

Abstract
Alzheimer's disease (AD) affects more than 18 million people worldwide and is characterized by progressive memory deficits, cognitive impairment and personality changes. The main cause of AD is generally attributed to the increased production and accumulation of amyloid-β (Aβ), in association with neurofibrillary tangle (NFT) formation. Increased levels of pro-inflammatory factors such as cytokines and chemokines, and the activation of the complement cascade occurs in the brains of AD patients and contributes to the local inflammatory response triggered by senile plaque. The existence of an inflammatory component in AD is now well known on the basis of epidemiological findings showing a reduced prevalence of the disease upon long-term medication with anti-inflammatory drugs, and evidence from studies of clinical materials that shows an accumulation of activated glial cells, particularly microglia and astrocytes, in the same areas as amyloid plaques. Glial cells maintain brain plasticity and protect the brain for functional recovery from injuries. Dysfunction of glial cells may promote neurodegeneration and, eventually, the retraction of neuronal synapses, which leads to cognitive deficits. The focus of this review is on glial cells and their diversity properties in AD.
Keywords: Alzheimer's disease, microglia, astrocyte, endothelial cells neurodegeneration, immunotherapy
Introduction
Alzheimer's disease (AD) has emerged as the most prevalent form (70%) of late-life mental failure in humans, which is characterized by progressive memory deficits, cognitive impairments and personality changes. At gross examination, the Alzheimer's brain shows severe atrophy and a reduction in brain weight of usually more than 35%. The histopathological hallmarks include the presence of extracellular deposition of Aβ, neurofibrillary tangles (NFT), progressive synaptic dysfunction and, much later, neuronal death, especially in the hippocampus [1–3].
Most cases of AD are sporadic, while less than 5% of cases are early onset familial Alzheimer's disease (EOFAD; age at onset < 65 years) with an autosomal dominant pattern of inheritance [4]. Among all the known cases of EOFAD, the genetic mutations are found to affect the expression levels of Aβ peptide, which was named amyloid after its starchy appearance following its aggregation. Aβpeptide is a 4-kD peptide, 40–43 aa, derived from proteolytic amyloid-cleavage of the amyloid precursor protein (APP), which maps to chromosome 21 and is found to be spread on neuronal cells [5–7]. The main role of Aβ as a mediator in AD is derived from the fact that it accumulates in the brain several decades before the disease is evident. Furthermore, Down syndrome patients that have tri-chromosome 21 suffer from AD symptoms in their late 30s. This evidence led to the establishment of the amyloid hypothesis in AD. While in early research the fibril form of Aβ was considered the toxic form, in more recent papers, the toxicity to the neurons seems also to be derived from soluble Aβ oligomers [8, 9]. The classical view is that Aβ is deposited extracellularly, however, emerging evidence from transgenic mice and human patients indicates that this peptide can also accumulate intraneuronally, which may contribute to disease progression [10].
Although Aβ is the major component of the amyloid deposits, other molecules are also associated with these deposits (e.g. ferritin, components of the complement pathway, α1-antichymotrypsin, α2-macroglobulin, low-density lipoprotein [LDL] receptor-related protein, APP, acetylcholinesterase, laminin, glycosaminoglycans and the apolipoproteins E and J) [11–13]. Aβ can adversely affect distinct molecular and cellular pathways, thereby facilitating tau phosphorylation, aggregation, mislocalization and accumulation. Intracellular abnormally phosphorylated tau and Aβ exhibit synergistic effects that finally lead to an acceleration of neurodegenerative mechanisms involved in metabolism, cellular detoxification, mitochondrial dysfunction and energy deficiency, which result in the formation of neuritic plaques [1]. Such plaques are also closely associated with microglia that express surface antigens associated with activation, and surrounded by reactive astrocytes displaying abundant glial filaments. The microglia are usually within and adjacent to the central amyloid core of the neuritic plaque, whereas the astrocytes often ring the outside of the plaque with some of these processes centripetal to the amyloid core [14] (Fig. 1). The time extent that it takes to develop such a neuritic plaque is unknown, but these lesions probably evolve very gradually over a substantial time, perhaps several years. The dialogue between the microglia and astro-cyte play an important role in shaping the immune response surrounding the amyloid plaques.
1.
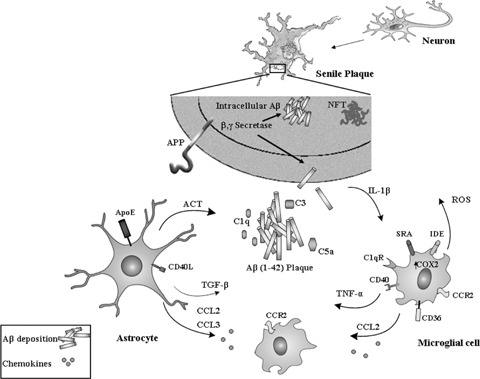
Activation of the glial cell response toward the formation of senile plaque in Alzheimer's disease. The overproduction and extracellular deposition of amyloid β-peptide (AβP) and an intracellular deposition of neurofibrillary tangles (NFT) initiates the pathogenesis of Alzheimer's disease (AD). The production of complement components (C1q, C3 and C5) is the first stage in response to Aβ deposition, resulting in the attraction and activation of microglial cells. Both microglial cells and astrocytes produce multiple pro-inflammatory and neurotoxic factors: transforming growth factor (TGF)-1; tumour necrosis factor (TNF)-α; interleukin-1 (IL-1); CC-chemokine ligand (CCL); antichymotrypsin (ACT); reactive oxygen species (ROS) and cyclooxygenase 2 (COX2). Activated microglial cells express various scavenger receptors (SRs) that mediate phagocytosis of Aβ, such as CD36, SR-A. Microglial cells can also degrade Aβ by releasing Aβ-degrading enzymes, such as insulin-degrading enzyme (IDE).
Innate immune response in the brain of AD
Although inflammation is not necessarily a hallmark of AD, an increased amount of findings suggests that Aβ deposition and NFT activate a potentially pathological innate immune response in the disease (Fig. 1). These investigations demonstrate that serum and cerebrospinal fluid (CSF) from AD patients contained antibodies that recognized human senile plaques in the central nervous system (CNS) [15, 16]. The mechanism underlying the innate immune system is based on the detection of different ligands that are not self-initiated and express highly conserved pattern recognition receptors (PRRs) to facilitate the clearance of debris (Table 1, Fig. 1). Among these highly conserved receptors, the most abundant factors expressed on glial cells are the complement receptors [17, 18], Toll-like receptors (TLRs) and scavenger receptors (SRs) [19–21] (Table 1).
1.
Glia and endothelial receptors that mediate interaction with Aβ
| Cell surface receptor | Microglia | Astrocyte | Endothel | Reference |
|---|---|---|---|---|
| Antibody receptor: | ||||
| FcRγ | + | [171–173] | ||
| Toll-like receptor: | ||||
| TLR2 | + | + | + | [174] |
| TLR3 | + | + | [19, 175, 176] | |
| TLR4 | + | + | [176, 177] | |
| TLR9 | + | + | [146] | |
| Scavenger receptor: | ||||
| SRA | + | + | + | [20, 178] |
| SRBI | + | + | [179, 180] | |
| SR-MACRO | + | + | [181] | |
| SRCL | + | [148] | ||
| CD36 | + | + | [53, 182, 183] | |
| Glycoprotein receptor: | ||||
| Lactadherin | + | + | + | [149] |
| Lipoprotein receptor: | ||||
| APOE | + | [93] | ||
| LRP | + | + | + | [99, 184] |
| CD14 | + | + | + | [185] |
| Advanced glycation end products receptor: | ||||
| RAGE | + | + | + | [104, 105] |
| Formyl peptide receptor: | ||||
| FPRL1 | + | [186–188] | ||
| Complement receptor: | ||||
| C1q receptor | + | + | [23, 25] | |
| CD11b (C3 receptor) | + | [31] | ||
| Chemokine receptor: | ||||
| CCR3 | + | + | [72] | |
| CCR2 | + | + | [65] | |
| CCR5 | + | + | [72] | |
| CXCR2 | + | + | [34] | |
| CXCR3 | + | + | [89] | |
| T-cell receptor: | ||||
| CD40 | + | [189] | ||
| CD40L | + | + | [190, 191] | |
| MHC-II | + | + | [62, 192] | |
| Mannose receptor: | ||||
| MR | + | + | [193] | |
The complement system provides an innate immune response against pathogenic features and contributes to the local inflammatory response triggered by Aβ deposition and NFT in AD [17, 22]. The CNS contains components of the complement system that are synthesized by astrocytes, microglia and neurons [17]. Complement component 1q (C1q) has been shown to colocalize with most of the amyloid deposits in brains of AD patients [23–25] and triggers a complement cascade, including the formation of the membrane attack complex (MAC), C5b–C9 [26]. In brain regions that have dense accumulations of plaques and tangles (e.g. hippocampus and cortex), C1q mRNA level is increased up to 80-fold compared to control levels [27]. Furthermore, the absence of C1q leads to less neuropathology in transgenic mouse models of AD [28]. Therefore, the activation of the classic complement pathway through C1q may provide a specific mechanism for recruiting reactive glial cells to the sites of the Aβ plaque and lead to inflammatory events, neuronal dysfunction and degeneration through production of pro-inflammatory cytokines and oxidative products such as nitric oxide [22, 29]. Nevertheless, the complement system serves in eliminating aggregated and toxic proteins associated with neurological disorders, and thus has a protective effect. Thereby activation of the complement system can lead to increased phagocytosis of Aβ . Inhibition of complement C3 activation in the brains of hAPP mice, by expressing soluble complement receptor-related protein y (sCrry), a complement inhibitor, led to marked increases in Aβ accumulation and neuronal degeneration compared to the hAPP mice [31]. Moreover, blocking complement activation diminished microglial cell activity in APP-transgenic mice and led to an increase in amyloid loads [31]. It is likely that a balanced complement activation is necessary for efficient clearance of Aβ without damaging the brain, and hence a successful clinical outcome.
Chemokines are secreted proteins that play an important role in mediating the innate immune response in the CNS. Chemokines can recruit immune cells from the blood or from within the brain.
The source of the chemokines in the CNS can be glia cells, especially microglia or astrocytes. Several chemokines and chemokine receptors have been found to be up-regulated in the AD brain [32]. Those chemokines were clusters from the CC and CXC families, of which most are related to glial cell activity [32]. Immunohistochemical analysis of human AD brains revealed an expression of four chemokine receptors: CCR3, CCR5, CXCR2 and CXCR3 (chemokine receptors of macrophage inflammatory protein-1α (MIP-1α; also known as CCL3), RANTES (also known as CCL5), MIP-2 and CXCL10, respectively [32–34]. Furthermore, increased levels of MCP-1 and IL-8/CXCL8 can be found in the CSF in the majority of AD patients, therefore chemokines in the CSF may provide a useful diagnosis for AD progression [35].
Pro-inflammatory cytokines, such as interleukin-1β (IL-1β/[36, 37], tumour necrosis factor-α (TNF-α) [38] and IL-6 [39], may play an important role in AD pathology and lead to an increased production of inducible nitric oxide synthase (iNOS), neuronal stress and further neuronal dysfunction and death [40]. Moreover, recent research of plasma signalling proteins suggests a correlation between the immune response mediating cytokines such as IL-1β and TNF-α within patients with pre-symptomatic Alzheimer's or mild cognitive impairment (MCI) and a greater risk of developing AD [41]. Interestingly, other evidence demonstrates that triggering localized hip-pocampal IL-1β overexpression mediates a substantial reduction in plaque pathology through microglial cell activation suggesting a dual effect of IL-1β in mediating inflammation in AD [42].
Transforming growth factor β (TGF-β) is a cytokine with neuroprotective features that can be released by both neurons and glia in response to stress [43]. Brain TGF-β1 is increased in AD brains compared with controls and positively correlates with the extent of cerebrovascular brain amyloid deposition and cerebral amyloid angiopathy (CAA) [44]. Interestingly, coexpression of TGF-β1 with hAPP/Aβ in bigenic mice resulted in reduction of brain Aβ while elevating cerebrovascular and meningeal amyloid deposition at 2–3 months of age [45].
Microglia
Activated microglia represent the major source of inflammatory factors in the brain in neurodegenerative disorders such as AD [46]. In AD, activated microglia diffusely situate throughout the cerebral cortex, and focally concentrate in Aβ plaques [47]. Microglia accumulate in plaques containing Aβ fibrils, but not in plaques containing amorphous non-fibrillar amyloid [47]. It is possible, however, that small soluble aggregates of Aβ trigger local activation of glial cells, as suggested by a recent study of the young thymus cell antigen 1 (Thy1)-APP mice that had not yet developed Aβ deposits [48]. Furthermore, studies found activated microglia in vivo (in the brains of AD patients), by using positron emission tomography and 11C®-PK111995, which is a marker of peripheral benzodiazepine binding sites that are upregulated on the activated microglia [49, 50].
An accumulation of damaged neuronal DNA fragments can be potent promoters of microglial activation in AD [51]. Activated microglia were found to contain fragmental DNA (fDNA) in their cytoplasm (AD brains and in culture) [51]. Furthermore, Aβ fib-ril interacts with numerous microglial membrane receptors including scavenger receptor A (SRA) [20, 52] and B [20], CD36 [53], CD40 [54], α6/β1 integrin-CD36-CD47 complex [55] and complement receptors [56] (Table 1). Soluble Aβ can be directly bound by microglial cell receptors, including the heparan sulphate proteoglycans (HSPGs) [57], insulin receptors [58], proteinase inhibitor (serpin)–enzyme complex receptor (SEC-R) [59] and an accumulation of the receptor for advanced glycation end products (RAGE) [60, 61].
Activated microglia bearing major histocompatibility complex (MHC) type II antigens on their surface are associated with Aβ fibrils in AD brain samples, and may interact with infiltrated T cells to induce inflammation as observed in autoimmune diseases [62]. Isolated microglia from postmortem AD and control (non-demented) patients’ brain tissue exhibit pro-inflammatory cytokine expression profiles, in the form of a significantly increased expression of IL-1β, TNF-α, IL-6 and IL-12, following an exposure to Aβ peptides. These results strongly suggest that either the Aβ peptides or the plaques can initiate the activation of microglia and subsequent release of pro-inflammatory molecules in the AD brain [62–64].
The possible function of the activated microglia may be scavenger cells, since they represent macrophage-derived cells in the CNS and therefore are capable of rapidly responding to the accumulation of foreign material, such as Aβ deposits [47] (Table 1). A deficiency in the activation of microglia in transgenic mouse models of AD promotes Aβ deposition and increases mortality in these mice [65]. Furthermore, a recent study reports that microglial cells exhibit significant telomere shortening and a reduction of telomerase activity in normal aging rats [66], and that in humans there is a tendency toward microglial cell telomere shortening with a presence of dementia [66]. Human brains containing high-amyloid loads demonstrate a significantly higher degree of microglial dystrophy than non-demented, amyloid-free control subjects [66]. This suggests that degeneration of microglia is a factor in the pathogenesis of AD [47].
Neurons can influence microglial functions, through direct cell-to-cell interactions as well as with the release of soluble mediators, such as neurotransmitters [67]. A recent article describes the up-regulation of nicotinic acetylcholine receptor α7 in cholinergic basal forebrain neurons in AD [68]. This receptor is also expressed by microglial cells and its activation weakens the pro-inflammatory response of microglial cultures, suggesting that acetylcholine may control brain inflammation [68].
Microglial cell activation can target Aβ plaques in the brain using many different mechanisms, and the source of the microglial cells can be brain resident or bone marrow-derived microglial cells [69]. Microglia cultured from rapid autopsies of AD and non-demented patients reveal an increased expression of chemokines such as IL-8, monocyte chemoattractant protein-1 (MCP-1; also known as CCL2) and macrophage inflammatory MIP-1α (CCL3) after experimental exposure to Aβ[70]. Neuropathological studies have found MCP-1 [71] and an increased expression of the chemokine receptors CCR3 and CCR5 in reactive microglia [72]. Recent evidence shows that a deficiency of CCR2, a chemokine receptor expressed on microglia and important for cell migration, accelerates early AD disease progression and markedly impairs microglial cell accumulation in a transgenic mouse model of AD. Furthermore, AD mice deficient in CCR2 accumulated Aβ earlier and died prematurely [65].
It is likely that once Aβ is deposited in an area of the brain, locally present microglia, which are constantly sampling their environment, interact with these Aβ peptides using one or more specific receptors. This interaction initiates a local inflammatory response that recruits additional microglia and astro cytes (Fig. 1). Microglial cells do not have uniform properties and, it is probable that there are different microglial cell phenotypes that have the potential to be beneficial or harmful in AD [73].
Astrocyte
Astrocytes are the most abundant cells in the CNS and play an important role in the homeostasis and maintenance of the brain [74, 75]. Astrocytes display an array of receptors involved in innate immunity, including TLRs, nucleotide-binding oligomer-ization domains, double-stranded RNA-dependent protein kinase, SRs, mannose receptor and components of the complement system [76] (Table 1). In an AD patient's brain, reactive astrocytes are integral components of neuritic plaques [77, 78]. Astrocyte activation seems to be particularly prominent around Aβ deposits both in the brain parenchyma and in the cerebrovasculature [63]. Astrocyte activity is marked by hypertrophy [79], resulting in an expression of proteins such as glial fibrillary acidic protein (GFAP), S100B [78, 80], adhesion molecules and antigen presenting capabilities, including MHC antigens [81, 82]. Tissue levels of S100B correlates with the number of neuritic plaques in the temporal lobe [78, 83] but not in the cerebellum where there is no evidence of neuritic plaques, suggesting a correlation between AD pathology and activated astrocyte.
Exposure of human astrocytes to LPS and IL-1β stimulates inflammatory mediators. These include TNF-α[84], IL-6 [85] and IL-12 [85]. Furthermore, an astrocytic response to IL-1β and TNF-α results in an elevation of colony stimulating factor 1 (CSF-1) [86], granulocyte-macrophage CSF (GM-CSF) [86], gran-ulocyte CSF (G-CSF) [86], MCP-1 and MIP-1α[33, 87], which can lead to the recruitment of microglial cells to Aβ plaques [88]. The CXCL10 ligand for CXCR3 can be present in subpopulations of astrocytes associated with senile plaques in AD brains compared with controls [89].
In AD, activated astrocytes can contain Aβ fragments, providing further evidence of their role in Aβ degradation [90]. Furthermore, Wyss-Coray et al.[91] demonstrate that astrocytes plated on Aβ brain sections (from a mouse model of AD) reduce overall Aβ levels in these sections. Apolipoprotein E (APOE), postulated to be a major lipid carrier protein in brain, is synthesized and secreted primarily by astrocytes and is involved in brain development and repair [92]. The isoform apolipoprotein E4 (APOE4) is a risk factor for developing AD at an earlier age and might contribute to this effect [92]. The putative clearance mechanism of Aβ by astrocyte may depend on their expression of APOE3 [93]. Furthermore, high expression of APOE4 in astrocytes can reduce their response to Aβ deposits [94, 95]. Thus, APOE3 seems to be important in the degradation and clearance of deposited Aβ species by astrocytes, a process that may be impaired in AD [93].
Glial cells mediate the peripheral immune response in AD
Glial cells are important cells in maintaining the integrity and homeostasis of the brain. However, upon stress, glia can mediate the permeability of the blood-brain barrier (BBB) to recruit specialized immune cells from the periphery, such as T cells (Fig. 2). Thus, understanding the dialogue between glial cells to peripheral immune cells is key to the success of any future immune intervention treatment of neurodegenerative diseases.
2.
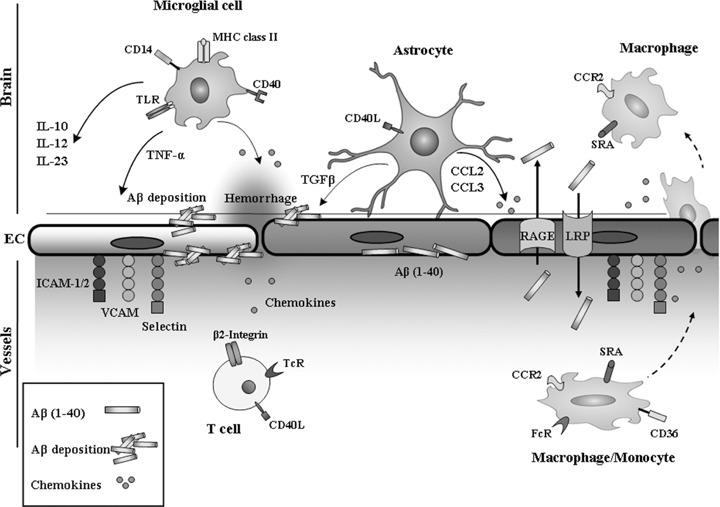
Glia–endothelia cell interaction in AD pathogenesis. Mediators released by astrocytes activate neighbouring cells and amplify the local, initial innate immune response further, modify BBB permeability and attract immune cells from the blood circulation into the neural tissue, thus supporting an adaptive immune response. Activated microglia up-regulate: MHC class II molecules, CD14, Toll-like receptors (TLRs) and CD40; and produce cytokines: transforming growth factor-β (TGF-β); tumour necrosis factor-α (TNF-α); interleukins and chemokines. TGF-β1 potentiates Aβ production in human astrocytes and thus favouring continuing deposition of Aβ, which in turn activates glial cell synthesis and the release of a number of cytokines. Low-density lipoprotein (LDL) receptor-related protein (LRP-1) and receptor for advanced glycation end products (RAGE) perform opposite functions in transporting Aβ. The weakening of the vessel wall is the likely cause of intracerebral haemorrhage.
Glial cell interaction with endothelial cells
The interaction of glial cells, particularly astrocyte, with endothelial cells, support and maintain the BBB [96]. The BBB is critical for mammalian brain home-ostasis. The BBB regulates Aβ transport to and from the brain, using two main receptors, the LDL receptor-related protein (LRP)-1 and RAGE (Table 1, Fig. 2) [97–99]. LRP-1 and RAGE perform opposite functions in transporting Aβ[97]; RAGE transports Aβ into the brain and LRP transports Aβ out of the brain.
LRP plays an important role in the balance between Aβ synthesis and clearance mechanisms [99]. LRP is expressed in reactive astrocytes and in the brain capillary endothelium [99, 100]. Glial cells secrete the LRP ligand, ApoE-containing lipoproteins (ApoE-LPs) [101], which have an important role in lipid homeostasis in the CNS. Glia-derived APOE-LPs also promote synaptogenesis and stimulate axon growth of CNS neurons [101]. In addition, binding APOE-LPs to LRP initiates the prevention of neuronal apoptosis [102]. The protective effect was greater for ApoE3 than for ApoE4, the expression of which is a risk factor for AD [103]. ApoE3 might be involved in the up-regulation of LRP and increase clearance of Aβ from the brain [103] (Table 1). These findings support the hypothesis that glial cells may play an important role in mediating Aβ transport through the endothelial cells.
RAGE is a multi-ligand receptor of the immunoglobulin super family of cell surface molecules and is expressed on endothelial and glial cells [104, 105]. RAGE is implicated in the development of the Alzheimer's neurovascular disorder by mediating circulating Aβ transcytosis across the BBB. After BBB transport, the circulating Aβ is rapidly taken up by neurons thereby inducing cellular stress. Hence, the RAGE–Aβ interaction in brain endothelium leads to an expansion of endothelin-1, a potent vasoconstrictor, resulting in a suppression of blood flow [97].
Dysfunction in the BBB clearance of Aβ through deregulated LRP1/RAGE-mediated transport, aberrant angiogenesis and arterial dysfunction may initiate neurovascular uncoupling, Aβ accumulation, cerebrovascular regression, brain hypoperfusion and neurovascular inflammation [106]. Severe AD is associated with significant changes in the relative distribution of RAGE and LRP-1 in the human hippocampus, as compared with age-matched controls [100].
TGF-β1 is a multi-functional cytokine that is a major regulator of the immune response and has profound affects on vasculogenesis, angiogenesis and the maintenance of vessel wall integrity [43]. Overexpression of TGF-β1 by the promoter of the GFAP, a specific marker of astrocytes, within the CNS can result in cerebrovascular amyloid and enhance immune cell infiltration from peripherally triggered autoimmune responses [107] (Fig. 2). In AD, the deposition of amyloid around cerebral blood vessels can lead to CAA which occurs in 80% of AD cases [108]. Aβ deposits in the cerebrovasculature are associated with the alteration of smooth muscle cells [109], the thinning of endothelial cells [110] and the loss of endothelial mitochondria [110]. The deposits cause intraparenchymal and subarachnoid bleeding, which leads to haemorrhage, stroke with hemosiderin deposits, multiple infarcts and cognitive impairment [109] (Fig. 2). Aβ increases endothelial monolayer permeability [111, 112], stimulates secretion of inflammatory cytokines [113] and up-regulates vascular cell adhesion molecules (Fig. 2).
Glial cell interaction with T cells
Although there is no clear T-cell response in the brains of AD patients, understanding T-cell responses and adaptive immunity has become important in AD immunotherapy. Studies show a stronger T-cell response to Aβ, in a proportion of healthy elderly individuals and patients with AD, than in middle-aged adults [114]. In the human trial of Aβ-peptide vaccination in which meningoencephalitis followed immunization with Aβ peptide [115, 116], the inflammatory response following immunization was composed of CD8+, CD4+ and CD3+ T cells. Recent works suggest that the final maturation of T cells might occur in the CNS through an interaction with resident antigen presented cells. Therefore, glial cells have an important role in facilitating the response to T cells. The glial cell–T cell interaction occurs through different receptors and the first type of signal mediates the fate of the immune response [117].
Different types of T cells would be expected to have different effects on microglial cell activation and therefore on amyloid clearance. Th1 cells can promote microglial cell activation through the secretion of IFN-γ, which also up-regulates the expression of MHC class II molecules by microglial cells, thereby enhancing the T cell–microglial cell interaction. Recent evidence has shown that the interaction of CD40 (expressed on the surface of microglia) with CD40L (T-cell receptor) promotes pro-inflammatory microglial cell activation in response to Aβ[118]. Transgenic mice overproducing Aβ, but deficient in CD40L, show reduced Aβ levels and Aβ plaque load [118]. This finding suggests several possible mechanisms underlying mitigation of AD pathology in response to CD40L and validates the CD40–CD40L interaction as a target for therapeutic intervention in AD [54, 118] (Table 1). As part of the defence mechanism of preventing brain inflammation, glial cells can facilitate apoptosis of the infiltrating T cells through signalling pathways initiated by CD95L, CD95L (FasL)/CD95 (Fas), perforin and the TNF receptor 1 (TNFR1) pathway [119–121].
T-cell responses might also be beneficial by aiding the clearance of Aβ, most likely through the activation of microglial cells. Hence, understanding the interaction between glial cells and T cells may reveal a new target for modulating brain inflammation and help in developing novel immunotherapeutic approaches to AD.
Glial cell activation as therapeutic target in AD
Glial cells provide a natural mechanism by which protein aggregates (including amyloid) and debris can be removed from the brain. In addition to their innate ability to phagocytose and clear Aβ, glial cells can also be activated to promote the clearance of Aβ and prevent neuronal death in AD [65, 117, 122, 123] (Table 1).
Epidemiological studies have documented a reduced prevalence of AD among users of non-steroidal anti-inflammatory drugs (NSAIDs) [124, 125]. The main known target of NSAIDs are the cyclooxygenase (COX) enzymes [125]. However, studies by Weggen et al.[126] demonstrate that a subset of NSAIDs lower amyloidogenic Aβ independently of COX activity and appear to operate by decreasing the activity of γ-secretase, an enzyme linked to increased production of Aβ. These findings support the view that decreasing the inflammatory response is not necessarily beneficial in AD and may not be linked to a reduction of Aβ in the brain. In support of this, Jantzen and colleagues [127] suggest that microglial activation may be beneficial based on results of their studies of a nitric oxide-releasing NSAID, NCX-2216. Thus, specifically activating microglia for Aβ phagocytosis, while reducing their pro-inflammatory response, is a major aim for therapeutic applications in AD that target microglial cells.
The classical mechanism for enhancing microglial phagocytosis is through the Fc receptor (FcR). Engagement of the FcR by immune IgG initiates phagocytosis [117, 128, 129], which is characterized by an extension of pseudopodia and an envelopment of the phagocytic target by a mechanism of tyrosine kinases, Rac and Cdc42 [130]. Active immunization with Aβ in mouse models of AD [131], and immunotherapy by passive administration of amyloid-specific antibodies [132] induce microglial cell activation and markedly reduces amyloid levels in the brain and reverses behavioural impairment in these mice [133, 134]. Furthermore, restricting Aβ-specific antibodies with anti-inflammatory drugs, such as dexamethasone, reduces microglial cell activation and inhibits the removal of fibrillar amyloid deposits [135]. There is also evidence that intravenous immunoglobulin (IVIg), an FDA-approved purified immunoglobulin fraction from normal human donor blood, shows promise as a passive immunotherapy for AD, which enhances microglial cell migration toward Aβ deposits, and mediates phagocytosis of Aβ[136].
The first human clinical trial of immunizing AD patients with Aβ peptide led to the activation of T cells and meningoencephalitis in some patients so the trial was discontinued [115]. Nevertheless, the cases of meningoencephalitis did not correlate with the presence of highly elevated concentrations of antibody titres to Aβ, and it is now generally believed that the meningoencephalitis was due to a T-cell response against Aβ resulting from the type of adjuvant that was used [117]. Furthermore, as it was suggested, since the Aβ peptide antibody may lead to local microhaemorrhages associated with amyloid-bearing vessels, the Aβ immunotherapy success rate might be improved by screening patients for the presence and severity of CAA (once this can be accomplished), before such therapies are undertaken. Constructing a specific antibody immune response against Aβ, and avoiding complications seen in the first clinical trial of a vaccine against Aβ remains a promising path for future therapeutic applications in AD [137]. Passive administration of an Aβ-specific humanized monoclonal antibody is currently in a phase II clinical trial of patients with AD [137, 138]. Additionally, there are also several other ongoing trials based on anti-Aβ antibodies, such as bapineuzumab (Elan/Wyeth), LY2062430 (Eli Lilly) and RN1219 (Pfizer) [138]. Furthermore, other routes of inducing autoantibodies toward Aβ, including the use of a viral envelope instead of an adjuvant or mucosal administration, might play an important role in shaping the beneficial types of glial cell activation toward the clearance of Aβ[139–141]. Nevertheless, a marked reduction of Aβ load in the brains of FcγR-deficient APP-transgenic mice following immunization with Aβ[142] indicates an additional in vivo mechanism besides FcR-mediated clearance.
Different types of T cells would be expected to have various effects on microglial cell activation and therefore on amyloid clearance [117]. A T-cell strategy would have to activate microglial cells without causing encephalitis. Recent reports [143, 144] suggest that this can be achieved in APP-transgenic mice by immunizing them with glatiramer acetate (GA) [145], which is a random amino acid copolymer of alanine, lysine, glutamic acid and tyrosine used for the treatment of multiple sclerosis that induces both Th2- and Th3-cell response. The immunization of APP-transgenic mice with GA activates microglial cells without inducing neurotoxicity and leads to Aβ clearance [143, 144]. Hence, understanding the interactions between glial cells and T cells may reveal a new target for modulating brain inflammation and help in developing novel immunotherapeutic approaches to AD.
TLRs are a family of PRRs in the innate immune system. Exogenous and endogenous TLR ligands activate microglia cell [146]. APP mouse models with TLR4 deficiency have an increase in insoluble Aβ in the cortex, as compared to TLR4 wild-type APP mouse models [146]. The activation of microglia with a TLR2, TLR4 or TLR9 ligand markedly boosted the ingestion of Aβin vitro[146]. These results suggest that the TLR signalling pathways may be involved in the clearance of Aβ deposits in the brain and that TLRs can be a therapeutic target for application in AD [146].
Activated glial cells express various SRs that mediate phagocytosis of Aβ fibrils and oligomers. Targeting SRs can mediate the activation of glial cells towards Aβ clearance [147]. A recent study found that there is a low expression of the scavenger receptor with C-type lectin (SRCL), a member of the SR family, in a double transgenic mouse model of AD and in AD patients. The presence of a large number of SRCL together with Aβ double-positive particles in the intracellular compartments of reactive astrocytes and vascular or perivascular cells in APP mice suggests a role for SRCL in Aβ clearance [148]. Lactadherin is a secreted extracellular matrix protein expressed in phagocytes and contributes to the removal of apoptotic cells. In addition, lactadherin can be expressed on vascular smooth muscle cells, astrocytes and microglia. One study found that there is less lactadherin found in the brains of AD patients [149]. This suggests that a lactadherin deficiency significantly limits Aβ phagocytosis. Hence, alterations in lactadherin production or function may contribute to the initiation or progression of AD [149].
Complement components may modulate the phagocytosis of Aβ by microglia [150]. Moreover, the blocking of complement activation diminishes microglial cell activity in APP-transgenic mice and leads to an increase of amyloid loads [31]. Thus, mechanisms that enhance the binding of complement to microglia might accelerate Aβ ingestion by microglia and may be of therapeutic value.
Considerable data has emerged indicating that several enzymes, such as insulin-degrading enzyme (IDE) and neprilysin (NEP) play a role in regulating the clearance of Aβ in the brain [151, 152]. Genetic studies have also shown that IDE and NEP gene variations are associated with the clinical symptoms of AD [151, 152]. Glial cells can also degrade Aβ by releasing other Aβ-degrading enzymes, such as metalloproteases, and gelatinase A [153, 154].
The innate immune response can include cellular activation following recognition of microbial components such as lipopolysaccharide (LPS) or other highly hydrophobic structures. In AD, Aβ is highly hydrophobic and therefore possibly induces innate immune responses similar to those triggered by LPS. Microglial cell activation due to LPS administration may mimic some features of AD [155–157]. In these studies, LPS was given intravenously [155], intrac-erebroventricularly [156] or intraperitoneally [157]. However, others studies showed enhanced Aβ clearance through microglial cell activation when LPS was injected intrahippocampally [158, 159] or intracranial [160] to older APP-transgenic mice. LPS might lead to microglial cell activation through an interaction with TLR4. Furthermore, nasal administration of a proteosome-based adjuvant (Protollin) [161], composed of purified outer membrane proteins of Neisseria meningitides and LPS, also reduces Aβ in an APP-transgenic mouse model in correlation with microglial cell activation [143]. Protollin might act by stimulating microglial cells both through LPS via TLR4 and through Por B, which makes up 70% of the proteosome protein and activates antigen-presenting cells via TLR2 [143].
Microtubules are the key component of the neuronal and glial cytoskeleton that regulates cell division, differentiation and protection. Tau proteins interact with tubulin to stabilize microtubules and promote tubulin assembly into microtubules. Increased tau hyperphosphorylation causes memory impairments in neurodegenerative diseases associated with tauopathies, including the most prevalent AD. Research of the activity-dependent neuroprotective protein (ADNP), a gene product essential for brain formation, shows that ADNP knockout is embryonic lethal and that an ADNP partial deficiency results in cognitive deficits that are paralleled by tau hyperphosphorylation [162]. Structure-activity studies identified a short peptide sequence in ADNP, known as a neuronal microtubule-interacting agent (NAP; amino acid sequence NAPVSIPQ), which mimics the neuropro-tective activity of the parent protein and crosses the BBB after systemic or intranasal administration and can support the integrity of the astrocyte and microglial microtubules [163, 164]. ADNP/NAP can also play an important role in immune regulation and down-regulates the key inflammatory cytokines such as TNF-α and IL-12 [165]. The use of NAP may open up new horizons in research and development of protective compounds to both neurons and the supporting glial cells [166, 167]. NAP is currently in phase II clinical trials by Allon Therapeutics to assess the affects on MCI [162].
Adult hippocampal neurogenesis, the process that creates neurons, dramatically decreases with increasing age and it has been proposed that this decline may contribute to age-related memory deficits, such as in AD [168, 169]. While the causes of age-associated decline in neurogenesis have yet to be fully determined, a loss in growth factors in the microenvironment of the subgranular zone (SGZ) and subventricular zone (SVZ) are prime contributors to the reduced neurogenic potential in the hippocam-pus and cortex regions affected in AD [170]. The ability of glia to produce neurotrophic factors is one aspect that makes glial cells attractive as therapeutic tools. Glial cells produce their own repertoire of neurotrophic factors, including nerve growth factor (NGF), brain-derived growth factor (BDGF), glial cell-derived neurotrophic factor (GDNF), insulin-like growth factor-I (IGF-1) and vascular endothelial growth factor (VEGF) [170]. Recent works suggest that glial cells can mediate neurogenesis in neurodegenerative diseases, such as AD [144].
Conclusion
Glial cells are not only essential for maintaining a healthy well-functioning brain, but they also protect and aid the brain in the functional recovery from injuries. The activation of glial cells in the CNS is the first defence mechanism against pathological abnormalities that occur in neurodegenerative diseases. Different signals can mediate and influence the fate of glial cell activation. As discussed in this review, a dysfunction of glial cell receptors, which alters the glial cells'sense of their environment, can lead to the development of neurological diseases. Emerging evidences in literature suggest that changes in the dividing process of glial cells during aging might lead to their abnormality and may provide clues to the pathogenesis of devastating diseases, such as AD. Because glial cells make up the largest cell population in the brain and continually renew during a human being's lifetime, this makes them a perfect target for therapeutic intervention in neurological dis-eases. Nevertheless, because there is such diversity among each glia family (astrocyte, microglia and oligodendrocyte), they are still a mystery that holds the key to their modification. However, glial cell plasticity and diversity may allow the maturation of new functional cells, which can help the neuronal repair process. Yet, the basis of successful therapeutic application in neurodegenerative diseases depends on understanding the neuronal–glial cell interaction. Advanced analysis may shed light on genes involved in glial cell function thus leading to the development of new drug-target candidates. Furthermore, understanding the glial cells’ continuous dialogue with cells from the peripheral immune response, may open up a new horizon for vaccine application. Hence, by modifying the activity of glial cells to reduce their neurotoxic properties and enhance their neuroprotective effects may pave the path for therapeutic interventions in neurodegenerative diseases, such as AD.
Acknowledgments
This work is supported by a Dana Foundation grant (to D.F.). We express our thanks to Rachel Barnett for editing this work.
References
- 1.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–8. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 2.Heinonen O, Soininen H, Sorvari H, Kosunen O, Paljärvi L, Koivisto E, Riekkinen PJ., Sr Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in Alzheimer's disease. Neuroscience. 1995;64:375–84. doi: 10.1016/0306-4522(94)00422-2. [DOI] [PubMed] [Google Scholar]
- 3.Jellinger KA, Bancher C. AD neuropathology. Neurology. 1996;46:1186–7. doi: 10.1212/wnl.46.4.1186-b. [DOI] [PubMed] [Google Scholar]
- 4.Goldman JS, Reed B, Gearhart R, Kramer JH, Miller BL. Very early-onset familial Alzheimer's disease: a novel presenilin 1 mutation. Int J Geriatr Psychiatry. 2002;17:649–51. doi: 10.1002/gps.657. [DOI] [PubMed] [Google Scholar]
- 5.Wisniewski T, Ghiso J, Frangione B. Alzheimer's disease and soluble A beta. Neurobiol Aging. 1994;15:143–52. doi: 10.1016/0197-4580(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. Alzheimer's disease: A central role for amyloid. J Neuropathol Exp Neurol. 1994;53:438–47. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 8.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 9.Klein WL. Abeta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–52. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 10.Li M, Chen L, Lee DH, Yu LC, Zhang Y. The role of intracellular amyloid beta in Alzheimer's disease. Prog Neurobiol. 2007;83:131–9. doi: 10.1016/j.pneurobio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:4725–7. doi: 10.1073/pnas.92.11.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bronfman FC, Garrido J, Alvarez A, Morgan C, Inestrosa NC. Laminin inhibits amyloid-beta-peptide fibrillation. Neurosci Lett. 1996;218:201–3. doi: 10.1016/s0304-3940(96)13147-5. [DOI] [PubMed] [Google Scholar]
- 13.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 14.Bamberger ME, Landreth GE. Microglial interaction with beta-amyloid: implications for the pathogenesis of Alzheimer's disease. Microsc Res Tech. 2001;54:59–70. doi: 10.1002/jemt.1121. [DOI] [PubMed] [Google Scholar]
- 15.Gaskin F, Finley J, Fang Q, Xu S, Fu SM. Human antibodies reactive with beta-amyloid protein in Alzheimer's disease. J Exp Med. 1993;177:1181–6. doi: 10.1084/jem.177.4.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyman BT, Smith C, Buldyrev I, Whelan C, Brown H, Tang MX, Mayeux R. Autoantibodies to amyloid-beta and Alzheimer's disease. Ann Neurol. 2001;49:808–10. doi: 10.1002/ana.1061. [DOI] [PubMed] [Google Scholar]
- 17.Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology. 2000;49:171–86. doi: 10.1016/s0162-3109(00)80302-1. [DOI] [PubMed] [Google Scholar]
- 18.McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging. 2001;22:799–809. doi: 10.1016/s0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 19.Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuro-protective mediators. Glia. 2006;53:688–95. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- 20.Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002;40:195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- 21.Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. J Neuroimmunol. 2002;130:86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- 22.Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Shen Y, Lue L, Yang L, Roher A, Kuo Y, Strohmeyer R, Goux WJ, Lee V, Johnson GV, Webster SD, Cooper NR, Bradt B, Rogers J. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1992;89:10016–20. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webster S, Rogers J. Relative efficacies of amyloid beta peptide (A beta) binding proteins in A beta aggregation. J Neurosci Res. 1996;46:58–66. doi: 10.1002/(SICI)1097-4547(19961001)46:1<58::AID-JNR8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 25.McGreal E, Gasque P. Structure-function studies of the receptors for complement C1q. Biochem Soc Trans. 2002;30:1010–4. doi: 10.1042/bst0301010. [DOI] [PubMed] [Google Scholar]
- 26.Hafer-Macko CE, Dyck PJ, Koski CL. Complement activation in acquired and hereditary amyloid neuropathy. J Peripher Nerv Syst. 2000;5:131–9. doi: 10.1046/j.1529-8027.2000.00018.x. [DOI] [PubMed] [Google Scholar]
- 27.Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer's disease brain. Am J Pathol. 1999;154:927–36. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci. 2004;24:6457–65. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGeer PL, McGeer EG. Mechanisms of cell death in Alzheimer disease–immunopathology. J Neural Transm Suppl. 1998;54:159–66. doi: 10.1007/978-3-7091-7508-8_15. [DOI] [PubMed] [Google Scholar]
- 30.Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Antibody-mediated phagocytosis of the amyloid beta-peptide in microglia is differentially modulated by C1q. J Immunol. 2001;166:7496–503. doi: 10.4049/jimmunol.166.12.7496. [DOI] [PubMed] [Google Scholar]
- 31.Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. 2002;99:10837–42. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia MQ, Hyman BT. Chemokines/chemokine receptors in the central nervous system and Alzheimer's disease. J Neurovirol. 1999;5:32–41. doi: 10.3109/13550289909029743. [DOI] [PubMed] [Google Scholar]
- 33.Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous sys-tem: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 34.Xia M, Qin S, McNamara M, Mackay C, Hyman BT. Interleukin-8 receptor B immunoreactivity in brain and neuritic plaques of Alzheimer's disease. Am J Pathol. 1997;150:1267–74. [PMC free article] [PubMed] [Google Scholar]
- 35.Galimberti D, Schoonenboom N, Scarpini E, Scheltens P. Chemokines in serum and cere-brospinal fluid of Alzheimer's disease patients. Ann Neurol. 2003;53:547–8. doi: 10.1002/ana.10531. [DOI] [PubMed] [Google Scholar]
- 36.Oprica M, Hjorth E, Spulber S, Popescu BO, Ankarcrona M, Winblad B, Schultzberg M. Studies on brain volume, Alzheimer-related proteins and cytokines in mice with chronic overexpression of IL-1 receptor antagonist. J Cell Mol Med. 2007;11:810–25. doi: 10.1111/j.1582-4934.2007.00074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging. 2001;22:903–8. doi: 10.1016/s0197-4580(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 38.Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–88. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata N, Ohnuma T, Takahashi T, Baba H, Ishizuka T, Ohtsuka M, Ueki A, Nagao M, Arai H. Effect of IL-6 polymorphism on risk of Alzheimer disease: genotype-phenotype association study in Japanese cases. Am J Med Genet. 2002;114:436–9. doi: 10.1002/ajmg.10417. [DOI] [PubMed] [Google Scholar]
- 40.Schultzberg M, Lindberg C, Aronsson AF, Hjorth E, Spulber SD, Oprica M. Inflammation in the nervous system–physiological and pathophysiological aspects. Physiol Behav. 2007;92:121–8. doi: 10.1016/j.physbeh.2007.05.050. [DOI] [PubMed] [Google Scholar]
- 41.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 42.Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK. Sustained hip-pocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117:1595–604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wyss-Coray T. Tgf-Beta pathway as a potential target in neurodegeneration and Alzheimer's. Curr Alzheimer Res. 2006;3:191–5. doi: 10.2174/156720506777632916. [DOI] [PubMed] [Google Scholar]
- 44.Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature. 1997;389:603–6. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- 45.Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–8. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 46.Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Martin Watterson D. Glia proinflam-matory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol. 2007;82:277–96. doi: 10.1016/S0074-7742(07)82015-0. [DOI] [PubMed] [Google Scholar]
- 47.Streit WJ. Microglia and Alzheimer's disease pathogenesis. J Neurosci Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- 48.Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation. 2005;2:22. doi: 10.1186/1742-2094-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cagnin A, Myers R, Gunn RN, Lawrence AD, Stevens T, Kreutzberg GW, Jones T, Banati RB. In vivo visualization of activated glia by [11C] (R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain. 2001;124:2014–27. doi: 10.1093/brain/124.10.2014. [DOI] [PubMed] [Google Scholar]
- 50.Cagnin A, Rossor M, Sampson EL, Mackinnon T, Banati RB. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol. 2004;56:894–7. doi: 10.1002/ana.20332. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Liu L, Liu D, Woodward S, Barger SW, Mrak RE, Griffin WS. Microglial activation by uptake of fDNA via a scavenger receptor. J Neuroimmunol. 2004;147:50–5. doi: 10.1016/j.jneuroim.2003.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–65. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 53.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–9. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 54.Townsend KP, Town T, Mori T, Lue LF, Shytle D, Sanberg PR, Morgan D, Fernandez F, Flavell RA, Tan J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur J Immunol. 2005;35:901–10. doi: 10.1002/eji.200425585. [DOI] [PubMed] [Google Scholar]
- 55.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eikelenboom P, Veerhuis R. The role of complement and activated microglia in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 1996;17:673–80. doi: 10.1016/0197-4580(96)00108-x. [DOI] [PubMed] [Google Scholar]
- 57.Giulian D, Haverkamp LJ, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher AE. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer's disease. J Biol Chem. 1998;273:29719–26. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 58.Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer's beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:RC221. doi: 10.1523/JNEUROSCI.22-10-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boland K, Behrens M, Choi D, Manias K, Perlmutter DH. The serpin-enzyme complex receptor recognizes soluble, nontoxic amyloid-beta peptide but not aggregated, cytotoxic amyloid-beta peptide. J Biol Chem. 1996;271:18032–44. doi: 10.1074/jbc.271.30.18032. [DOI] [PubMed] [Google Scholar]
- 60.Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of microglial receptor for advanced glycation endprod-ucts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 61.Munch G, Thome J, Foley P, Schinzel R, Riederer P. Advanced glycation endproducts in ageing and Alzheimer's disease. Brain Res Brain Res Rev. 1997;23:134–43. doi: 10.1016/s0165-0173(96)00016-1. [DOI] [PubMed] [Google Scholar]
- 62.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meda L, Baron P, Scarlato G. Glial activation in Alzheimer's disease: the role of Abeta and its associated proteins. Neurobiol Aging. 2001;22:885–93. doi: 10.1016/s0197-4580(01)00307-4. [DOI] [PubMed] [Google Scholar]
- 64.Weiner HL, Selkoe DJ. Inflammation and therapeutic vaccination in CNS diseases. Nature. 2002;420:879–84. doi: 10.1038/nature01325. [DOI] [PubMed] [Google Scholar]
- 65.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 66.Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007;10:61–74. doi: 10.1089/rej.2006.9096. [DOI] [PubMed] [Google Scholar]
- 67.Carnevale D, De Simone R, Minghetti L. Microglia-neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS Neurol Disord Drug Targets. 2007;6:388–97. doi: 10.2174/187152707783399193. [DOI] [PubMed] [Google Scholar]
- 68.De Simone R, Ajmone-Cat MA, Carnevale D, Minghetti L. Activation of alpha7 nicotinic acetyl-choline receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation. 2005;2:4. doi: 10.1186/1742-2094-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 70.Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiol Aging. 2001;22:945–56. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 71.Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of mono-cyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer's disease. Psychiatry Clin Neurosci. 1997;51:135–8. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- 72.Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer's disease brains. Am J Pathol. 1998;153:31–7. doi: 10.1016/s0002-9440(10)65542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–37. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 74.Haydon PG. Neuroglial networks: neurons and glia talk to each other. Curr Biol. 2000;10:R712–4. doi: 10.1016/s0960-9822(00)00708-9. [DOI] [PubMed] [Google Scholar]
- 75.Sofroniew MV, Bush TG, Blumauer N, Lawrence K, Mucke L, Johnson MH. Genetically-targeted and conditionally-regulated ablation of astroglial cells in the central, enteric and peripheral nervous systems in adult transgenic mice. Brain Res. 1999;835:91–5. doi: 10.1016/s0006-8993(99)01639-x. [DOI] [PubMed] [Google Scholar]
- 76.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–45. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 77.Shao Y, Gearing M, Mirra SS. Astrocyte-apolipopro-tein E associations in senile plaques in Alzheimer disease and vascular lesions: a regional immunohis-tochemical study. J Neuropathol Exp Neurol. 1997;56:376–81. doi: 10.1097/00005072-199704000-00006. [DOI] [PubMed] [Google Scholar]
- 78.Marshak DR, Pesce SA, Stanley LC, Griffin WS. Increased S100 beta neurotrophic activity in Alzheimer's disease temporal lobe. Neurobiol Aging. 1992;13:1–7. doi: 10.1016/0197-4580(92)90002-f. [DOI] [PubMed] [Google Scholar]
- 79.Da Cunha A, Jefferson JJ, Tyor WR, Glass JD, Jannotta FS, Vitkovic L. Gliosis in human brain: relationship to size but not other properties of astrocytes. Brain Res. 1993;600:161–5. doi: 10.1016/0006-8993(93)90415-j. [DOI] [PubMed] [Google Scholar]
- 80.Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ, Griffin WS. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. 1996;17:761–6. doi: 10.1016/0197-4580(96)00104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Edwards MM, Robinson SR. TNF alpha affects the expression of GFAP and S100B: implications for Alzheimer's disease. J Neural Transm. 2006;113:1709–15. doi: 10.1007/s00702-006-0479-5. [DOI] [PubMed] [Google Scholar]
- 82.Frohman EM, Van Den Noort S, Gupta S. Astrocytes and intracerebral immune responses. J Clin Immunol. 1989;9:1–9. doi: 10.1007/BF00917121. [DOI] [PubMed] [Google Scholar]
- 83.Sheng JG, Mrak RE, Griffin WS. S100 beta protein expression in Alzheimer disease: potential role in the pathogenesis of neuritic plaques. J Neurosci Res. 1994;39:398–404. doi: 10.1002/jnr.490390406. [DOI] [PubMed] [Google Scholar]
- 84.Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J Immunol. 1990;144:2999–3007. [PubMed] [Google Scholar]
- 85.Bsibsi M, Bajramovic JJ, Van Duijvenvoorden E, Persoon C, Ravid R, Van Noort JM, Vogt MH. Identification of soluble CD14 as an endogenous agonist for Toll-like receptor 2 on human astrocytes by genome-scale functional screening of glial cell derived proteins. Glia. 2007;55:473–82. doi: 10.1002/glia.20473. [DOI] [PubMed] [Google Scholar]
- 86.Aloisi F, Carè A, Borsellino G, Gallo P, Rosa S, Bassani A, Cabibbo A, Testa U, Levi G, Peschle C. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1 beta and tumor necrosis factor-alpha. J Immunol. 1992;149:2358–66. [PubMed] [Google Scholar]
- 87.Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4:590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hurwitz AA, Lyman WD, Berman JW. Tumor necrosis factor alpha and transforming growth factor beta upreg-ulate astrocyte expression of monocyte chemoattrac-tant protein-1. J Neuroimmunol. 1995;57:193–8. doi: 10.1016/0165-5728(95)00011-p. [DOI] [PubMed] [Google Scholar]
- 89.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer's disease. J Neuroimmunol. 2000;108:227–35. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 90.DeWitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer's disease. Exp Neurol. 1998;149:329–40. doi: 10.1006/exnr.1997.6738. [DOI] [PubMed] [Google Scholar]
- 91.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 92.Fagan AM, Holtzman DM. Astrocyte lipoproteins, effects of apoE on neuronal function, and role of apoE in amyloid-beta deposition in vivo. Microsc Res Tech. 2000;50:297–304. doi: 10.1002/1097-0029(20000815)50:4<297::AID-JEMT9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 93.Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–26. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 94.Guo S, Wang S, Kim WJ, Lee SR, Frosch MP, Bacskai BJ, Greenberg SM, Lo EH. Effects of apoE isoforms on beta-amyloid-induced matrix metallopro-teinase-9 in rat astrocytes. Brain Res. 2006;1111:222–6. doi: 10.1016/j.brainres.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 95.Dolev I, Michaelson DM. A nontransgenic mouse model shows inducible amyloid-beta (Abeta) peptide deposition and elucidates the role of apolipoprotein E in the amyloid cascade. Proc Natl Acad Sci USA. 2004;101:13909–14. doi: 10.1073/pnas.0404458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang J, Aschner M. Developmental aspects of blood-brain barrier (BBB) and rat brain endothelial (RBE4) cells as in vitro model for studies on chlorpyrifos transport. Neurotoxicology. 2003;24:741–5. doi: 10.1016/S0161-813X(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 97.Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–31. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 98.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–13. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 99.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–99. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Donahue JE, Flaherty SL, Johanson CE, Duncan JA, 3rd, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q, Sabo E, Hovanesian V, Stopa EG. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 2006;112:405–15. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 101.Han X. The role of apolipoprotein E in lipid metabolism in the central nervous system. Cell Mol Life Sci. 2004;61:1896–906. doi: 10.1007/s00018-004-4009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hayashi H, Campenot RB, Vance DE, Vance JE. Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J Neurosci. 2007;27:1933–41. doi: 10.1523/JNEUROSCI.5471-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Levy-Lahad E, Bird TD. Genetic factors in Alzheimer's disease: a review of recent advances. Ann Neurol. 1996;40:829–40. doi: 10.1002/ana.410400604. [DOI] [PubMed] [Google Scholar]
- 104.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A, Migheli A, Stern D. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–712. [PMC free article] [PubMed] [Google Scholar]
- 105.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–91. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 106.Deane R, Zlokovic BV. Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2007;4:191–7. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- 107.Wyss-Coray T, Borrow P, Brooker MJ, Mucke L. Astroglial overproduction of TGF-beta 1 enhances inflammatory central nervous system disease in transgenic mice. J Neuroimmunol. 1997;77:45–50. doi: 10.1016/s0165-5728(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 108.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–36. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 109.Weller RO, Nicoll JA. Cerebral amyloid angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res. 2003;25:611–6. doi: 10.1179/016164103101202057. [DOI] [PubMed] [Google Scholar]
- 110.Szpak GM, Lewandowska E, Wierzba-Bobrowicz T, Bertrand E, Pasennik E, Mendel T, Stepien T, Leszczynska A, Rafalowska J. Small cerebral vessel disease in familial amyloid and non-amyloid angiopathies: FAD-PS-1 (P117L) mutation and CADASIL. Immunohistochemical and ultrastructural studies. Folia Neuropathol. 2007;45:192–204. [PubMed] [Google Scholar]
- 111.Blanc EM, Toborek M, Mark RJ, Hennig B, Mattson MP. Amyloid beta-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells. J Neurochem. 1997;68:1870–81. doi: 10.1046/j.1471-4159.1997.68051870.x. [DOI] [PubMed] [Google Scholar]
- 112.Strazielle N, Ghersi-Egea JF, Ghiso J, Dehouck MP, Frangione B, Patlak C, Fenstermacher J, Gorevic P. In vitro evidence that beta-amyloid peptide 1–40 diffuses across the blood-brain barrier and affects its permeability. J Neuropathol Exp Neurol. 2000;59:29–38. doi: 10.1093/jnen/59.1.29. [DOI] [PubMed] [Google Scholar]
- 113.Suo Z, Tan J, Placzek A, Crawford F, Fang C, Mullan M. Alzheimer's beta-amyloid peptides induce inflammatory cascade in human vascular cells: the roles of cytokines and CD40. Brain Res. 1998;807:110–7. doi: 10.1016/s0006-8993(98)00780-x. [DOI] [PubMed] [Google Scholar]
- 114.Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe DJ, Weiner HL. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415–22. doi: 10.1172/JCI18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 116.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 117.Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer's disease. Nat Rev Immunol. 2006;6:404–16. doi: 10.1038/nri1843. [DOI] [PubMed] [Google Scholar]
- 118.Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–93. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- 119.Gasque P, Jones J, Singhrao SK, Morgan B. Identification of an astrocyte cell population from human brain that expresses perforin, a cytotoxic protein implicated in immune defense. J Exp Med. 1998;187:451–60. doi: 10.1084/jem.187.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Magnus T, Chan A, Grauer O, Toyka KV, Gold R. Microglial phagocytosis of apoptotic inflammatory T cells leads to down-regulation of microglial immune activation. J Immunol. 2001;167:5004–10. doi: 10.4049/jimmunol.167.9.5004. [DOI] [PubMed] [Google Scholar]
- 121.Medana I, Li Z, Flugel A, Tschopp J, Wekerle H, Neumann H. Fas ligand (CD95L) protects neurons against perforin-mediated T lymphocyte cytotoxicity. J Immunol. 2001;167:674–81. doi: 10.4049/jimmunol.167.2.674. [DOI] [PubMed] [Google Scholar]
- 122.Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev. 2005;48:234–9. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 123.Takata K, Kitamura Y, Yanagisawa D, Morikawa S, Morita M, Inubushi T, Tsuchiya D, Chishiro S, Saeki M, Taniguchi T, Shimohama S, Tooyama I. Microglial transplantation increases amyloid-beta clearance in Alzheimer model rats. FEBS Lett. 2007;581:475–8. doi: 10.1016/j.febslet.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 124.McGeer PL, McGeer EG. Anti-inflammatory drugs in the fight against Alzheimer's disease. Ann N Y Acad Sci. 1996;777:213–20. doi: 10.1111/j.1749-6632.1996.tb34421.x. [DOI] [PubMed] [Google Scholar]
- 125.Hoozemans JJ, Veerhuis R, Rozemuller AJ, Eikelenboom P. Non-steroidal anti-inflammatory drugs and cyclooxygenase in Alzheimer's disease. Curr Drug Targets. 2003;4:461–8. doi: 10.2174/1389450033490902. [DOI] [PubMed] [Google Scholar]
- 126.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–6. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 127.Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002;22:2246–54. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–21. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- 129.Kaplan G. Differences in the mode of phagocytosis with Fc and C3 receptors in macrophages. Scand J Immunol. 1977;6:797–807. doi: 10.1111/j.1365-3083.1977.tb02153.x. [DOI] [PubMed] [Google Scholar]
- 130.Ohsawa K, Imai Y, Kanazawa H, Sasaki Y, Kohsaka S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci. 2000;113:3073–84. doi: 10.1242/jcs.113.17.3073. [DOI] [PubMed] [Google Scholar]
- 131.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 132.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 133.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–82. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 134.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–5. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 135.Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol Dis. 2004;15:11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 136.Solomon B. Antibody-mediated immunotherapy for Alzheimer's disease. Curr Opin Investig Drugs. 2007;8:519–24. [PubMed] [Google Scholar]
- 137.Solomon B. Beta-amyloidbased immunotherapy as a treatment of Alzheimers disease. Drugs Today. 2000;43:333–42. doi: 10.1358/dot.2007.43.5.1062670. [DOI] [PubMed] [Google Scholar]
- 138.Melnikova I. Therapies for Alzheimer's disease. Nat Rev Drug Discov. 2007;6:341–2. doi: 10.1038/nrd2314. [DOI] [PubMed] [Google Scholar]
- 139.Frenkel D, Dewachter I, Van Leuven F, Solomon B. Reduction of beta-amyloid plaques in brain of transgenic mouse model of Alzheimer's disease by EFRH-phage immunization. Vaccine. 2003;21:1060–5. doi: 10.1016/s0264-410x(02)00609-6. [DOI] [PubMed] [Google Scholar]
- 140.Hara H, Monsonego A, Yuasa K, Adachi K, Xiao X, Takeda S, Takahashi K, Weiner HL, Tabira T. Development of a safe oral Abeta vaccine using recombinant adeno-associated virus vector for Alzheimer's disease. J Alzheimers Dis. 2004;6:483–8. doi: 10.3233/jad-2004-6504. [DOI] [PubMed] [Google Scholar]
- 141.Mouri A, Noda Y, Hara H, Mizoguchi H, Tabira T, Nabeshima T. Oral vaccination with a viral vector containing Abeta cDNA attenuates age-related Abeta accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J. 2007;21:2135–48. doi: 10.1096/fj.06-7685com. [DOI] [PubMed] [Google Scholar]
- 142.Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE. Amyloid-beta immunization effectively reduces amyloid deposition in FcRgamma-/- knockout mice. J Neurosci. 2003;23:8532–8. doi: 10.1523/JNEUROSCI.23-24-08532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Frenkel D, Maron R, Burt DS, Weiner HL. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. J Clin Invest. 2005;115:2423–33. doi: 10.1172/JCI23241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci USA. 2006;103:11784–9. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aharoni R, Kayhan B, Eilam R, Sela M, Arnon R. Glatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situ. Proc Natl Acad Sci USA. 2003;100:14157–62. doi: 10.1073/pnas.2336171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–19. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Verdier Y, Penke B. Binding sites of amyloid beta-peptide in cell plasma membrane and implications for Alzheimer's disease. Curr Protein Pept Sci. 2004;5:19–31. doi: 10.2174/1389203043486937. [DOI] [PubMed] [Google Scholar]
- 148.Nakamura K, Ohya W, Funakoshi H, Sakaguchi G, Kato A, Takeda M, Kudo T, Nakamura T. Possible role of scavenger receptor SRCL in the clearance of amyloid-beta in Alzheimer's disease. J Neurosci Res. 2006;84:874–90. doi: 10.1002/jnr.20992. [DOI] [PubMed] [Google Scholar]
- 149.Boddaert J, Kinugawa K, Lambert JC, Boukhtouche F, Zoll J, Merval R, Blanc-Brude O, Mann D, Berr C, Vilar J, Garabedian B, Journiac N, Charue D, Silvestre JS, Duyckaerts C, Amouyel P, Mariani J, Tedgui A, Mallat Z. Evidence of a role for lactadherin in Alzheimer's disease. Am J Pathol. 2007;170:921–9. doi: 10.2353/ajpath.2007.060664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol. 2000;161:127–38. doi: 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- 151.Eckman EA, Eckman CB. Abeta-degrading enzymes: modulators of Alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans. 2005;33:1101–5. doi: 10.1042/BST20051101. [DOI] [PubMed] [Google Scholar]
- 152.Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–93. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 153.Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–8. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 154.Yamada T, Miyazaki K, Koshikawa N, Takahashi M, Akatsu H, Yamamoto T. Selective localization of gelatinase A, an enzyme degrading beta-amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol. 1995;89:199–203. doi: 10.1007/BF00309334. [DOI] [PubMed] [Google Scholar]
- 155.Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, Carter DB, Chin JE. Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer's disease. Brain Res Bull. 2001;56:581–8. doi: 10.1016/s0361-9230(01)00730-4. [DOI] [PubMed] [Google Scholar]
- 156.Qiao X, Cummins DJ, Paul SM. Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur J Neurosci. 2001;14:474–82. doi: 10.1046/j.0953-816x.2001.01666.x. [DOI] [PubMed] [Google Scholar]
- 157.Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroin-flammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–45. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- 158.DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–12. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 159.Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, Gordon MN. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–53. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 160.Quinn J, Montine T, Morrow J, Woodward WR, Kulhanek D, Eckenstein F. Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer's disease. J Neuroimmunol. 2003;137:32–41. doi: 10.1016/s0165-5728(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 161.Jones T, Cyr S, Allard F, Bellerose N, Lowell GH, Burt DS. Protollin: a novel adjuvant for intranasal vaccines. Vaccine. 2004;22:3691–7. doi: 10.1016/j.vaccine.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 162.Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, Gozes I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. 2007;323:438–49. doi: 10.1124/jpet.107.129551. [DOI] [PubMed] [Google Scholar]
- 163.Divinski I, Mittelman L, Gozes I. A femtomolar acting octapeptide interacts with tubulin and protects astrocytes against zinc intoxication. J Biol Chem. 2004;279:28531–8. doi: 10.1074/jbc.M403197200. [DOI] [PubMed] [Google Scholar]
- 164.Zaltzman R, Alexandrovich A, Trembovler V, Shohami E, Gozes I. The influence of the peptide NAP on Mac-1-deficient mice following closed head injury. Peptides. 2005;26:1520–7. doi: 10.1016/j.peptides.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 165.Quintana FJ, Zaltzman R, Fernandez-Montesinos R, Herrera JL, Gozes I, Cohen IR, Pozo D. NAP, a peptide derived from the activity-dependent neuro-protective protein, modulates macrophage function. Ann N Y Acad Sci. 2006;1070:500–6. doi: 10.1196/annals.1317.069. [DOI] [PubMed] [Google Scholar]
- 166.Gozes I, Divinski I. The femtomolar-acting NAP interacts with microtubules: Novel aspects of astrocyte protection. J Alzheimers Dis. 2004;6:S37–41. doi: 10.3233/jad-2004-6s605. [DOI] [PubMed] [Google Scholar]
- 167.Gozes I, Morimoto BH, Tiong J, Fox A, Sutherland K, Dangoor D, Holser-Cochav M, Vered K, Newton P, Aisen PS, Matsuoka Y, Van Dyck CH, Thal L. NAP: research and development of a peptide derived from activity-dependent neuroprotective protein (ADNP) CNS Drug Rev. 2005;11:353–68. doi: 10.1111/j.1527-3458.2005.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kempermann G, Wiskott L, Gage FH. Functional significance of adult neurogenesis. Curr Opin Neurobiol. 2004;14:186–91. doi: 10.1016/j.conb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 169.Spencer B, Rockenstein E, Crews L, Marr R, Masliah E. Novel strategies for Alzheimer's disease treatment. Expert Opin Biol Ther. 2007;7:1853–67. doi: 10.1517/14712598.7.12.1853. [DOI] [PubMed] [Google Scholar]
- 170.Siegel GJ, Chauhan NB. Neurotrophic factors in Alzheimer's and Parkinson's disease brain. Brain Res Brain Res Rev. 2000;33:199–227. doi: 10.1016/s0165-0173(00)00030-8. [DOI] [PubMed] [Google Scholar]
- 171.Rogers J, Strohmeyer R, Kovelowski CJ, Li R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia. 2002;40:260–9. doi: 10.1002/glia.10153. [DOI] [PubMed] [Google Scholar]
- 172.Peress NS, Fleit HB, Perillo E, Kuljis R, Pezzullo C. Identification of Fc gamma RI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer's disease. J Neuroimmunol. 1993;48:71–9. doi: 10.1016/0165-5728(93)90060-c. [DOI] [PubMed] [Google Scholar]
- 173.Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, Wu Z, Holtzman DM, Zlokovic BV. IgG-assisted age-dependent clearance of Alzheimer's amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci. 2005;25:11495–503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–9. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 175.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–24. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 176.Bsibsi M, Ravid R, Gveric D, Van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–21. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 177.Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer's Disease. Cell Physiol Biochem. 2007;20:947–56. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 178.Alarcon R, Fuenzalida C, Santibanez M, Von Bernhardi R. Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J Biol Chem. 2005;280:30406–15. doi: 10.1074/jbc.M414686200. [DOI] [PubMed] [Google Scholar]
- 179.Husemann J, Loike JD, Kodama T, Silverstein SC. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J Neuroimmunol. 2001;114:142–50. doi: 10.1016/s0165-5728(01)00239-9. [DOI] [PubMed] [Google Scholar]
- 180.Husemann J, Silverstein SC. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer's disease brain. Am J Pathol. 2001;158:825–32. doi: 10.1016/S0002-9440(10)64030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Granucci F, Petralia F, Urbano M, Citterio S, Di Tota F, Santambrogio L, Ricciardi-Castagnoli P. The scavenger receptor MARCO mediates cytoskeleton rearrangements in dendritic cells and microglia. Blood. 2003;102:2940–7. doi: 10.1182/blood-2002-12-3651. [DOI] [PubMed] [Google Scholar]
- 182.Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–12. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–66. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang X, Lee SR, Arai K, Lee SR, Tsuji K, Rebeck GW, Lo EH. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–7. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 185.Fassbender K, Walter S, Khl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ, Rivest S, Lentschat A, Gulbins E, Jucker M, Staufenbiel M, Brechtel K, Walter J, Multhaup G, Penke B, Adachi Y, Hartmann T, Beyreuther K. The LPS receptor (CD14) links innate immunity with Alzheimer's disease. FASEB J. 2004;18:203–5. doi: 10.1096/fj.03-0364fje. [DOI] [PubMed] [Google Scholar]
- 186.Cui Y, Le Y, Yazawa H, Gong W, Wang JM. Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer's disease. J Leukoc Biol. 2002;72:628–35. [PubMed] [Google Scholar]
- 187.Iribarren P, Zhou Y, Hu J, Le Y, Wang JM. Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol Res. 2005;31:165–76. doi: 10.1385/IR:31:3:165. [DOI] [PubMed] [Google Scholar]
- 188.Le Y, Gong W, Tiffany HL, Tumanov A, Nedospasov S, Shen W, Dunlop NM, Gao JL, Murphy PM, Oppenheim JJ, Wang JM. Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci. 2001;21:RC123. doi: 10.1523/JNEUROSCI.21-02-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Togo T, Akiyama H, Kondo H, Ikeda K, Kato M, Iseki E, Kosaka K. Expression of CD40 in the brain of Alzheimer's disease and other neurological diseases. Brain Res. 2000;885:117–21. doi: 10.1016/s0006-8993(00)02984-x. [DOI] [PubMed] [Google Scholar]
- 190.Mach F, Schönbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA. 1997;94:1931–6. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Calingasan NY, Erdely HA, Altar CA. Identification of CD40 ligand in Alzheimer's disease and in animal models of Alzheimer's disease and brain injury. Neurobiol Aging. 2002;23:31–9. doi: 10.1016/s0197-4580(01)00246-9. [DOI] [PubMed] [Google Scholar]
- 192.Lehmann DJ, Wiebusch H, Marshall SE, Johnston C, Warden DR, Morgan K, Schappert K, Poirier J, Xuereb J, Kalsheker N, Welsh KI, Smith AD. HLA class I, II & III genes in confirmed late-onset Alzheimer's disease. Neurobiol Aging. 2001;22:71–7. doi: 10.1016/s0197-4580(00)00180-9. [DOI] [PubMed] [Google Scholar]
- 193.Burudi EM, Riese S, Stahl PD, Regnier-Vigouroux A. Identification and functional characterization of the mannose receptor in astrocytes. Glia. 1999;25:44–55. doi: 10.1002/(sici)1098-1136(19990101)25:1<44::aid-glia5>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]