

Abstract
Many different external and intrinsic apoptotic stimuli induce the accumulation in the cells of a set of proteins known as stress or heat shock proteins (HSPs). HSPs are conserved proteins present in both prokaryotes and eukaryotes. These proteins play an essential role as molecular chaperones by assisting the correct folding of nascent and stress-accumulated misfolded proteins, and by preventing their aggregation. HSPs have a protective function, that is they allow the cells to survive to otherwise lethal conditions. Various mechanisms have been proposed to account for the cytoprotective functions of HSPs. Several of these proteins have demonstrated to directly interact with components of the cell signalling pathways, for example those of the tightly regulated caspasedependent programmed cell death machinery, upstream, downstream and at the mitochondrial level. HSPs can also affect caspase-independent apoptosis-like process by interacting with apoptogenic factors such as apoptosis-inducing factor (AIF) or by acting at the lysosome level. This review will describe the different key apoptotic proteins interacting with HSPs and the consequences of these interactions in cell survival, proliferation and apoptotic processes. Our purpose will be illustrated by emerging strategies in targeting these protective proteins to treat haematological malignancies.
Keywords: heat shock proteins, apoptosis, caspases, mitochondria, lysosomes, death receptors, cell signalling, haematopoietic malignancies, emerging chemotherapeutic treatments
Introduction
Heat shock proteins (HSPs), also called stress proteins, are highly conserved proteins whose expression is induced by different kinds of stresses [1]. HSPs have strong cytoprotective effects and behave as molecular chaperones for other cellular proteins. Inappropriate activation of signalling pathways occurs during acute or chronic stress as a result of protein misfolding, protein aggregation or disruption of regulatory complexes. The action of chaperones, through their properties in protein homeostasis, is thought to restore the balance. Mammalian HSPs have been classified in two groups according to their size: high molecular weight HSPs and small molecular weight HSPs. The first group includes three major families: HSP90, HSP70 and HSP60. Some of them are expressed constitutively; whereas, expression of the others is induced by stressful conditions. These proteins can be targeted to different subcellular compartments. High molecular weight HSPs are adenosine-5′-triphosphate (ATP)-dependent chaperones and require co-chaperones to modulate their conformation and ATP binding. In contrast, small HSPs, such as HSP27, are ATP-independent chaperones [2]. Among the different HSPs, HSP27 and HSP70 are the most strongly induced after stresses such as anticancer drugs, oxidative stress or irradiation. While hardly expressed in non-transformed cells, both HSP27 and HSP70 are abundantly expressed in cancer cells and therefore have been suggested as important prognostic factors in malignant diseases [1].
Apoptosis is a programmed cell death induced by stimuli-like growth factor withdrawal, hypoxia, DNA damage or cytotoxic drugs. Interestingly, these same stimuli induce the expression and accumulation of HSPs, demonstrating that a death stimulus can elicit a protective response in the cell. Many data accumulated in the last 10 years clearly demonstrated that HSPs have essential anti-apoptotic properties. HSPs associate with key stress signalling and apoptotic molecules, thereby blocking cell death and promoting survival, proliferation or differentiation. In this review, we will discuss the role of the main HSPs (HSP27, HSP60, HSP70, HSP90α and HSP90β) in the different apoptotic pathways. As defaults in apoptosis and cell proliferation are at the basis of tumour development, the pathophysiological functions of HSPs in haematological malignancies will be developed as an example.
Main HSPs
Small HSPs are a group of proteins that vary in size from 15 to 30 kD and share sequence homologies and biochemical properties such as phosphorylation and oligomerization. HSP27, probably the most studied member of this family, is an ATP-independent chaperone whose main function is protection against protein aggregation [2]. HSP27 can form oligomers up to 1000 kD. The HSP27 dimer might be the building block for the multimeric complexes. HSP27 oligomerization is a dynamic process that depends on the phosphorylation status of the protein and exposure to stress [3]. Human HSP27 can be phosphorylated at three serine residues and its dephosphorylation enhances oligomerization. This phosphorylation is a reversible process catalysed by the MAPKAP kinases 2 and 3 in response to a variety of stresses including differentiating agents, mitogens, inflammatory cytokines such as tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), hydrogen peroxide and other oxidants. HSP27 is expressed in many cell types and tissues, at specific stages of development and differentiation [3]. It is a late inducible HSP that accumulates in the cells after several different stresses such as oxidative stress, serum deprivation, anticancer drugs or radiation. In cancer cells, the basal expression of HSP27 is abnormally high and associates with cell resistance to anticancer therapy.
Mammalian HSP60, also called chaperonin, is mostly contained within the mitochondrial matrix, although it can also be detected in extra-mitochondr-ial sites including the cytosol [4, 5]. HSP60 is a con-stitutively expressed HSP, although its expression can slightly increase after certain stresses, mainly heat [6]. HSP60 participates in the folding of mitochondrial proteins and facilitates the proteolytic degradation of misfolded or denatured proteins in an ATP-dependent manner. The chaperone function of HSP60 is regulated by HSP10, which binds to HSP60 and regulates its substrate binding and ATPase activity. In the presence of ADP, two HSP10 molecules bind to one HSP60 molecule. HSP60 and HSP10 do not always act as a single functional unit: only newly mitochondria imported proteins are severely affected by inactivation of HSP10 [7].
The HSP70 family constitutes the most conserved and best-studied class of HSPs. It encompasses proteins ranging from 66 to 78 kD that are encoded by a multi-gene family consisting of at least 11 genes in humans. Some have mainly a cytosolic localization like the major inducible HSP70 (called simply HSP70 or HSP72) or the constitutively expressed HSC70, one is located into the mitochondria (mtHSP70) and one in the endoplasmic reticulum (GRP78/Bip) [8]. Eukaryotic HSP70s contain two functional domains: the NH2-terminal ATP-binding domain (ABD) and the COOH-terminal peptide-binding domain (PBD). Under normal conditions, HSP70 proteins function as ATP-dependent molecular chaperones by assisting the folding of newly synthesized polypeptides, the assembly of multi-protein complexes and the transport of proteins across cellular membranes [9]. Proteins like HSP40, CHIP, HOP, HIP, BAG-1 and BAG-3 are HSP70 co-chaperones. They can bind either the PBD or the ABD of HSP70, thereby modulating the chaperone activity of the protein. A range of stimuli, including anticancer agents, rapidly induces the synthesis of stress-inducible HSP70. Gene ablation studies show that inducible HSP70 plays an important role in apoptosis. Mouse embryonic cells lacking the two genes that encode for inducible HSP70, hsp70.1 and hsp70.3, are very sensitive to apoptosis induced by a wide range of lethal stimuli [10]. Further, disruption of the testis specific isoform of HSP70 (hsp70.2) results in germ cell apoptosis [11].
Prominent members of the HSP90 family of proteins are HSP90α and HSP90β[12]. These two HSP90 isoforms are essential for the viability of eukaryotic cells. They are constitutively abundant, make up 1–2% of cytosolic proteins, and can be further stimulated in their expression level by stress. HSP90 associates with a number of signalling proteins including ligand-dependent transcription factors, such as steroid receptor [13], ligand-independent transcription factors, such as MyoD [14], tyrosine kinases, such as v-Src [15], and serine/threonine kinases, such as Raf-1 [16]. The stability of these HSP90-binding proteins, called HSP90 client proteins, is ensured by HSP90. The inhibition of the chaperone induces their degradation by the protea-some. HSP90 binds ATP and undergoes a conformational change upon ATP binding that is needed for its chaperone function. Co-chaperones of HSP90 include Cdc37, p23, Aha1, PP5, HOP and CHIP.
HSPs, cell signalling and apoptosis
Apoptosis, or programmed cell death, is a type of death essential during embryogenesis and, latter on in the organism, to assure cell homeostasis. Apoptosis is also a very frequent type of cell death observed after treatment with cytotoxic drugs [17]. Two pathways characterize apoptotic processes, both mediated by a family of cysteine proteases known as caspases: the intrinsic or mitochondrial pathway and the extrinsic or death receptors path-way. The two signal-transducing cascades converge at the level of capase-3, an effector caspase that leads to the typical morphologic and biochemical changes of the apoptotic cell.
The intrinsic pathway involves the production or activation of pro-apoptotic molecules upon intracellular stress signals. These molecules converge on the mitochondria to trigger the release of mitochondrial apoptogenic molecules under control of the Bcl-2 (B-cell lymphocytic-leukaemia proto-oncogene) family of proteins. Bcl-2 proteins include anti-apoptotic members such as Bcl-2 and Bcl-xL, multi-domain pro-apoptotic members mainly Bax and Bak [18, 19] and a series of BH3 domain-only pro-apoptotic proteins, such as Bid [20], that function upstream of Bax and Bak [21]. One of the released mitochondrial molecule is cytochrome c, which interacts with cytosolic apoptotic protease activation factor-1 (Apaf-1) and pro-caspase-9 to form the apoptosome, the caspase-3 activation complex [22]. Two other mitochondrial proteins, Smac/Diablo and Htra2/Omi, activate apoptosis by neutralizing the inhibitory activity of the inhibitory apoptotic proteins (IAPs) that associate with and inhibit some of the activated caspases [23].
The extrinsic pathway is triggered through plasma membrane proteins of the TNF receptor family known as death receptors and leads to the direct activation of the receptor-proximal caspase-8 or caspase-10 in the death-inducing signalling complex (DISC). Caspase-8 either directly activates the downstream cascade of caspases or cleaves Bid into an active truncated form named tBid that connects the extrinsic to the intrinsic apoptotic pathways through mitochondria permeabilization [24].
HSPs have been shown to block apoptosis by interfering with caspase activation. Overexpression of either HSP27, HSP70, HSP60 or HSP90, inhibits apoptosis and prevents caspase activation in many different cellular models upon a variety of cellular stresses, including accumulation of misfolded proteins, reactive oxygen species (ROS) or DNA damage [1, 25, 26, 27]. On the contrary, depletion of HSP27, HSP60, HSP70 or HSP90, either by antisense constructions or siRNA strategies, increases the cells'sensitivity to apoptotic stimuli [28, 29–31]. In some cellular contexts, HSP70 depletion is sufficient to trigger apoptosis through caspase-3 activation, in the absence of any additional stressful stimulus [32, 33]. Therefore, HSPs are involved, either directly or indirectly, in the modulation of caspase activities. HSPs can block both the intrinsic and the extrinsic apoptotic pathways through the interaction with key proteins at three levels: (i) upstream the mitochondria, thereby modulating signalling pathways;(ii) at the mitochondrial level, controlling the release of apoptogenic molecules (iii) and at the post-mitochondrial level, by blocking apoptosis from a later phase than any known survival enhancing drug or protein.
HSPs’ targets in upstream signalling pathways
Growth factors, such as nerve growth factor or platelet-derived growth factor, induce cell survival by activating the phosphatidylinositol 3-kinase pathway (PI3-K). Activated PI3-K phosphorylates inositol lipids in the plasma membrane that attract the serine/threonine kinase Akt/PKB, a protein that generates a survival signal in response to growth factor stimulation. Akt targets multiple proteins of the apoptotic machinery [34, 35], including Bad [36] and caspase-9 [37]. HSP27 has been described to have prosurvival effects through its interaction with the protein kinase Akt, an association that is necessary for Akt activation in stressed cells (Fig. 1A). In turn, Akt can phosphorylate HSP27, thus leading to the disruption of HSP27-Akt complexes [38]. HSP27 can also bind to F-actin and prevent the disruption of the cytoskeleton resulting either from heat shock, cytochalasin D or from other stresses. This has been reported to affect mitochondria membrane structure and thereby the release of mitochondrial apoptogenic molecules such as cytochrome c [39]. Recently, we have demonstrated that HSP27 protection to apoptosis induced by etoposide or TNF-α in different cancer cell lines can result from an increase in the activity of the survival transcription factor nuclear factor-κB (NF-κB). NF-κB is involved in the expression of several anti-apoptotic proteins, such as Bcl-2, Bcl-xL and c-IAPs. HSP27 effect on NF-κB results from an increase in the ubiquitination and proteasomal degradation of the NF-κB inhibitor I-κBα[40] (Fig. 1B). HSP27 also increases the ubiquitination and proteasomal degradation of the cell cycle protein p27kip1 under stress conditions. As a consequence, cell proliferation, once the stress conditions are over, is facilitated [41] (Fig. 1B).
1.
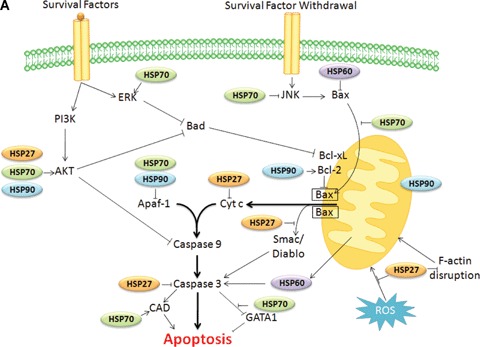
Schematic representation of HSPs regulatory function in the intrinsic (A), extrinsic and caspase independent pathways to death (B). (A) HSPs can block the mitochondrial intrinsic pathway of apoptosis by interacting with key proteins at three levels: (i) upstream the mitochondria, thereby modulating signalling pathways (HSP70 modulates the activation of stress-activated kinases such as Akt, JNK or ERK); (ii) at the mitochondrial level, controlling the release of cytochrome c (HSP27 by its interaction with the actin, HSP70 or HSP60 with Bax, and HSP90 with Bcl2) and (iii) at the post-mitochondrial level, by blocking apoptosis by their interaction with cytochrome c (HSP27), Apaf-1 (HSP70 or HSP90) or caspase-3 (HSP27). (B) At the death receptors level, HSP70 and HSP90 can interact with RIP favoring cell survival. HSP70 (through TRAIL and FADD recruitment) and HSP90 (through FLIP) block caspase 8 activation. HSP90 and HSP70 inhibit Bid cleavage while HSP27 affects Bid mitochondrial re-distribution. At the caspase-independent pathways level, HSP70 neutralizes AIF and inhibits cathepsines release from lysosomes. AKS1 pathway is affected directly by HSP70 or through Daxx by HSP27. Inhibition: −|, activation: →.
HSP70 also inhibits stress-activated kinases, such as apoptosis signal-regulating kinase 1 (Ask1), for example HSP70 down-regulation facilitates H2O2-induced Ask1 activation and subsequent apoptosis in NIH3T3 cells [42] (Fig. 1B). HSP70 also binds to c-Jun N-terminal kinase (JNK), which does not require its ATPase domain [26, 43] and prevents its activation [44] (Fig. 1A). For example, MEF hsp70.1−/− resist to JNK-mediated apoptosis induced by hyperosmolarity [45]; whereas, AEG3482, a promising novel compound, inhibits JNK activity through increased expression of HSP70 [46]. HSP70 also negatively interferes with p38 kinase activity [47].
HSP70 binds to non-phosphorylated protein kinase C (PKC) via the kinase's unphosphorylated carboxyl-terminus, priming the kinase for re-phos-phorylation and stabilizing the protein [48]. HSP70 also binds and stabilizes protein kinase B or Akt [48] (Fig. 1A). Interestingly, the endothelial-specific HSPA12B, a distant member of HSP70 family that is required for zebrafish vasculature development, is involved in endothelial cell migration and tube formation through sustaining Akt activity [49]. Thus, HSP70 family members could play a role both in the control of cell survival and differentiation.
HSP70 has also been shown to affect some transcription factors involved in the expression of Bcl-2 proteins. Bcl-2 and Bax are transcriptional targets of the tumour suppressor protein p53: the transcription of Bcl-2 is repressed by p53 whereas that of Bax is induced. As a consequence, p53 expression induces apoptosis in response to DNA damage. Many tumour cells have a mutated p53 and either HSP70 or HSC70 could form stable complexes with this mutant protein. HSP70 could also mask the nuclear localization sequence (NLS) of p53, thus preventing its nuclear import [50, 51].
The role of HSP70 in regulating NF-κB function is more controversial. Cytosolic HSP70 could inhibit NF-κB while plasma membrane-associated HSP70 could activate this transcription factor [52, 53], but both cytosolic and membrane-associated HSP70 usually accumulate together upon stressful stimuli [54]. Anyway, in endothelial cells, elevation of HSP70 to a significant level favours TNF-α-mediated apoptosis via inhibition of the NF-κB survival pathway [55].
HSP70 could block NF-κB activation through inhibition of both I-κBα Kinase (IKK) activation and subsequent degradation of I-κBα[56, 57]. This effect could mediate a function of HSP70 in promoting elimination of cells with damaged DNA. Inhibitors of growth (ING) proteins, which are tumour suppressors whose expression is down-regulated in a variety of human cancers, transduce stress signals after DNA damage and bind histones, thus regulating chromatin structure and p53 activity. These proteins induce HSP70 that in turn promotes TNF-α receptor-mediated apoptosis by binding I-κBα Kinase (IKK) and impairing NF-κB survival signalling [58] (Fig. 1B).
HSP90 regulates the activity and stability of many transcription factors and kinases implicated in apoptosis such as NF-κB, p53, Akt, Raf-1 and JNK [59]. HSP90 can affect NF-κB survival activity via the IKK complex, which is composed of two catalytic and one regulatory subunits (Fig. 1B). HSP90 and Cdc37 are also present, association mediated through the kinase domain of the catalytic subunits [60]. HSP90 associates to mutated p53 and stabilizes the protein. In chronic leukaemia cells, inhibition of HSP90 by geldanamycin down-regulates mutated p53 protein while up-regulating wild-type p53 [61]. On the other hand, p53 could repress HSP90α gene expression in UV irradiated cells [62]. Other transcription factors whose function could be modulated by HSP90 include Her2 and HIF1α[63, 64].
HSP90 interacts with and stabilizes phosphorylated Akt (Fig. 1A). In turn, phosphorylated Akt can phosphorylate the pro-apoptotic Bcl-2 family protein Bad and the caspase-9 [37], leading to their inactivation and to cell survival. Akt has also been shown to phosphorylate the I-κB kinase, which results in promotion of NF-κB-mediated cell survival [35]. HSP90 also interacts with Raf-1 and the dissociation of this complex results in apoptosis in mast cells [65] and in B-lymphocytes [66]. JNK is another client protein of HSP90. Finally, several cytokines, such as IL-6 and type I and II interferons, could up-regulate HSP90 expression, leading to a survival loop. IL-6 transgenic mice exhibit a transcriptional up-regulation of HSP90 mRNA. The transcription factors STAT3 and CCAAT/enhancer-binding protein β (C/EBPβ), which are under the control of the IL-6 receptor and the MAPK pathway, co-operatively bind and activate the HSP90β promoter [67, 68].
HSPs’ targets atthe mitochondrial level
HSP27, in L929 murine fibrosarcoma cells exposed to cytochalasin D or staurosporine, prevents the cytoskeletal disruption and Bid re-distribution to the mitochondria that precedes cytochrome c release [39]. HSP27 has also been shown to inhibit the mitochondrial release of Smac and thereby to confer resistance of multiple myeloma cells to dexamethasone [69] (Fig. 1A).
HSP70, coupled to HSP40, blocks Bax translocation preventing mitochondrial outer membrane permeabilization and thereby inhibiting the release of cytochrome c and that of other mitochondrial apoptogenic molecules such as apoptosis inducing factor (AIF) [70]. This HSP70 function depends on both its chaperone and its ATP hydrolytic domains [71] (Fig. 1A).
Cytosolic HSP60 could form a complex with the pro-apoptotic protein Bax [72]. Under hypoxic conditions HSP60 and Bax dissociate, whereupon Bax translocates to the mitochondria and induces apoptosis. The interaction of HSP60 and Bax may therefore prevent apoptosis. Accordingly, recent studies show that reducing HSP60 expression with antisense oligonucleotides in cardiomyocytes correlated with an increase in Bax and a reduction in Bcl-2 and resulted in induction of apoptosis [73, 74]. In neuroblastoma cells, mitochondrial HSP60 has been shown to protect mitochondrial normal oxidative phosphorylation functions by selectively acting at the complex IV activity level, thereby preserving ATP generation and decreasing cytochrome c release [75] (Fig. 1A).
In tumour cells, HSP90 is present in the mitochondria in combination with its related molecule TRAP-1. This mitochondria-located HSP90 regulates mitochondrial membrane permeabilization and cytochrome c release. Its inhibition by the peptidomimetic Shepherdin (which contains a cell-penetrating sequence) provokes a depolarisation of the mitochondrial membrane and a dose-dependent release of cytochrome c. This might have important consequences in cancer therapy since HSP90 chaperone was identified in tumour cells’ mitochondria, not in mitochondria from normal tissues [76]. Some reports describe the formation of a Bcl-2/HSP90β complex that could prevent the release of cytochrome c from mitochondria and caspase-3 activation in mast cells [77]. This effect was recently confirmed in monocytes/macrophages and dendritic cells treated with an unmethylated CpG motif of bacterial DNA and radicicol can inhibit this association [78] (Fig. 1A).
HSPs’ targets at the post-mitochondrial level
We, and other groups, have demonstrated that HSP27 can prevent the activation of caspases by directly sequestering cytochrome c when released from the mitochondria into the cytosol [79, 80] (Fig. 1A). The heme group of cytochrome c is necessary but not sufficient for this interaction that involves amino acids 51 and 141 of HSP27 and requires dimerization of the stress protein. In human monocytes undergoing spontaneous apoptosis, HSP27 has been shown to associate to and block active caspase-3 [81]. HSP27 can also increase the antioxidant defence of cells by decreasing ROS cell content [82] and neutralizes the toxic effects of oxidized proteins [83]. This latter effect may occur more specifically in neuronal cells and involves phosphorylated HSP27 [84]. HSP27 may also affect membrane blebs formation, one of the morphological changes that take place on the late phase of apoptosis. The actin–myosin system has been proposed as a source of contractile force for bleb formation [85] and HSP27 is a cap-binding protein that plays a role in F-actin reorganization [86] (Fig. 1A).
Li et al.[87] found HSP70-inhibited apoptosis downstream of the release of cytochrome c and upstream of the activation of caspase-3. Indeed, HSP70 has been demonstrated to directly bind to Apaf-1, thereby preventing the recruitment of procas-pase-9 to the apoptosome [88]. The ATPase domain of HSP70 was described to be necessary for this interaction [89]. In TNF-induced apoptosis, HSP70 does not preclude the activation of caspase-3 but prevents downstream morphological changes that are characteristic of dying cells like activation of phospholipase A2 and changes in nuclear morphology [8]. During the final phases of apoptosis, chromosomal DNA is digested by the DNAse CAD (Caspase Activated DNAse) following activation by caspase-3. The enzymatic activity and proper folding of CAD has been reported to be regulated by HSP70, its co-chaperone HSP40 and ICAD, the inhibitor of CAD. ICAD recognizes an intermediate folding state conferred by HSP70-HSP40 [90]. It has also been reported in T cell receptor (TCR)-stimulated T cells that HSP70 binds CAD and enhances its activity [91]. Another final target of caspase-3 is the transcription factor GATA-1. We have demonstrated in human primary erythroid precursors that HSP70 can protect GATA-1 from caspase-3 cleavage. As a consequence, erythroid cells do not die by apoptosis but instead differentiate [92] (Fig. 1A).
HSP90 could negatively affect Apaf-1 function in apoptosis. HSP90 directly binds Apaf-1 and inhibits its oligomerization and further recruitment of procas-pase-9 [93]. Vimentin, a major component of intermediate filament, is degraded in response to apoptotic inducers. The cleavage of vimentin by caspases probably results in disrupting of its filamentous structure which may facilitate nuclear condensation and subsequent fragmentation. HSP90 binds to vimentin and protects it from apoptotic cleavage [94] (Fig. 1A).
A pro-apoptotic role for HSP60 and HSP10 has been demonstrated by two independent groups. In both HeLa and Jurkat cells, activation of caspase-3 by camptothecin or staurosporine was observed to occur simultaneously with HSP60 and HSP10 release from the mitochondria (Fig. 1). The authors demonstrated in vivo and in vitro that HSP60 and HSP10 associated to procaspase-3 and favoured its activation by cytochrome c in an ATP-dependent manner, suggesting that the chaperone function of HSP60 was involved in this process [95, 96] (Fig. 1A).
HSPs and the extrinsic death receptor pathway
HSP27 inhibits Fas-induced apoptosis [97]. The phosphorylated form of HSP27 directly interacts with Daxx. This latter protein connects Fas signalling to the protein kinase Ask1 that mediates a caspase-independent cell death [98] (Fig. 1B).
HSP70 inhibits TNF-α-induced cell death and this protective effect is lost in Bid homozygous-deleted mouse embryonic fibroblast (MEF) cells [99]. HSP70 can block the cleavage of Bid by activated caspase-8 [99]. Exposure of haematopoietic cells to TNF-α induces the activity of the pro-apoptotic double-stranded RNA-dependent protein kinase (PKR). An inhibitor of PKR is the Fanconi anaemia complemen-tation group C gene product (FANCC). HSP70 interacts with the FANCC protein via its ATPase domain and, together with HSP40, inhibits TNF-induced apoptosis through the ternary complex HSP70, FANCC and PKR [100, 101].
In human chronic myeloid leukaemia (CML) cells, HSP70 can mediate Bcr-Abl-induced resistance to TNFα-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by preventing the formation of DISCss involving DR4 and DR5 [102]. The role of HSP70 in Fas-mediated apoptsis is more controversial with opposite effects that depend on the cell context [103, 104] (Fig. 1B).
A key regulator in apoptosis induced by TRAIL is FLIP. It has been shown in glioma cells that HSP90α associates with FLIP(S) in a manner dependent on the ATP-binding NH2-terminal domain of the chaperone. Following TRAIL exposure, HSP90α and its client protein FLIP(S) are recruited to the DISC. HSP90α depletion blocked the recruitment of FLIP(S) to the DISC and thereby sensitized resistant glioma cells to TRAIL-induced apoptosis [105]. HSP90 has also been shown to interact with and stabilize the Receptor Interacting protein (RIP). Upon ligation of tumor necrosis factor receptor (TNFR)-1, receptor interacting protein (RIP)-1 is recruited to the receptor and promotes the activation of NF-κB and JNK. Degradation of RIP-1 in the absence of HSP90 precludes activation of NF-κB mediated by TNF-α and sensitizes cells to apoptosis [106]. Finally, in NIH3T3 fibroblasts, HSP90 has been shown to suppress TNF-α-induced apoptosis by directly associating to Bid, thereby preventing its cleavage [107] (Fig. 1B).
HSPs and alternatives, caspase-independent, apoptosis-like pathways
Upon activation of the intrinsic pathway not only cytochrome c is released from the mitochondria. AIF and endonuclease G (EndoG) are also released upon an apoptotic stimulus. These two mitochondria intermembrane proteins translocate to the nucleus and triggers caspase-independent nuclear changes [108].
HSP70 has been shown to prevent cell death in conditions in which caspase activation does not occur, due to the addition of exogenous caspase inhibitors [109], or in cells in which Apaf-1 or caspase-9 were genetically inactivated [110], indicating that the cytochrome c/Apaf-1/caspase was not the sole pathway of the anti-apoptotic action of HSP70. Indeed, HSP70 directly binds to AIF and inhibits AIF-induced chromatin condensation. HSP70 neutralizes the apoptogenic effects of AIF in cell-free systems, in intact cells microinjected with recombinant HSP70 and/or AIF protein, as well as in cells transiently transfected with AIF cDNA. Of note, endogenous levels of HSP70 seem to be sufficiently high to control AIF-mediated apoptosis since down-regulation of HSP70 by an antisense construct sensitized the cells to serum withdrawal and AIF [110]. This protective effect of HSP70 might be physiologically relevant as AIF sequestration by HSP70 reduces neonatal hypoxic/ischaemic brain injury [111]. In addition, HSP70 inhibits erythroblast apoptotis by blocking AIF nuclear import [112]. HSP70 associates also with EndoG to prevent DNA fragmentation [113], but this association could involve AIF as a molecular bridge (Fig. 1B).
Lysosomes also functions as integrators of cell death signals in many different cell death scenarios [114]. Lysosomal proteases, of which the most studied are the cathepsins, translocate from the lysosomal lumen to the cytosol in response to a wide variety of apoptotic stimuli, such as TNF-α[115, 116], Fas [117], microtubule stabilizing agents [118], stau-rosporine [119], p53 activation [120], oxidative stress [117, 121], growth factor deprivation [117] and lyso-somotropic agents [121, 122]. Upon release to the cytosol, cathepsins can trigger mitochondrial outer membrane permeabilization [115, 119, 120, 122, 123]. Cathepsins release can also ensue an apoptotic morphology that is independent of the mitochondria release of cytochrome c or AIF [116, 124]. HSP70 is found in the endolysosomal membranes of many tumours and stressed cells where it inhibits the release of lysosomal cathepsins into the cytosol [125, 126]. Lysosomes positive for HSP70 display an increased size and resistance against chemical and physical membrane destabilization [125] (Fig. 1B).
HSP90β is involved in signalling prolactin-induced apoptosis during spermatogenesis. Prolactin receptor is a client protein of HSP90β and its inhibition promotes spermatogonial apoptosis [127] by a still unknown mechanism.
What directs the interaction of a HSP with a given apoptotic partner?
HSPs are strong cytoprotective proteins that can associate to multiple apoptotic partners and thereby can block apoptosis at different levels. The cellular survival needs, which depend on the experimental conditions (i.e. apoptotic stimulus and experimental model), might dictate which are the apoptotic targets that must be inactivated by the chaperones. HSPs post-translational modifications and/or their presence in a given cellular compartment are important variables in determining the interaction of a HSP with a given apoptotic partner. In this way HSP27 seems to modulate its different protective properties by changing its oligomerization pattern, which is regulated by the phosphorylation status of the protein. We have demonstrated in vitro and in vivo that HSP27-mediated inhibition of caspasedependent apoptosis involved large non-phosphorylated oligomers of HSP27 [128]. In contrast, the phospho-rylated form of the protein directly interacts with Daxx [98]. These results suggest that the oligomerization/phosphorylation of the protein alters HSP27 conformation and hence determine its capacity to interact with different apoptotic proteins. The essential HSP27 anti-apoptotic function seems to take place in the cytosol [79]. Accordingly, leptomycin B, which is an inhibitor of the nuclear export of proteins, triggers nuclear accumulation of HSP27, which prevents its protective functions [129].
In contrast to HSP27, little is known about HSP70 post-translational modifications. HSP70 is one of the most powerful anti-apoptotic proteins since it blocks almost all identified cell death pathways. Interaction of HSP70 with a given partner may be determined by HSP70 co-chaperones and its cellular localization. For example, the presence of HSP70 in the lysosomal membrane is necessary for inhibiting the lysosomal pathway to death. During red blood cell formation, HSP70 migrates into the nucleus where it interacts with the transcription factor GATA-1 to prevent its cleavage by caspase-3, whose activation is required for the erythroid differentiation process. Thereby, the migration of HSP70 in the nucleus is essential to determine whether an erythroblast is going to die by apoptosis or to differentiate into a red blood cell [92]. In tumour cells, HSP70 expression at the plasma membrane facilitates their clearance by the immune system. Membranary HSP70 facilitates granzyme B penetration in a perforin-independent way and ensures cytotoxic T lymphocytes killing through FasL/Fas-mediated contact [54, 130].
HSP90 anti-apoptotic function can be explained because some of its client proteins, to whom HSP90 assure their stability, are anti-apoptotic proteins. The first explanation of why HSP90 associates to a given partner can be found in the different isoforms of HSP90, the α and β that most studies unfortunately do not differentiate. Evidence accumulates that these isofoms may have different functions in the cell. For instance, HSP90β isoform has a function in cell differentiation that the α isoform does not have [131]. The α and β isoforms might also have a different function in the regulation of apoptosis. Post-translational modifications of HSP90 could also account for the differential affinity of the chaperone for its multiple partners. HSP90α can be acetylated on its lysine 294, which inhibits its binding to several client proteins. HSP90β can be phosphorylated on Ser 254, which favours tumour regression and tumour immunogenicity [132]. However, how the acetylation or phosphorylation of HSP90 isoforms affect their interaction with different protein partners, and how these events affect their anti-apoptotic functions remain to be determined. HSP90 is the only chaperone targeted in cancer therapy to sensitize tumour cells to chemotherapy-induced death. Geldanamycin derivatives [133] bind the ATP pocket of HSP90 to inhibit their chaperone function. In turn, HSP90 ‘client’ proteins are not anymore stabilized by HSP90 and are degraded by the proteasome. These inhibitors equally block HSP90 α and β isoforms which precludes their experimental use to determine the relative contribution of each of them in apoptosis control.
HSP60 role in apoptosis is rather contradictory. Overexpression of HSP60 and/or HSP10 increases the survival rate of cardiac myocytes undergoing ischaemia/reperfusion injury [134]; whereas, reduction in HSP60 level is sufficient to precipitate apoptosis in myocytes [73]. Nevertheless, expression of HSP60 was identified as a good prognostic indicator in human oesophageal squamous cell carcinoma, reflecting the ambiguous role of this chaperone in tumour cell death process [135]. HSP60 can be pro-apoptotic by facilitating caspase-3 activation or anti-apoptotic through inhibiting Bax. These controversial results might be re-counciled on the basis on HSP60 localization and experimental variables. Cytosolic HSP60 might have a protective function whereas mitochondrial HSP60, released to the cytosol upon an apoptotic stimulus (in other words, in cells already committed to apoptosis and that can not be anymore recuperated), facilitates cell death. Thus, interaction of cytosolic HSP60 with active caspase-3 could dictate its effect on cell death [136]. HSP60 can also be found in the plasma membrane where its expression correlates with increased apoptosis in adult cardiac myocytes [137]. These opposite HSP60 chaperone functions and/or its cellular localization could be regulated by the phosphorylation/dephosphorylation of the protein [5].
Role of HSPs in haematological malignancies
In contrast to normal, non-transformed, cells and tissues where the expression of stress-inducible HSPs is, at the basal level, rather low or non-existent, HSPs are abundantly expressed in most cancer cells, in particular in several haematological malignancies, including lymphoid diseases and chronic or acute myeloid leukaemias. Overexpression of HSP27, HSP70 and HSP90 correlates with a poor prognosis in acute myeloid leukaemias and myelodysplasic syndromes [138, 139]. In acute myeloid leukaemia cells, HSP27 prevents drug-induced apoptosis [140]. In multiple myeloma cells, down-regulation of HSP27 can restore the apoptotic response to dexamethasone through activation of the intrinsic, caspasedependent pathway [69].
Importantly, up-regulated HSP70 or HSP90 could stimulate cell proliferation through the control of tyrosine kinase functions. In CML cells, Bcr-Abl tyrosine kinase activity results in the phosphorylation and activation of Akt, which inactivates Bad, caspase-9 and FOXO3, and in the phosphorylation and DNA-binding activity of STAT5, which results in increased expression of the anti-apoptotic protein Bcl-xL. Increased expression of HSP70 results in up-regulation of STAT5 level and activity. Inhibition of the Bcr-Abl tyrosine kinase activity with imatinib or inhibition of PI-3K activity with wortmannin, both result in a decreased HSP70 expression and a down-regulation of STAT5 activity [102]. Thus, uncontrolled cell signalling may result in the transcriptional up-regulation of HSP70.
Targeting HSP90 also provides evidence for the role of HSP90 in the control of haematopoietic malignant cell apoptosis, survival and proliferation. In mantle cell lymphomas, the HSP90 inhibitor, 17-allylamino-demethoxygeldanamycin (17-AAG), induces cell cycle arrest and apoptosis by activating caspase-9 [141]. In HTLV1-associated adult T cell leukaemias, HSP90 inhibition abrogates cell cycle progression and induces apoptosis through inhibition of the NF-κB pathway [142]. In malignant mast cells, 17-AAG inhibits the constitutive activity of the receptor tyrosine kinase c-kit and downstream signalling molecules Akt and STAT3 [143]. In acute myeloid leukaemia cell lines, 17-AAG reduces the level of the constitutively activated tyrosine kinase FLT-3 with internal tandem repeat mutation and downstream STAT5 activity [144]. HSP90 also functions as a chaperone for ZAP-70 (Zeta-associated of 70 kD), a protein that is abnormally expressed in patients with aggressive chronic lymphocytic leukaemia (CLL), and its inhibition leads to ZAP-70 degradation and leukaemic cell death [145, 146].
Recently, a novel HSP90 inhibitor, IPI-504, was tested in mice with Bcr-Abl leukaemias that resist to the tyrosine kinase inhibitor imatinib because of a T315I mutation in the fusion gene. The combination of IPI-405 and imatinib increases mice survival and appears to eliminate leukaemic stem cells [147]. A combination of 17-AAG and cytarabine also renders acute myeloid leukaemia cells more sensitive to death [148].
In multiple myeloma, HSP90 demonstrated dual functions. First, inhibition of Hsp90α and Hsp90β with siRNA or 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), an analogue of 17-AAG, both induce apoptosis in IL6-independent and dependent myeloma cell lines as well as in primary CD138+ multiple myeloma cells co-cultured with their bone marrow microenvironment.17-DMAG attenuates the level of STAT3 and phospho-extracellular signal regulated kinase (ERK), which inhibits cell survival [149]. Secondly, the anti-leukaemic activity of bortezomib, a proteasome inhibitor, in multiple myeloma was shown to involve the exposure of HSP90 at the cell surface of dead cells, which facilitates contacts with dendritic cells and stimulates the antitumour immune response. In such a situation, the combined use of bortezomib and a HSP90 inhibitor abrogates myeloma cell immunogenicity and could be deleterious [150].
The expression level of HSPs may not be the only parameter that modulates their oncogenic function. Histone deacetylase 6 (HDAC6) controls HSP90 deacetylation [151] and inhibitors of HDAC (HDACi) capable of inhibiting HDAC6 induce the hyperacetylation of HSP90. In turn, proteins such as Akt, Bcr-Abl or Flt-3 could be degraded by the proteasome in acute and CML cells [152, 153]. For example, siRNA-mediated knockdown of HDAC6 in Bcr-Abl-positive, K562 leukaemic cells results in HSP90 hyperacetylation, loss of its chaperone function and degradation of Bcr-Abl, Akt and c-Raf. However, the effects of this strategy on tumour cell growth and survival remains limited [154], suggesting that a combination of HDAC inhibitors with specific tyrosine kinase inhibitors might be necessary. A combination of 17-AAG and HDAC6i (LBH589) is also efficient in inducing cell death in various leukaemic cell lines and primary leukaemic cells through functional inhibition and degradation of HSP90, leading to proteasomal degradation of client Bcr-Abl and/or Flt-3 [155]. In leukaemic cells in response to the antitumour drug alkyl-lysophospholipid analogue edelfosine, JNK and HSP90 are recruited in lipid rafts. Inhibition of HSP90 decreases edelfosine-induced JNK activation which is Ask1 independent [156].
HSPs inhibitors: drugs of the future?
Because of their anti-apoptotic and tumourigenic properties, HSPs are interesting targets in cancer therapy. The receptor tyrosine kinase FLT3 involved in acute myeloid leukaemias, the chimeric proteins Bcr-Abl and NPM-ALK involved in CML and anaplastic large-cell lymphomas respectively, are client proteins for HSP90 [157, 158]. HSP90 inhibitory agents, currently under clinical investigations, are therefore promising drugs in these diseases. These agents include natural products such as benzoquinone ansamycins (such as the geldamicyn derivatives 17-AAG and 17-DMAG, already in phase II clinical trials) and many emerging synthetic compounds. Interestingly, HSP90 inhibitors seem specific for cancer cells. The molecular basis for the selective antitumoural activity of the HSP90 inhibitors might be the conformation of the HSP90 complex, as HSP90 isolated from tumour cells has a 20 to 200 times higher binding affinity for the inhibitors than HSP90 isolated from normal cells [159–161].
Studies in Bcr-Abl human leukaemia cells suggest that HSP70 is also a promising therapeutic target for reversing drug resistance, probably due to its ability to inhibit death pathways both upstream and downstream of the mitochondrial signalling [102, 162]. An inhibitor of HSP70 would be very useful alone and in combination with other drugs since most of them, including the already mentioned inhibitors of HSP90, enhance HSP70 expression in malignant cells, which it is believed to reduce the drug efficacy [163]. Unfortunately, thus far, no small or soluble molecule that would selectively inhibit HSP70 is available. Some inhibitors of the heat shock response, acting at the level of the heat shock transcription factor 1 (HSF1) to block the transcription of HSPs genes, have been experimentally tested. These include the flavonoid quercetin [30] and the diterpene triepoxide, triptolide [164] but these molecules are rather unspecific. We, and others, have extensively reported that HSP70 antisense constructs demonstrated chemosensitizing properties and even killed cancer cell lines in the absence of additional stimulus [32, 165]. The cytotoxic effect of HSP70 down-modulation is particularly strong in transformed cells whereas the targeted protein is undetectable in normal cells [10]. The selective efficacy of HSP70 depletion could be related to the constitutively stressed phenotype of tumour cells, as compared to normal cells, with an enhanced dependency on the cytoprotective action of HSP70 [33, 166].
We have recently demonstrated that rationally engineered decoy targets of HSP70 derived from the protein AIF, called ADD70 (for AIF Derived Decoy for HSP70), can sensitize cancer cells to apoptosis induction by neutralizing HSP70 function. These all peptides carry the AIF region from aa 150 to aa 228, previously defined as required for HSP70 binding [167]. These constructs bind to HSP70 but lack an apoptotic function. Experiments using different malignant cell types demonstrate that some of these AIF derivatives inhibitors of HSP70 strongly increased the sensitivity of cancer cells to chemotherapy in vitro. This effect was merely related to their ability to neutralize endogenous HSP70 since this pro-apoptotic activity was lost in HSP70-negative cells [10]. In vivo, in a syngeneic rat colon cancer cell model and in a syngeneic mouse melanoma model (B16F10), ADD70 decreased the size of the tumours or at least delayed in their growth. In addition, ADD70 sensitized both the rat colon cancer cells and the mouse melanoma cells to the chemotherapeutic agent cisplatin. This ADD70 antitumourigenic effect was not observed in immunodeficient animals and were associated with an increase in tumour-infiltrating cytotoxic CD8+ T cells [163]. Therefore a positive strategy aimed at interfering with HSP70, as opposed to negative strategies based on antisense constructs or RNA interference, is feasible for chemosensitization, at least in vitro and in vivo in experimental models. Future will tell whether these peptides can be permeated (i.e. by its fusion with a membrane translocation domain) and directly injected in the tumour. That could open new perspectives in their use as local chemosensitizing agents in HSP70-expressing human tumours.
HSP27 is undoubtedly also an interesting target in cancer therapy, as HSP27 depletion experiments in cancer cells demonstrate [28, 29, 168]. However, the structural complexity of this molecule makes difficult the search for therapeutic molecules that could neutralize it [3].
Although the repercussions at the long term of HSPs inhibition deserves to be evaluated (for instance the genome stability for HSP90 [169]) or the evolution of neuronal degenerative diseases for HSP27 [84, 170], they might be relevant drugs in the future, not only in malignant diseases but also in other pathologies in which a high level of HSPs is observed. For example, our unpublished results show that HSP70 and HSP90 are abundant in the extracellular medium of animals that develop a graft versus host disease and demonstrate an immunogenic function. These HSPs could be a danger signal that increases the T cell-dependent immune response, which would open a new future for HSPs inhibitors in the setting of haematopoietic stem cell transplantation.
Acknowledgments
Our work is supported by the INSERM and grants from the ‘Ligue Nationale Contre le Cancer’(and its Nièvre committee) and ‘Conseil Règional de Bourgogne’. DL and MB are recipients of a post-doctoral fellowship from the ‘Ministère de la Education Nationale’.
References
- 1.Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- 2.Parcellier A, Gurbuxani S, Schmitt E, Solary E, Garrido C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem Biophys Res Commun. 2003;304:505–12. doi: 10.1016/s0006-291x(03)00623-5. [DOI] [PubMed] [Google Scholar]
- 3.Garrido C. Size matters: of the small HSP27 and its large oligomers. Cell Death Differ. 2002;9:483–5. doi: 10.1038/sj.cdd.4401005. [DOI] [PubMed] [Google Scholar]
- 4.Ellis RJ. Molecular chaperones: pathways and networks. Curr Biol. 1999;9:R137–9. doi: 10.1016/s0960-9822(99)80082-7. [DOI] [PubMed] [Google Scholar]
- 5.Khan IU, Wallin R, Gupta RS, Kammer GM. Protein kinase A-catalyzed phosphorylation of heat shock protein 60 chaperone regulates its attachment to histone 2B in the T lymphocyte plasma membrane. Proc Natl Acad Sci USA. 1998;95:10425–30. doi: 10.1073/pnas.95.18.10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vargas-Parada L, Solis CF, Laclette JP. Heat shock and stress response of Taenia solium and T. crassiceps (Cestoda) Parasitology. 2001;122:583–8. doi: 10.1017/s0031182001007764. [DOI] [PubMed] [Google Scholar]
- 7.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–66. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 8.Jaattela M, Wissing D, Kokholm K, Kallunki T, Egeblad M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998;17:6124–34. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y, Thomas JO. The transport of proteins into the nucleus requires the 70-kilodalton heat shock protein or its cytosolic cognate. Mol Cell Biol. 1992;12:2186–92. doi: 10.1128/mcb.12.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmitt E, Parcellier A, Gurbuxani S, Cande C, Hammann A, Morales MC, Hunt CR, Dix DJ, Kroemer RT, Giordanetto F, Jaattela M, Penninger JM, Pance A, Kroemer G, Garrido C. Chemosensitization by a non-apoptogenic heat shock protein 70-binding apoptosis-inducing factor mutant. Cancer Res. 2003;63:8233–40. [PubMed] [Google Scholar]
- 11.Dix DJ, Allen JW, Collins BW, Mori C, Nakamura N, Poorman-Allen P, Goulding EH, Eddy EM. Targeted gene disruption of Hsp70–2 results in failed meiosis, germ cell apoptosis, and male infertility. Proc Natl Acad Sci USA. 1996;93:3264–8. doi: 10.1073/pnas.93.8.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004;562:11–5. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 13.Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–25. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaknovich R, Shue G, Kohtz DS. Conformational activation of a basic helix-loop-helix protein (MyoD1) by the C-terminal region of murine HSP90 (HSP84) Mol Cell Biol. 1992;12:5059–68. doi: 10.1128/mcb.12.11.5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartson SD, Matts RL. Association of Hsp90 with cellular Src-family kinases in a cell-free system correlates with altered kinase structure and function. Biochemistry. 1994;33:8912–20. doi: 10.1021/bi00196a008. [DOI] [PubMed] [Google Scholar]
- 16.Wartmann M, Davis RJ. The native structure of the activated Raf protein kinase is a membrane-bound multi-subunit complex. J Biol Chem. 1994;269:6695–701. [PubMed] [Google Scholar]
- 17.Solary E, Droin N, Bettaieb A, Corcos L, Dimanche-Boitrel MT, Garrido C. Positive and negative regulation of apoptotic pathways by cytotoxic agents in hematological malignancies. Leukemia. 2000;14:1833–49. doi: 10.1038/sj.leu.2401902. [DOI] [PubMed] [Google Scholar]
- 18.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–6. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breckenridge DG, Xue D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol. 2004;16:647–52. doi: 10.1016/j.ceb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 22.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 23.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 24.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 25.Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. Faseb J. 1999;13:2061–70. doi: 10.1096/fasebj.13.14.2061. [DOI] [PubMed] [Google Scholar]
- 26.Mosser DD, Caron AW, Bourget L, Meriin AB, Sherman MY, Morimoto RI, Massie B. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol. 2000;20:7146–59. doi: 10.1128/mcb.20.19.7146-7159.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mosser DD, Morimoto RI. Molecular chaperones and the stress of oncogenesis. Oncogene. 2004;23:2907–18. doi: 10.1038/sj.onc.1207529. [DOI] [PubMed] [Google Scholar]
- 28.Kamada M, So A, Muramaki M, Rocchi P, Beraldi E, Gleave M. Hsp27 knockdown using nucleotide-based therapies inhibit tumor growth and enhance chemotherapy in human bladder cancer cells. Mol Cancer Ther. 2007;6:299–308. doi: 10.1158/1535-7163.MCT-06-0417. [DOI] [PubMed] [Google Scholar]
- 29.Choi DH, Ha JS, Lee WH, Song JK, Kim GY, Park JH, Cha HJ, Lee BJ, Park JW. Heat shock protein 27 is associated with irinotecan resistance in human col-orectal cancer cells. FEBS Lett. 2007;581:1649–56. doi: 10.1016/j.febslet.2007.02.075. [DOI] [PubMed] [Google Scholar]
- 30.Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D, Sharif R, Dawra R, Lerch MM, Saluja A. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–25. doi: 10.1158/0008-5472.CAN-06-1567. [DOI] [PubMed] [Google Scholar]
- 31.Compton SA, Elmore LW, Haydu K, Jackson-Cook CK, Holt SE. Induction of nitric oxide synthase-dependent telomere shortening after functional inhibition of Hsp90 in human tumor cells. Mol Cell Biol. 2006;26:1452–62. doi: 10.1128/MCB.26.4.1452-1462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurbuxani S, Bruey JM, Fromentin A, Larmonier N, Parcellier A, Jaattela M, Martin F, Solary E, Garrido C. Selective depletion of inducible HSP70 enhances immunogenicity of rat colon cancer cells. Oncogene. 2001;20:7478–85. doi: 10.1038/sj.onc.1204948. [DOI] [PubMed] [Google Scholar]
- 33.Nylandsted J, Rohde M, Brand K, Bastholm L, Elling F, Jaattela M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc Natl Acad Sci USA. 2000;97:7871–6. doi: 10.1073/pnas.97.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–6. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–5. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 36.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 37.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 38.Rane MJ, Pan Y, Singh S, Powell DW, Wu R, Cummins T, Chen Q, McLeish KR, Klein JB. Heat shock protein 27 controls apoptosis by regulating Akt activation. J Biol Chem. 2003;278:27828–35. doi: 10.1074/jbc.M303417200. [DOI] [PubMed] [Google Scholar]
- 39.Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP. Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol. 2002;22:816–34. doi: 10.1128/MCB.22.3.816-834.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parcellier A, Schmitt E, Gurbuxani S, Seigneurin-Berny D, Pance A, Chantome A, Plenchette S, Khochbin S, Solary E, Garrido C. HSP27 is a ubiq-uitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol Cell Biol. 2003;23:5790–802. doi: 10.1128/MCB.23.16.5790-5802.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parcellier A, Brunet M, Schmitt E, Col E, Didelot C, Hammann A, Nakayama K, Nakayama KI, Khochbin S, Solary E, Garrido C. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 2006;20:1179–81. doi: 10.1096/fj.05-4184fje. [DOI] [PubMed] [Google Scholar]
- 42.Park HS, Cho SG, Kim CK, Hwang HS, Noh KT, Kim MS, Huh SH, Kim MJ, Ryoo K, Kim EK, Kang WJ, Lee JS, Seo JS, Ko YG, Kim S, Choi EJ. Heat shock protein hsp72 is a negative regulator of apoptosis signal-regulating kinase 1. Mol Cell Biol. 2002;22:7721–30. doi: 10.1128/MCB.22.22.7721-7730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meriin AB, Yaglom JA, Gabai VL, Zon L, Ganiatsas S, Mosser DD, Sherman MY. Protein-damaging stresses activate c-Jun N-terminal kinase via inhibition of its dephosphorylation: a novel pathway controlled by HSP72. Mol Cell Biol. 1999;19:2547–55. doi: 10.1128/mcb.19.4.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park HS, Lee JS, Huh SH, Seo JS, Choi EJ. Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. EMBO J. 2001;20:446–56. doi: 10.1093/emboj/20.3.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JS, Lee JJ, Seo JS. HSP70 deficiency results in activation of c-Jun N-terminal Kinase, extracellular signal-regulated kinase, and caspase-3 in hyperos-molarity-induced apoptosis. J Biol Chem. 2005;280:6634–41. doi: 10.1074/jbc.M412393200. [DOI] [PubMed] [Google Scholar]
- 46.Salehi AH, Morris SJ, Ho WC, Dickson KM, Doucet G, Milutinovic S, Durkin J, Gillard JW, Barker PA. AEG3482 is an anti-apoptotic compound that inhibits Jun kinase activity and cell death through induced expression of heat shock protein 70. Chem Biol. 2006;13:213–23. doi: 10.1016/j.chembiol.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 47.Gabai VL, Yaglom JA, Volloch V, Meriin AB, Force T, Koutroumanis M, Massie B, Mosser DD, Sherman MY. Hsp72-mediated suppression of c-Jun N-terminal kinase is implicated in development of tolerance to caspase-independent cell death. Mol Cell Biol. 2000;20:6826–36. doi: 10.1128/mcb.20.18.6826-6836.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao T, Newton AC. The turn motif is a phosphorylation switch that regulates the binding of Hsp70 to protein kinase C. J Biol Chem. 2002;277:31585–92. doi: 10.1074/jbc.M204335200. [DOI] [PubMed] [Google Scholar]
- 49.Hu G, Tang J, Zhang B, Lin Y, Hanai J, Galloway J, Bedell V, Bahary N, Han Z, Ramchandran R, Thisse B, Thisse C, Zon LI, Sukhatme VP. A novel endothelial-specific heat shock protein HspA12B is required in both zebrafish development and endothelial functions in vitro. J Cell Sci. 2006;119:4117–26. doi: 10.1242/jcs.03179. [DOI] [PubMed] [Google Scholar]
- 50.Akakura S, Yoshida M, Yoneda Y, Horinouchi S. A role for Hsc70 in regulating nucleocytoplasmic transport of a temperature-sensitive p53 (p53Val-135) J Biol Chem. 2001;276:14649–57. doi: 10.1074/jbc.M100200200. [DOI] [PubMed] [Google Scholar]
- 51.Zylicz M, King FW, Wawrzynow A. Hsp70 interactions with the p53 tumour suppressor protein. EMBO J. 2001;20:4634–8. doi: 10.1093/emboj/20.17.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 53.Mijatovic T, Mathieu V, Gaussin JF, De Neve N, Ribaucour F, Van Quaquebeke E, Dumont P, Darro F, Kiss R. Cardenolide-induced lysosomal membrane permeabilization demonstrates therapeutic benefits in experimental human non-small cell lung cancers. Neoplasia. 2006;8:402–12. doi: 10.1593/neo.05850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. J Leukoc Biol. 2007;81:15–27. doi: 10.1189/jlb.0306167. [DOI] [PubMed] [Google Scholar]
- 55.Ran R, Lu A, Zhang L, Tang Y, Zhu H, Xu H, Feng Y, Han C, Zhou G, Rigby AC, Sharp FR. Hsp70 promotes TNF-mediated apoptosis by binding IKK gamma and impairing NF-kappa B survival signaling. Genes Dev. 2004;18:1466–81. doi: 10.1101/gad.1188204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shanley TP, Ryan MA, Eaves-Pyles T, Wong HR. Heat shock inhibits phosphorylation of I-kappaBalpha. Shock. 2000;14:447–50. doi: 10.1097/00024382-200014040-00005. [DOI] [PubMed] [Google Scholar]
- 57.Yoo CG, Lee S, Lee CT, Kim YW, Han SK, Shim YS. Anti-inflammatory effect of heat shock protein induction is related to stabilization of I kappa B alpha through preventing I kappa B kinase activation in respiratory epithelial cells. J Immunol. 2000;164:5416–23. doi: 10.4049/jimmunol.164.10.5416. [DOI] [PubMed] [Google Scholar]
- 58.Feng X, Bonni S, Riabowol K. HSP70 induction by ING proteins sensitizes cells to tumor necrosis factor alpha receptor-mediated apoptosis. Mol Cell Biol. 2006;26:9244–55. doi: 10.1128/MCB.01538-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med. 2004;82:488–99. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 60.Chen G, Cao P, Goeddel DV. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol Cell. 2002;9:401–10. doi: 10.1016/s1097-2765(02)00450-1. [DOI] [PubMed] [Google Scholar]
- 61.Lin K, Rockliffe N, Johnson GG, Sherrington PD, Pettitt AR. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene. 2008;27:2445–55. doi: 10.1038/sj.onc.1210893. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Wang JS, Chen LL, Cheng XK, Heng FY, Wu NH, Shen YF. Repression of hsp90beta gene by p53 in UV irradiation-induced apoptosis of Jurkat cells. J Biol Chem. 2004;279:42545–51. doi: 10.1074/jbc.M314213200. [DOI] [PubMed] [Google Scholar]
- 63.Munster PN, Marchion DC, Basso AD, Rosen N. Degradation of HER2 by ansamycins induces growth arrest and apoptosis in cells with HER2 overexpression via a HER3, phosphatidylinositol 3’-kinase-AKT-dependent pathway. Cancer Res. 2002;62:3132–7. [PubMed] [Google Scholar]
- 64.Hur E, Kim HH, Choi SM, Kim JH, Yim S, Kwon HJ, Choi Y, Kim DK, Lee MO, Park H. Reduction of hypoxia-induced transcription through the repression of hypoxia-inducible factor-1alpha/aryl hydrocarbon receptor nuclear translocator DNA binding by the 90-kDa heat-shock protein inhibitor radicicol. Mol Pharmacol. 2002;62:975–82. doi: 10.1124/mol.62.5.975. [DOI] [PubMed] [Google Scholar]
- 65.Cissel DS, Beaven MA. Disruption of Raf-1/heat shock protein 90 complex and Raf signaling by dexamethasone in mast cells. J Biol Chem. 2000;275:7066–70. doi: 10.1074/jbc.275.10.7066. [DOI] [PubMed] [Google Scholar]
- 66.Piatelli MJ, Doughty C, Chiles TC. Requirement for a hsp90 chaperone-dependent MEK1/2-ERK pathway for B cell antigen receptor-induced cyclin D2 expression in mature B lymphocytes. J Biol Chem. 2002;277:12144–50. doi: 10.1074/jbc.M200102200. [DOI] [PubMed] [Google Scholar]
- 67.Sato N, Yamamoto T, Sekine Y, Yumioka T, Junicho A, Fuse H, Matsuda T. Involvement of heat-shock protein 90 in the interleukin-6-mediated signaling pathway through STAT3. Biochem Biophys Res Commun. 2003;300:847–52. doi: 10.1016/s0006-291x(02)02941-8. [DOI] [PubMed] [Google Scholar]
- 68.Kalvakolanu DV, Roy SK. CCAAT/enhancer binding proteins and interferon signaling pathways. J Interferon Cytokine Res. 2005;25:757–69. doi: 10.1089/jir.2005.25.757. [DOI] [PubMed] [Google Scholar]
- 69.Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, Catley L, Tai YT, Hayashi T, Shringarpure R, Burger R, Munshi N, Ohtake Y, Saxena S, Anderson KC. Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance. Blood. 2003;102:3379–86. doi: 10.1182/blood-2003-05-1417. [DOI] [PubMed] [Google Scholar]
- 70.Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–39. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- 71.Ruchalski K, Mao H, Li Z, Wang Z, Gillers S, Wang Y, Mosser DD, Gabai V, Schwartz JH, Borkan SC. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem. 2006;281:7873–80. doi: 10.1074/jbc.M513728200. [DOI] [PubMed] [Google Scholar]
- 72.Gupta S, Knowlton AA. Cytosolic heat shock protein 60, hypoxia, and apoptosis. Circulation. 2002;106:2727–33. doi: 10.1161/01.cir.0000038112.64503.6e. [DOI] [PubMed] [Google Scholar]
- 73.Kirchhoff SR, Gupta S, Knowlton AA. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation. 2002;105:2899–904. doi: 10.1161/01.cir.0000019403.35847.23. [DOI] [PubMed] [Google Scholar]
- 74.Shan YX, Liu TJ, Su HF, Samsamshariat A, Mestril R, Wang PH. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol. 2003;35:1135–43. doi: 10.1016/s0022-2828(03)00229-3. [DOI] [PubMed] [Google Scholar]
- 75.Veereshwarayya V, Kumar P, Rosen KM, Mestril R, Querfurth HW. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular beta-amyloid-induced inhibition of complex IV and limit apoptosis. J Biol Chem. 2006;281:29468–78. doi: 10.1074/jbc.M602533200. [DOI] [PubMed] [Google Scholar]
- 76.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific hsp90 chaperone network. Cell. 2007;131:257–70. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 77.Cohen-Saidon C, Carmi I, Keren A, Razin E. Anti-Apoptotic function of Bcl-2 in mast cells is dependent on its association with heat shock protein 90beta. Blood. 2006;107:1413–20. doi: 10.1182/blood-2005-07-2648. [DOI] [PubMed] [Google Scholar]
- 78.Kuo CC, Liang CM, Lai CY, Liang SM. Involvement of heat shock protein (Hsp)90 beta but not Hsp90 alpha in anti-apoptotic effect of CpG-B oligodeoxynucleotide. J Immunol. 2007;178:6100–8. doi: 10.4049/jimmunol.178.10.6100. [DOI] [PubMed] [Google Scholar]
- 79.Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–52. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 80.Samali A, Robertson JD, Peterson E, Manero F, Van Zeijl L, Paul C, Cotgreave IA, Arrigo AP, Orrenius S. Hsp27 protects mitochondria of thermotolerant cells against apoptotic stimuli. Cell Stress Chaperones. 2001;6:49–58. doi: 10.1379/1466-1268(2001)006<0049:hpmotc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voss OH, Batra S, Kolattukudy SJ, Gonzalez-Mejia ME, Smith JB, Doseff AI. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J Biol Chem. 2007;282:25088–99. doi: 10.1074/jbc.M701740200. [DOI] [PubMed] [Google Scholar]
- 82.Mehlen P, Kretz-Remy C, Preville X, Arrigo AP. Human hsp27, Drosophila hsp27 and human alphaB-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against TNFalpha-induced cell death. EMBO J. 1996;15:2695–706. [PMC free article] [PubMed] [Google Scholar]
- 83.Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo AP, Buchner J, Gaestel M. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem. 1999;274:18947–56. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- 84.Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–51. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 85.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–45. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 86.Pivovarova AV, Chebotareva NA, Chernik IS, Gusev NB, Levitsky DI. Small heat shock protein Hsp27 prevents heat-induced aggregation of F-actin by forming soluble complexes with denatured actin. FEBS J. 2007;274:5937–48. doi: 10.1111/j.1742-4658.2007.06117.x. [DOI] [PubMed] [Google Scholar]
- 87.Li CY, Lee JS, Ko YG, Kim JI, Seo JS. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem. 2000;275:25665–71. doi: 10.1074/jbc.M906383199. [DOI] [PubMed] [Google Scholar]
- 88.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 89.Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2:476–83. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- 90.Sakahira H, Nagata S. Co-translational folding of caspase-activated DNase with Hsp70, Hsp40, and inhibitor of caspase-activated DNase. J Biol Chem. 2002;277:3364–70. doi: 10.1074/jbc.M110071200. [DOI] [PubMed] [Google Scholar]
- 91.Liu QL, Kishi H, Ohtsuka K, Muraguchi A. Heat shock protein 70 binds caspase-activated DNase and enhances its activity in TCR-stimulated T cells. Blood. 2003;102:1788–96. doi: 10.1182/blood-2002-11-3499. [DOI] [PubMed] [Google Scholar]
- 92.Ribeil JA, Zermati Y, Vandekerckhove J, Cathelin S, Kersual J, Dussiot M, Coulon S, Moura IC, Zeuner A, Kirkegaard-Sorensen T, Varet B, Solary E, Garrido C, Hermine O. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature. 2007;445:102–5. doi: 10.1038/nature05378. [DOI] [PubMed] [Google Scholar]
- 93.Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, Weichselbaum R, Nalin C, Alnemri ES, Kufe D, Kharbanda S. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000;19:4310–22. doi: 10.1093/emboj/19.16.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang MH, Lee JS, Kim HJ, Jin DI, Kim JI, Lee KJ, Seo JS. HSP90 protects apoptotic cleavage of vimentin in geldanamycin-induced apoptosis. Mol Cell Biochem. 2006;281:111–21. doi: 10.1007/s11010-006-0638-x. [DOI] [PubMed] [Google Scholar]
- 95.Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 1999;18:2040–8. doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xanthoudakis S, Roy S, Rasper D, Hennessey T, Aubin Y, Cassady R, Tawa P, Ruel R, Rosen A, Nicholson DW. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 1999;18:2049–56. doi: 10.1093/emboj/18.8.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mehlen P, Schulze-Osthoff K, Arrigo AP. Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1- and staurosporine-induced cell death. J Biol Chem. 1996;271:16510–4. doi: 10.1074/jbc.271.28.16510. [DOI] [PubMed] [Google Scholar]
- 98.Charette SJ, Lavoie JN, Lambert H, Landry J. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol Cell Biol. 2000;20:7602–12. doi: 10.1128/mcb.20.20.7602-7612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gabai VL, Mabuchi K, Mosser DD, Sherman MY. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol Cell Biol. 2002;22:3415–24. doi: 10.1128/MCB.22.10.3415-3424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pang Q, Keeble W, Christianson TA, Faulkner GR, Bagby GC. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-gamma/TNF-alphamediated cytotoxicity. EMBO J. 2001;20:4478–89. doi: 10.1093/emboj/20.16.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pang Q, Christianson TA, Keeble W, Koretsky T, Bagby GC. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. J Biol Chem. 2002;277:49638–43. doi: 10.1074/jbc.M209386200. [DOI] [PubMed] [Google Scholar]
- 102.Guo F, Sigua C, Bali P, George P, Fiskus W, Scuto A, Annavarapu S, Mouttaki A, Sondarva G, Wei S, Wu J, Djeu J, Bhalla K. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood. 2005;105:1246–55. doi: 10.1182/blood-2004-05-2041. [DOI] [PubMed] [Google Scholar]
- 103.Schett G, Steiner CW, Groger M, Winkler S, Graninger W, Smolen J, Xu Q, Steiner G. Activation of Fas inhibits heat-induced activation of HSF1 and up-regulation of hsp70. FASEB J. 1999;13:833–42. doi: 10.1096/fasebj.13.8.833. [DOI] [PubMed] [Google Scholar]
- 104.Liossis SN, Ding XZ, Kiang JG, Tsokos GC. Overexpression of the heat shock protein 70 enhances the TCR/CD3- and Fas/Apo-1/CD95-mediated apoptotic cell death in Jurkat T cells. J Immunol. 1997;158:5668–75. [PubMed] [Google Scholar]
- 105.Panner A, Murray JC, Berger MS, Pieper RO. Heat shock protein 90alpha recruits FLIPS to the death-inducing signaling complex and contributes to TRAIL resistance in human glioma. Cancer Res. 2007;67:9482–9. doi: 10.1158/0008-5472.CAN-07-0569. [DOI] [PubMed] [Google Scholar]
- 106.Lewis J, Devin A, Miller A, Lin Y, Rodriguez Y, Neckers L, Liu ZG. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J Biol Chem. 2000;275:10519–26. doi: 10.1074/jbc.275.14.10519. [DOI] [PubMed] [Google Scholar]
- 107.Zhao C, Wang E. Heat shock protein 90 suppresses tumor necrosis factor alpha induced apoptosis by preventing the cleavage of Bid in NIH3T3 fibroblasts. Cell Signal. 2004;16:313–21. doi: 10.1016/j.cellsig.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 108.Cande C, Vahsen N, Garrido C, Kroemer G. Apoptosis-inducing factor (AIF): caspase-independent after all. Cell Death Differ. 2004;11:591–5. doi: 10.1038/sj.cdd.4401400. [DOI] [PubMed] [Google Scholar]
- 109.Creagh EM, Carmody RJ, Cotter TG. Heat shock protein 70 inhibits caspasedependent and -independent apoptosis in Jurkat T cells. Exp Cell Res. 2000;257:58–66. doi: 10.1006/excr.2000.4856. [DOI] [PubMed] [Google Scholar]
- 110.Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–43. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 111.Matsumori Y, Hong SM, Aoyama K, Fan Y, Kayama T, Sheldon RA, Vexler ZS, Ferriero DM, Weinstein PR, Liu J. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:899–910. doi: 10.1038/sj.jcbfm.9600080. [DOI] [PubMed] [Google Scholar]
- 112.Lui JC, Kong SK. Heat shock protein 70 inhibits the nuclear import of apoptosis-inducing factor to avoid DNA fragmentation in TF-1 cells during erythropoiesis. FEBS Lett. 2007;581:109–17. doi: 10.1016/j.febslet.2006.11.082. [DOI] [PubMed] [Google Scholar]
- 113.Kalinowska M, Garncarz W, Pietrowska M, Garrard WT, Widlak P. Regulation of the human apoptotic DNase/RNase Endonuclease G: involvement of Hsp70 and ATP. Apoptosis. 2005;10:821–30. doi: 10.1007/s10495-005-0410-9. [DOI] [PubMed] [Google Scholar]
- 114.Jaattela M, Tschopp J. Caspase-independent cell death in T lymphocytes. Nat Immunol. 2003;4:416–23. doi: 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- 115.Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH, Gores GJ. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106:1127–37. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M, Elling F, Leist M, Jaattela M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol. 2001;153:999–1010. doi: 10.1083/jcb.153.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brunk UT, Svensson I. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep. 1999;4:3–11. doi: 10.1179/135100099101534675. [DOI] [PubMed] [Google Scholar]
- 118.Broker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004;64:27–30. doi: 10.1158/0008-5472.can-03-3060. [DOI] [PubMed] [Google Scholar]
- 119.Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–11. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 120.Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L, Brunk UT. Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci USA. 2002;99:6286–91. doi: 10.1073/pnas.092135599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brunk UT, Dalen H, Roberg K, Hellquist HB. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic Biol Med. 1997;23:616–26. doi: 10.1016/s0891-5849(97)00007-5. [DOI] [PubMed] [Google Scholar]
- 122.Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T, Perfettini JL, Kroemer G. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003;22:3927–36. doi: 10.1038/sj.onc.1206622. [DOI] [PubMed] [Google Scholar]
- 123.Roberg K, Kagedal K, Ollinger K. Microinjection of cathepsin d induces caspasedependent apoptosis in fibroblasts. Am J Pathol. 2002;161:89–96. doi: 10.1016/S0002-9440(10)64160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vancompernolle K, Van Herreweghe F, Pynaert G, Van de Craen M, De Vos K, Totty N, Sterling A, Fiers W, Vandenabeele P, Grooten J. Atractyloside-induced release of cathepsin B, a protease with cas-pase-processing activity. FEBS Lett. 1998;438:150–8. doi: 10.1016/s0014-5793(98)01275-7. [DOI] [PubMed] [Google Scholar]
- 125.Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G, Rohde M, Jaattela M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–35. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bivik C, Rosdahl I, Ollinger K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis. 2007;28:537–44. doi: 10.1093/carcin/bgl152. [DOI] [PubMed] [Google Scholar]
- 127.Saribek B, Jin Y, Saigo M, Eto K, Abe S. HSP90beta is involved in signaling prolactin-induced apoptosis in newt testis. Biochem Biophys Res Commun. 2006;349:1190–7. doi: 10.1016/j.bbrc.2006.08.143. [DOI] [PubMed] [Google Scholar]
- 128.Bruey JM, Paul C, Fromentin A, Hilpert S, Arrigo AP, Solary E, Garrido C. Differential regulation of HSP27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene. 2000;19:4855–63. doi: 10.1038/sj.onc.1203850. [DOI] [PubMed] [Google Scholar]
- 129.Tsuchiya A, Tashiro E, Yoshida M, Imoto M. Involvement of nuclear accumulation of heat shock protein 27 in leptomycin B-induced apoptosis in HeLa cells. J Antibiot. 2005;58:810–6. doi: 10.1038/ja.2005.108. [DOI] [PubMed] [Google Scholar]
- 130.Sashchenko LP, Dukhanina EA, Shatalov YV, Yashin DV, Lukyanova TI, Kabanova OD, Romanova EA, Khaidukov SV, Galkin AV, Gnuchev NV, Georgiev GP. Cytotoxic T lymphocytes carrying a pattern recognition protein Tag7 can detect evasive, HLA-negative but Hsp70-exposing tumor cells, thereby ensuring FasL/Fas-mediated contact killing. Blood. 2007;110:1997–2004. doi: 10.1182/blood-2006-12-064444. [DOI] [PubMed] [Google Scholar]
- 131.Voss AK, Thomas T, Gruss P. Mice lacking HSP90beta fail to develop a placental labyrinth. Development. 2000;127:1–11. doi: 10.1242/dev.127.1.1. [DOI] [PubMed] [Google Scholar]
- 132.Negroni L, Samson M, Guigonis JM, Rossi B, Pierrefite-Carle V, Baudoin C. Treatment of colon cancer cells using the cytosine deaminase/5-fluorocytosine suicide system induces apoptosis, modulation of the proteome, and Hsp90beta phosphorylation. Mol Cancer Ther. 2007;6:2747–56. doi: 10.1158/1535-7163.MCT-07-0040. [DOI] [PubMed] [Google Scholar]
- 133.Chiosis G, Caldas Lopes E, Solit D. Heat shock protein-90 inhibitors: a chronicle from geldanamycin to today's agents. Curr Opin Investig Drugs. 2006;7:534–41. [PubMed] [Google Scholar]
- 134.Lin KM, Lin B, Lian IY, Mestril R, Scheffler IE, Dillmann WH. Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia-reoxy-genation. Circulation. 2001;103:1787–92. doi: 10.1161/01.cir.103.13.1787. [DOI] [PubMed] [Google Scholar]
- 135.Faried A, Sohda M, Nakajima M, Miyazaki T, Kato H, Kuwano H. Expression of heat-shock protein Hsp60 correlated with the apoptotic index and patient prognosis in human oesophageal squamous cell carcinoma. Eur J Cancer. 2004;40:2804–11. doi: 10.1016/j.ejca.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 136.Chandra D, Choy G, Tang DG. Cytosolic Accumulation of HSP60 during Apoptosis with or without Apparent Mitochondrial Release: evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J Biol Chem. 2007;282:31289–301. doi: 10.1074/jbc.M702777200. [DOI] [PubMed] [Google Scholar]
- 137.Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, Torre-Amione G, Knowlton AA. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293:H2238–47. doi: 10.1152/ajpheart.00740.2007. [DOI] [PubMed] [Google Scholar]
- 138.Thomas X, Campos L, Mounier C, Cornillon J, Flandrin P, Le QH, Piselli S, Guyotat D. Expression of heat-shock proteins is associated with major adverse prognostic factors in acute myeloid leukemia. Leuk Res. 2005;29:1049–58. doi: 10.1016/j.leukres.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 139.Duval A, Olaru D, Campos L, Flandrin P, Nadal N, Guyotat D. Expression and prognostic significance of heat-shock proteins in myelodysplastic syndromes. Haematologica. 2006;91:713–4. [PubMed] [Google Scholar]
- 140.Schepers H, Geugien M, Van Der Toorn M, Bryantsev AL, Kampinga HH, Eggen BJ, Vellenga E. HSP27 protects AML cells against VP-16-induced apoptosis through modulation of p38 and c-Jun. Exp Hematol. 2005;33:660–70. doi: 10.1016/j.exphem.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 141.Georgakis GV, Li Y, Younes A. The heat shock protein 90 inhibitor 17-AAG induces cell cycle arrest and apoptosis in mantle cell lymphoma cell lines by depleting cyclin D1, Akt, Bid and activating caspase 9. Br J Haematol. 2006;135:68–71. doi: 10.1111/j.1365-2141.2006.06247.x. [DOI] [PubMed] [Google Scholar]
- 142.Kawakami H, Tomita M, Okudaira T, Ishikawa C, Matsuda T, Tanaka Y, Nakazato T, Taira N, Ohshiro K, Mori N. Inhibition of heat shock protein-90 modulates multiple functions required for survival of human T-cell leukemia virus type I-infected T-cell lines and adult T-cell leukemia cells. Int J Cancer. 2007;120:1811–20. doi: 10.1002/ijc.22403. [DOI] [PubMed] [Google Scholar]
- 143.Fumo G, Akin C, Metcalfe DD, Neckers L. 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood. 2004;103:1078–84. doi: 10.1182/blood-2003-07-2477. [DOI] [PubMed] [Google Scholar]
- 144.Yao Q, Nishiuchi R, Kitamura T, Kersey JH. Human leukemias with mutated FLT3 kinase are synergistically sensitive to FLT3 and Hsp90 inhibitors: the key role of the STAT5 signal transduction pathway. Leukemia. 2005;19:1605–12. doi: 10.1038/sj.leu.2403881. [DOI] [PubMed] [Google Scholar]
- 145.Castro JE, Prada CE, Loria O, Kamal A, Chen L, Burrows FJ, Kipps TJ. ZAP-70 is a novel conditional heat shock protein 90 (Hsp90) client: inhibition of Hsp90 leads to ZAP-70 degradation, apoptosis, and impaired signaling in chronic lymphocytic leukemia. Blood. 2005;106:2506–12. doi: 10.1182/blood-2005-03-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jones DT, Addison E, North JM, Lowdell MW, Hoffbrand AV, Mehta AB, Ganeshaguru K, Folarin NI, Wickremasinghe RG. Geldanamycin and her-bimycin A induce apoptotic killing of B chronic lymphocytic leukemia cells and augment the cells'sensitivity to cytotoxic drugs. Blood. 2004;103:1855–61. doi: 10.1182/blood-2003-05-1603. [DOI] [PubMed] [Google Scholar]
- 147.Peng C, Brain J, Hu Y, Goodrich A, Kong L, Grayzel D, Pak R, Read M, Li S. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood. 2007;110:678–85. doi: 10.1182/blood-2006-10-054098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mesa RA, Loegering D, Powell HL, Flatten K, Arlander SJ, Dai NT, Heldebrant MP, Vroman BT, Smith BD, Karp JE, Eyck CJ, Erlichman C, Kaufmann SH, Karnitz LM. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood. 2005;106:318–27. doi: 10.1182/blood-2004-09-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chatterjee M, Jain S, Stuhmer T, Andrulis M, Ungethum U, Kuban RJ, Lorentz H, Bommert K, Topp M, Kramer D, Muller-Hermelink HK, Einsele H, Greiner A, Bargou RC. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood. 2007;109:720–8. doi: 10.1182/blood-2006-05-024372. [DOI] [PubMed] [Google Scholar]
- 150.Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–45. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 152.Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R, Atadja P, Bhalla K. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126–35. [PubMed] [Google Scholar]
- 153.Bali P, George P, Cohen P, Tao J, Guo F, Sigua C, Vishvanath A, Scuto A, Annavarapu S, Fiskus W, Moscinski L, Atadja P, Bhalla K. Superior activity of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 kinase inhibitor PKC412 against human acute myelogenous leukemia cells with mutant FLT-3. Clin Cancer Res. 2004;10:4991–7. doi: 10.1158/1078-0432.CCR-04-0210. [DOI] [PubMed] [Google Scholar]
- 154.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 155.George P, Bali P, Annavarapu S, Scuto A, Fiskus W, Guo F, Sigua C, Sondarva G, Moscinski L, Atadja P, Bhalla K. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood. 2005;105:1768–76. doi: 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- 156.Nieto-Miguel T, Gajate C, Gonzalez-Camacho F, Mollinedo F. Proapoptotic role of Hsp90 by its interaction with c-Jun N-terminal kinase in lipid rafts in edelfosine-mediated antileukemic therapy. Oncogene. 2008;27:1779–87. doi: 10.1038/sj.onc.1210816. [DOI] [PubMed] [Google Scholar]
- 157.Yao Q, Nishiuchi R, Li Q, Kumar AR, Hudson WA, Kersey JH. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res. 2003;9:4483–93. [PubMed] [Google Scholar]
- 158.Nimmanapalli R, O’Bryan E, Bhalla K. Geldanamycin and its analogue 17-allylamino-17-demethoxygel-danamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 2001;61:1799–804. [PubMed] [Google Scholar]
- 159.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 160.Workman P. Combinatorial attack on multistep onco-genesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004;206:149–57. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 161.Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, Rosen N, Spampinato C, Modrich P, Chiosis G. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol. 2004;11:787–97. doi: 10.1016/j.chembiol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 162.Ray S, Lu Y, Kaufmann SH, Gustafson WC, Karp JE, Boldogh I, Fields AP, Brasier AR. Genomic mechanisms of p210BCR-ABL signaling: induction of heat shock protein 70 through the GATA response element confers resistance to paclitaxel-induced apoptosis. J Biol Chem. 2004;279:35604–15. doi: 10.1074/jbc.M401851200. [DOI] [PubMed] [Google Scholar]
- 163.Schmitt E, Maingret L, Puig PE, Rerole AL, Ghiringhelli F, Hammann A, Solary E, Kroemer G, Garrido C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006;66:4191–7. doi: 10.1158/0008-5472.CAN-05-3778. [DOI] [PubMed] [Google Scholar]
- 164.Westerheide SD, Kawahara TL, Orton K, Morimoto RI. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J Biol Chem. 2006;281:9616–22. doi: 10.1074/jbc.M512044200. [DOI] [PubMed] [Google Scholar]
- 165.Zhao ZG, Shen WL. Heat shock protein 70 antisense oligonucleotide inhibits cell growth and induces apoptosis in human gastric cancer cell line SGC-7901. World J Gastroenterol. 2005;11:73–8. doi: 10.3748/wjg.v11.i1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jaattela M. Heat shock proteins as cellular lifeguards. Ann Med. 1999;31:261–71. doi: 10.3109/07853899908995889. [DOI] [PubMed] [Google Scholar]
- 167.Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E, Kouranti I, Spahr C, Pance A, Kroemer G, Garrido C. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene. 2003;22:6669–78. doi: 10.1038/sj.onc.1206794. [DOI] [PubMed] [Google Scholar]
- 168.Rocchi P, Jugpal P, So A, Sinneman S, Ettinger S, Fazli L, Nelson C, Gleave M. Small interference RNA targeting heat-shock protein 27 inhibits the growth of prostatic cell lines and induces apoptosis via caspase-3 activation in vitro. BJU Int. 2006;98:1082–9. doi: 10.1111/j.1464-410X.2006.06425.x. [DOI] [PubMed] [Google Scholar]
- 169.Sangster TA, Bahrami A, Wilczek A, Watanabe E, Schellenberg K, McLellan C, Kelley A, Kong SW, Queitsch C, Lindquist S. Phenotypic diversity and altered environmental plasticity in Arabidopsis thaliana with reduced Hsp90 levels. PLoS ONE. 2007;2:e648. doi: 10.1371/journal.pone.0000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Arrigo AP, Simon S, Gibert B, Kretz-Remy C, Nivon M, Czekalla A, Guillet D, Moulin M, Diaz-Latoud C, Vicart P. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007;581:3665–74. doi: 10.1016/j.febslet.2007.04.033. [DOI] [PubMed] [Google Scholar]