–97. doi: 10.1128/MCB.00915-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 91.Fardel O. Cytokines as molecular targets for aryl hydrocarbon receptor ligands: implications for toxicity and xenobiotic detoxification. Expert Opin Drug Metab Toxicol. 2013;9:141–52. doi: 10.1517/17425255.2013.738194. [DOI] [PubMed] [Google Scholar]
- 92.Haarmann-Stemmann T, Bothe H, Abel J. Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem Pharmacol. 2009;77:508–20. doi: 10.1016/j.bcp.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 93.Sekine H, et al. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol Cell Biol. 2009;29:6391–400. doi: 10.1128/MCB.00337-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lahoti TS, et al. Aryl hydrocarbon receptor antagonism attenuates growth factor expression, proliferation, and migration in fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Pharmacol Exp Ther. 2014;348:236–45. doi: 10.1124/jpet.113.209726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Apetoh L, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11:854–61. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hollingshead BD, Beischlag TV, Dinatale BC, Ramadoss P, Perdew GH. Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF-7 cells. Cancer Res. 2008;68:3609–17. doi: 10.1158/0008-5472.CAN-07-6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Furman DP, Oshchepkova EA, Oshchepkov DY, Shamanina MY, Mordvinov VA. Promoters of the genes encoding the transcription factors regulating the cytokine gene expression in macrophages contain putative binding sites for aryl hydrocarbon receptor. Comput Biol Chem. 2009;33:465–8. doi: 10.1016/j.compbiolchem.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 98.Vogel CF, et al. Aryl hydrocarbon receptor signaling regulates NF-kappaB RelB activation during dendritic-cell differentiation. Immunol Cell Biol. 2013;91:568–75. doi: 10.1038/icb.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vogel CF, et al. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol. 2007;171:1538–48. doi: 10.2353/ajpath.2007.070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3'-diindolylmethane in breast cancer cells. J Nutr. 2009;139:26–32. doi: 10.3945/jn.108.099259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274:510–5. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- 102.Vogel CF, et al. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol. 2007;21:2941–55. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim DW, et al. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene. 2000;19:5498–506. doi: 10.1038/sj.onc.1203945. [DOI] [PubMed] [Google Scholar]
- 104.Vogel CF, Sciullo E, Matsumura F. Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines. Biochem Biophys Res Commun. 2007;363:722–6. doi: 10.1016/j.bbrc.2007.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vogel CF, et al. Interaction of aryl hydrocarbon receptor and NF-kappaB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys. 2011;512:78–86. doi: 10.1016/j.abb.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 107.Corthay A. Does the Immune System Naturally Protect Against Cancer. Front Immunol. 2014;5:197. doi: 10.3389/fimmu.2014.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Darrasse-Jeze G, Podsypanina K. How Numbers, Nature, and Immune Status of Foxp3 Regulatory T-Cells Shape the Early Immunological Events in Tumor Development. Front Immunol. 2013;4:292. doi: 10.3389/fimmu.2013.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marshall NB, Vorachek WR, Steppan LB, Mourich DV, Kerkvliet NI. Functional characterization and gene expression analysis of CD4+ CD25+ regulatory T cells generated in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Immunol. 2008;181:2382–91. doi: 10.4049/jimmunol.181.4.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gandhi R, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol. 2010;11:846–53. doi: 10.1038/ni.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Funatake CJ, Marshall NB, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation of alloreactive CD8+ T cells toward a regulatory T cell phenotype by a mechanism that is dependent on aryl hydrocarbon receptor in CD4+ T cells. J Immunotoxicol. 2008;5:81–91. doi: 10.1080/15476910802019037. [DOI] [PubMed] [Google Scholar]
- 112.Kerkvliet NI, Shepherd DM, Baecher-Steppan L. T lymphocytes are direct, aryl hydrocarbon receptor (AhR)-dependent targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): AhR expression in both CD4+ and CD8+ T cells is necessary for full suppression of a cytotoxic T lymphocyte response by TCDD. Toxicol Appl Pharmacol. 2002;185:146–52. doi: 10.1006/taap.2002.9537. [DOI] [PubMed] [Google Scholar]
- 113.Kerkvliet NI, et al. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy. 2009;1:539–47. doi: 10.2217/imt.09.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Benson JM, Shepherd DM. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn's disease. Toxicol Sci. 2011;120:68–78. doi: 10.1093/toxsci/kfq360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Singh NP, et al. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One. 2011;6:e23522. doi: 10.1371/journal.pone.0023522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Quintana FJ, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 117.Punj S, et al. Benzimidazoisoquinolines: a new class of rapidly metabolized aryl hydrocarbon receptor (AhR) ligands that induce AhR-dependent Tregs and prevent murine graft-versus-host disease. PLoS One. 2014;9:e88726. doi: 10.1371/journal.pone.0088726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang HK, et al. Dietary flavonoid naringenin induces regulatory T cells via an aryl hydrocarbon receptor mediated pathway. J Agric Food Chem. 2012;60:2171–8. doi: 10.1021/jf204625y. [DOI] [PubMed] [Google Scholar]
- 119.Luster MI, et al. 1-amino-3,7,8-trichlorodibenzo-p-dioxin: a specific antagonist for TCDD-induced myelotoxicity. Biochem Biophys Res Commun. 1986;139:747–56. doi: 10.1016/s0006-291x(86)80054-7. [DOI] [PubMed] [Google Scholar]
- 120.Merchant M, Arellano L, Safe S. The mechanism of action of alpha-naphthoflavone as an inhibitor of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced CYP1A1 gene expression. Arch Biochem Biophys. 1990;281:84–9. doi: 10.1016/0003-9861(90)90416-v. [DOI] [PubMed] [Google Scholar]
- 121.Gasiewicz TA, Rucci G. Alpha-naphthoflavone acts as an antagonist of 2,3,7, 8-tetrachlorodibenzo-p-dioxin by forming an inactive complex with the Ah receptor. Mol Pharmacol. 1991;40:607–12. [PubMed] [Google Scholar]
- 122.Henry EC, et al. Flavone antagonists bind competitively with 2,3,7, 8-tetrachlorodibenzo-p-dioxin (TCDD) to the aryl hydrocarbon receptor but inhibit nuclear uptake and transformation. Mol Pharmacol. 1999;55:716–25. [PubMed] [Google Scholar]
- 123.Murray IA, et al. Antagonism of aryl hydrocarbon receptor signaling by 6,2',4'-trimethoxyflavone. J Pharmacol Exp Ther. 2010;332:135–44. doi: 10.1124/jpet.109.158261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ciolino HP, Daschner PJ, Yeh GC. Resveratrol inhibits transcription of CYP1A1 in vitro by preventing activation of the aryl hydrocarbon receptor. Cancer Res. 1998;58:5707–12. [PubMed] [Google Scholar]
- 125.Kim SH, et al. Novel compound 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol Pharmacol. 2006;69:1871–8. doi: 10.1124/mol.105.021832. [DOI] [PubMed] [Google Scholar]
- 126.Boitano AE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–8. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smith KJ, et al. Identification of a high affinity ligand that exhibits complete Ah receptor antagonism. J Pharmacol Exp Ther. 2011;338:318–27. doi: 10.1124/jpet.110.178392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brembilla NC, et al. In vivo dioxin favors interleukin-22 production by human CD4+ T cells in an aryl hydrocarbon receptor (AhR)-dependent manner. PLoS One. 2011;6:e18741. doi: 10.1371/journal.pone.0018741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao B, Degroot DE, Hayashi A, He G, Denison MS. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol Sci. 2010;117:393–403. doi: 10.1093/toxsci/kfq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lewis JS, Jordan VC. Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat Res. 2005;591:247–63. doi: 10.1016/j.mrfmmm.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 131.Safe S, McDougal A. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for treatment of hormone-dependent cancers (Review) Int J Oncol. 2002;20:1123–8. [PubMed] [Google Scholar]
- 132.Steffan RJ, et al. Synthesis and activity of substituted 4-(indazol-3-yl)phenols as pathway-selective estrogen receptor ligands useful in the treatment of rheumatoid arthritis. J Med Chem. 2004;47:6435–8. doi: 10.1021/jm049194+. [DOI] [PubMed] [Google Scholar]
- 133.Chadwick CC, et al. Identification of pathway-selective estrogen receptor ligands that inhibit NF-kappaB transcriptional activity. Proc Natl Acad Sci U S A. 2005;102:2543–8. doi: 10.1073/pnas.0405841102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Murray IA, et al. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol Pharmacol. 2010;77:247–54. doi: 10.1124/mol.109.061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Murray IA, et al. Development of a selective modulator of aryl hydrocarbon (Ah) receptor activity that exhibits anti-inflammatory properties. Chem Res Toxicol. 2010;23:955–66. doi: 10.1021/tx100045h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Murray IA, et al. Suppression of cytokine-mediated complement factor gene expression through selective activation of the Ah receptor with 3',4'-dimethoxy-alpha-naphthoflavone. Mol Pharmacol. 2011;79:508–19. doi: 10.1124/mol.110.069369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Astroff B, et al. 6-Methyl-1,3,8-trichlorodibenzofuran as a 2,3,7,8-tetrachlorodibenzo-p-dioxin antagonist: inhibition of the induction of rat cytochrome P-450 isozymes and related monooxygenase activities. Mol Pharmacol. 1988;33:231–6. [PubMed] [Google Scholar]
- 138.Zacharewski T, et al. 6-Methyl-1,3,8-trichlorodibenzofuran (MCDF) as an antiestrogen in human and rodent cancer cell lines: evidence for the role of the Ah receptor. Toxicol Appl Pharmacol. 1992;113:311–8. doi: 10.1016/0041-008x(92)90130-k. [DOI] [PubMed] [Google Scholar]
- 139.McDougal A, Wilson C, Safe S. Inhibition of 7,12-dimethylbenz[a]anthracene-induced rat mammary tumor growth by aryl hydrocarbon receptor agonists. Cancer Lett. 1997;120:53–63. doi: 10.1016/s0304-3835(97)00299-1. [DOI] [PubMed] [Google Scholar]
- 140.Zhang S, et al. The aryl hydrocarbon receptor as a target for estrogen receptor-negative breast cancer chemotherapy. Endocr Relat Cancer. 2009;16:835–44. doi: 10.1677/ERC-09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang S, et al. Aryl hydrocarbon receptor agonists induce microRNA-335 expression and inhibit lung metastasis of estrogen receptor negative breast cancer cells. Mol Cancer Ther. 2012;11:108–18. doi: 10.1158/1535-7163.MCT-11-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Manchester DK, Gordon SK, Golas CL, Roberts EA, Okey AB. Ah receptor in human placenta: stabilization by molybdate and characterization of binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin, 3-methylcholanthrene, and benzo(a)pyrene. Cancer Res. 1987;47:4861–8. [PubMed] [Google Scholar]
- 143.Flaveny CA, Murray IA, Perdew GH. Differential gene regulation by the human and mouse aryl hydrocarbon receptor. Toxicol Sci. 2010;114:217–25. doi: 10.1093/toxsci/kfp308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Forgacs AL, Dere E, Angrish MM, Zacharewski TR. Comparative analysis of temporal and dose-dependent TCDD-elicited gene expression in human, mouse, and rat primary hepatocytes. Toxicol Sci. 2013;133:54–66. doi: 10.1093/toxsci/kft028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Black MB, et al. Cross-species comparisons of transcriptomic alterations in human and rat primary hepatocytes exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2012;127:199–215. doi: 10.1093/toxsci/kfs069. [DOI] [PubMed] [Google Scholar]
- 146.Flaveny C, Reen RK, Kusnadi A, Perdew GH. The mouse and human Ah receptor differ in recognition of LXXLL motifs. Arch Biochem Biophys. 2008;471:215–23. doi: 10.1016/j.abb.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ramadoss P, Perdew GH. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol Pharmacol. 2004;66:129–36. doi: 10.1124/mol.66.1.129. [DOI] [PubMed] [Google Scholar]
- 148.Flaveny CA, Murray IA, Chiaro CR, Perdew GH. Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol Pharmacol. 2009;75:1412–20. doi: 10.1124/mol.109.054825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Saito R, et al. Aryl hydrocarbon receptor in breast cancer-a newly defined prognostic marker. Horm Cancer. 2014;5:11–21. doi: 10.1007/s12672-013-0160-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Richmond O, et al. The aryl hydrocarbon receptor is constitutively active in advanced prostate cancer cells. PLoS One. 2014;9:e95058. doi: 10.1371/journal.pone.0095058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yin XF, Chen J, Mao W, Wang YH, Chen MH. Downregulation of aryl hydrocarbon receptor expression decreases gastric cancer cell growth and invasion. Oncol Rep. 2013;30:364–70. doi: 10.3892/or.2013.2410. [DOI] [PubMed] [Google Scholar]
- 152.Su JM, Lin P, Chang H. Prognostic value of nuclear translocation of aryl hydrocarbon receptor for non-small cell lung cancer. Anticancer Res. 2013;33:3953–61. [PubMed] [Google Scholar]
- 153.Liu Z, et al. AhR expression is increased in hepatocellular carcinoma. J Mol Histol. 2013;44:455–61. doi: 10.1007/s10735-013-9495-6. [DOI] [PubMed] [Google Scholar]
- 154.Tanaka G, et al. Induction and activation of the aryl hydrocarbon receptor by IL-4 in B cells. Int Immunol. 2005;17:797–805. doi: 10.1093/intimm/dxh260. [DOI] [PubMed] [Google Scholar]
- 155.Vogel CF, et al. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-kappaB. J Biol Chem. 2014;289:1866–75. doi: 10.1074/jbc.M113.505578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18:207–50. doi: 10.1615/critreveukargeneexpr.v18.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mellor AL, et al. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171:1652–5. doi: 10.4049/jimmunol.171.4.1652. [DOI] [PubMed] [Google Scholar]
- 158.Litzenburger UM, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget. 2014;5:1038–51. doi: 10.18632/oncotarget.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kolluri SK, Weiss C, Koff A, Gottlicher M. p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev. 1999;13:1742–53. doi: 10.1101/gad.13.13.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pang PH, et al. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J Cell Physiol. 2008;215:161–71. doi: 10.1002/jcp.21299. [DOI] [PubMed] [Google Scholar]
- 161.Puga A, et al. Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J Biol Chem. 2000;275:2943–50. doi: 10.1074/jbc.275.4.2943. [DOI] [PubMed] [Google Scholar]
- 162.Barhoover MA, Hall JM, Greenlee WF, Thomas RS. Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol Pharmacol. 2010;77:195–201. doi: 10.1124/mol.109.059675. [DOI] [PubMed] [Google Scholar]
- 163.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–34. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 164.Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem. 1976;251:4936–46. [PubMed] [Google Scholar]
- 165.Farrell K, Safe L, Safe S. Synthesis and aryl hydrocarbon receptor binding properties of radiolabeled polychlorinated dibenzofuran congeners. Arch Biochem Biophys. 1987;259:185–95. doi: 10.1016/0003-9861(87)90485-1. [DOI] [PubMed] [Google Scholar]
- 166.Jensen BA, Reddy CM, Nelson RK, Hahn ME. Developing tools for risk assessment in protected species: Relative potencies inferred from competitive binding of halogenated aromatic hydrocarbons to aryl hydrocarbon receptors from beluga (Delphinapterus leucas) and mouse. Aquat Toxicol. 2010;100:238–45. doi: 10.1016/j.aquatox.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kolasa E, Houlbert N, Balaguer P, Fardel O. AhR- and NF-kappaB-dependent induction of interleukin-6 by co-exposure to the environmental contaminant benzanthracene and the cytokine tumor necrosis factor-alpha in human mammary MCF-7 cells. Chem Biol Interact. 2013;203:391–400. doi: 10.1016/j.cbi.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 168.Gillner M, Bergman J, Cambillau C, Fernstrom B, Gustafsson JA. Interactions of indoles with specific binding sites for 2,3,7,8-tetrachlorodibenzo-p-dioxin in rat liver. Mol Pharmacol. 1985;28:357–63. [PubMed] [Google Scholar]
- 169.Jin UH, Lee SO, Safe S. Aryl hydrocarbon receptor (AHR)-active pharmaceuticals are selective AHR modulators in MDA-MB-468 and BT474 breast cancer cells. J Pharmacol Exp Ther. 2012;343:333–41. doi: 10.1124/jpet.112.195339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.O'Donnell EF, et al. The anti-inflammatory drug leflunomide is an agonist of the aryl hydrocarbon receptor. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Quattrochi LC, Tukey RH. Nuclear uptake of the Ah (dioxin) receptor in response to omeprazole: transcriptional activation of the human CYP1A1 gene. Mol Pharmacol. 1993;43:504–8. [PubMed] [Google Scholar]
- 172.Ciolino HP, Daschner PJ, Yeh GC. Dietary flavonols quercetin and kaempferol are ligands of the aryl hydrocarbon receptor that affect CYP1A1 transcription differentially. Biochem J. 1999;340:715–22. Pt 3. [PMC free article] [PubMed] [Google Scholar]
- 173.Oberg M, Bergander L, Hakansson H, Rannug U, Rannug A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci. 2005;85:935–43. doi: 10.1093/toxsci/kfi154. [DOI] [PubMed] [Google Scholar]
- 174.Savouret JF, et al. 7-ketocholesterol is an endogenous modulator for the arylhydrocarbon receptor. J Biol Chem. 2001;276:3054–9. doi: 10.1074/jbc.M005988200. [DOI] [PubMed] [Google Scholar]