

Abstract
Oxidative stress has been implicated in the pathogenesis of Alzheimer's disease (AD). Both AD and arguably its earlier form, mild cognitive impairment (MCI), have elevated membrane oxidative damage in brain. The tumor suppressor and transcription factor p53 plays a pivotal function in neuronal apoptosis triggered by oxidative stress. Apoptosis contributes to neuronal death in many neurological disorders, including AD. In this study, we investigated p53 expression in a specific region of the cerebral cortex, namely the inferior parietal lobule (IPL), in MCI and AD brain, to test the hypothesis that alterations of this pro-apoptotic protein may be involved in neuronal death in the progression of AD. By immunoprecipitation assay, we also investigated whether 4-hydroxy-2-transnonenal (HNE), an aldehydic product of lipid peroxidation, was bound in excess to p53 in IPL from subjects with MCI and AD compared to control. Overall, the data provide evidence that p53 is involved in the neuronal death in both MCI and AD, suggesting that the observed alterations are early events in the progression of AD. In addition, HNE may be a novel non-protein mediator of oxidative stress-induced neuronal apoptosis.
Keywords: mild cognitive impairment (MCI), Alzheimer's disease (AD), apoptosis, oxidative stress, 4-hydroxynonenal, HNE, p53
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by progressive cognitive impairment, deficits in acquiring memory, disordered spatiotemporal relationships and altered personality effects [1]. Characteristic neuropathological features of AD include: neuronal loss, gliosis, neurofibrillary tangles, proliferated and dystrophic neurites, neuritic plaques and fibrils and deposits of amyloid β-peptide in the cerebral cortex [2–4]. The symptoms of this disease result from degeneration and death of specific populations of synapses and neurons [1, 5]. A wide variety of cellular and molecular processes have been proposed to underlie the relationship between neuronal death and AD, but the actual correlation has yet to be elucidated.
There is much evidence to suggest that p53, a tumor suppressor protein, is a key mediator of neuronal death, as this protein plays an essential role in the death of many cell types. Data from in vivo and in vitro models documented an increase of p53 levels in affected neurons, supporting a potentially pivotal role of this protein in neuronal loss in neurodegenerative conditions [6–9].
p53 is a multifunctional protein whose main role is to maintain genomic integrity [10]. Exposure to different signals such as radiation, genotoxic chemicals, hypoxia, oxidative stress, and depletion of ribonucleotides, induces p53 to accumulate in the nucleus, to bind to specific DNA sequences and to transactivate several genes (transcriptional-dependent pathways), most of which are involved in cell-cycle control, DNA repair and apoptosis [11]. However, evidence for transcription-independent pathways of p53-mediated apoptosis is growing [12, 13]. This evidence shows that p53 might contribute to apoptosis by direct signaling to mitochondria [14], thus leading to cytochrome c (cyt-c) release and caspase activation [15]. Lack of p53 activity could be essentially due to gene mutations, as it often occurs in tumors, or post-transcriptional modification, both possibilities potentially altering p53 tertiary structure and preventing its binding to specific DNA sequences [16–18].
Oxidative stress has been implicated as an important factor in the etiology and pathogenesis of neurodegenerative diseases, including AD [19–22]. Increased protein oxidation, protein nitration and lipid peroxidation occur in neurofibrillary tangles and neuritic plaques of AD patients, and levels of oxidation products are also increased in cerebrospinal fluid of AD patients [23]. In particular, one product of lipid peroxidation, 4-hydroxy-2-nonenal (HNE), has been shown to be elevated in AD brain [24, 25]. HNE is highly reactive with cellular components, including proteins and DNA. In particular, HNE-DNA adducts are a potent mutagen in human cells and are preferentially formed at codon 249 of the p53 gene, a mutational hotspot in human cancers [26]. HNE causes disruption of Ca2+ homeostasis, membrane damage and cell death [27]. Several authors have investigated the mechanisms by which HNE causes cell death in human colorectal carcinoma cells. These authors found out that such mechanisms lead to apoptosis by inducing alterations of mitochondrial function, yielding release of cyt-c and subsequent activation of the caspase cascade [28]. HNE can also activate the Fas/FasL signaling-mediated apoptotic pathway in epithelial cells [29]. In addition, it has been shown that exogenous application of HNE induces apoptotic death in PC12 cells and hippocampal neuronal culture [30].
Mild cognitive impairment (MCI) represents a transition stage between the cognitive changes of normal aging and the more serious problems caused by AD [31]. Therefore, for those patients who convert to AD, MCI arguably is the earliest stage of this dementing disorder. MCI is characterized by a memory deficit without loss of general cognitive and functional abilities. Recently, data have suggested that there are increased HNE levels in MCI brain, including the inferior parietal lobule (IPL) [32, 33]. These observations suggest that oxidative stress occurs as an early event in the progression of AD.
The aim of the current study was to investigate whether p53 levels were altered in AD and MCI IPL, thereby implicating p53 in apoptosis in MCI and AD, and to know whether HNE is implicated mechanistically in the activation of the apoptosis mediated by p53. The identification of HNE as a trigger of p53 signaling may provide new clues to apoptotic mechanisms in neurodegenerative diseases.
Materials and methods
Materials
All chemicals used were purchased from Sigma-Aldrich (St. Louis, MO, USA) with exceptions of nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), electrophoretic transfer system (Transblot semi-dry Transfer Cell; Bio-Rad), anti-p53 monoclonal antibody used for immunoprecipitation and Western blotting (Calbiochem, LA Jolla, CA, USA), anti-HNE polyclonal antibody (Alpha diagnostic International, St. Antonio, TX, USA), and anti-mouse IgG alkaline phosphatase secondary antibody (Chemicon International, Temulca, CA, USA).
Patients
Frozen IPL samples from MCI, AD and age-matched controls were obtained from the University of Kentucky Rapid Autopsy Program of the Alzheimer's Disease Clinical Center (UK ADC). The diagnosis of probable AD was made according to criteria developed by the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer's Disease and Related Disorders Association (ADRDA) [34]. All AD patients displayed progressive intellectual decline. Control subjects were without history of dementia or other neurological disorders and underwent annual mental status testing and semi-annual physical and neurological exams as part of the UK ADC normal volunteer longitudinal aging study. In addition, patients had test scores in the normal range (Table 2). Samples and demographics used for the AD study were described previously [35]. Other demographic parameters of control, MCI, and AD subjects are provided in Tables 1 and 2.
2.
Characteristics of control and AD patients (mean ± SD)
| Demographic variables | Control subjects | AD subjects |
|---|---|---|
| Number of subjects | 5 | 5 |
| Gender (male/female) | 3/2 | 3/2 |
| Age at death (yrs) | 87.0 ± 3.94 | 85.8 ± 6.02 |
| Postmortem interval (hr) | 2.9 ± 0.70 | 3.4 ± 1.4 |
| MMSE; number of months prior to death test taken | 28 ± 0.8;6.6 ±1.4 | 15.7 ± 2.6;19.7 ± 1.0 |
| APOE genotype, if known (N) | 3/3 (3) 3/4 (2) | ND |
AD, Alzheimer's disease; MMSE, mini-mental state examination;APOE, apolipoprotein E; ND, not determined; N, number of individuals; SD, standard deviation; COPD, chronic obstructive pulmonary disease (adapted from [35]).
1.
Characteristics of control and MCI patients (mean ± SD)
| Demographic variables | Control subjects | MCI subjects |
|---|---|---|
| Number of subjects | 7 | 7 |
| Gender (male/female) | 3/4 | 3/4 |
| Postmortem interval (hr) | 2.87 ± 1.14 | 3.125 ± 1.033 |
| Brain weight (g) | 1260 ± 120 | 1120 ± 61 |
| Braak stage | I–II | III–V |
MCI, mild cognitive impairment.
Sample preparation
Samples were prepared as described previously [36], with small changes. The brain tissues (IPL) from control, MCI and AD were sonicated in lysis buffer (pH 7.4) containing 10 mM HEPES buffer, 137 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4 and 0.6 mM MgSO4, as well as proteinase inhibitors leupeptin (0.5 mg/ml), pepstatin (0.7 mg/ml), type II S soybean trypsin inhibitor (0.5 mg/ml) and PMSF (40 mg/ml). Homogenates were centrifuged at 14,000 (g for 10 min to remove debris. Protein concentration in the super-natant was determined by the Pierce BCA method (Pierce, Rockford, IL, USA).
Western blotting analysis
For immunoblot analysis, 75 μg of protein extracts were electrophoresed on 10% SDS-PAGE, and transferred to nitrocellulose paper (Bio-Rad Trans-blot semi-dry Transfer Cell) at 45 mA for 2 hrs. Subsequently, the membranes were blocked at 4°C for 1 hr with fresh blocking buffer made of 5% non-fat dried milk in phosphate-buffered saline containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 (PBST). The membranes were incubated for 3 hrs at room temperature with primary antibodies: Anti-p53 monoclonal antibody (1:500). After washing the blots three times in PBST for 5 min each, blots were incubated with anti-mouse IgG alkaline phosphatase secondary antibody (1:2000) in PBST for 1 hr at room temperature. Membranes were then washed three times in PBST for 5 min and developed using 5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium (BCIP/NBT) color developing reagent. Blots were dried and scanned with Adobe Photoshop and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image) software.
Immunoprecipitation analysis
For immunoprecipitation experiments, 150 μg of protein extracts were resuspended in 500 μl RIPA buffer (10 mM Tris, pH 7.6;140 mM NaCl;0.5% NP40 including protease inhibitors) and then incubated with 1 μg of conformation-specific antibody against p53 protein (wild-type specific—PAb11) at 4°C overnight. Immunocomplexes were collected by using protein A/G suspension and washed five times with immunoprecipitation buffer. Immunoprecipitated p53 was recovered by resuspending the pellets in loading buffer, and protein was detected by Western blotting with rabbit antibody against HNE (1:2000).
Statistics
All statistical analysis was performed using a two-tailed Student's t-test. P < 0.05 was considered significantly different from control.
Results and discussion
A major hallmark of AD is the progressive neuronal loss associated with reduced memory function. Previous studies suggested an involvement of p53 in degenerating neurons in AD. For example, de la Monte et al. [7], showed increased p53 and Fas expression in specific populations of cortical neurons. Kitamura et al.[8] reported increased levels of p53 in temporal cortex, in particular in glial cells. The present study was conducted in another region of cerebral cortex, namely the IPL, in MCI, AD and control brains.
The IPL region exhibits severe histopathologic alterations in AD, that correlate to oxidative damage [37], including elevated lipid peroxidation [25, 38]. To determine whether p53 levels were elevated in MCI (Fig. 1A) and AD (Fig. 1B), the results of Western blotting of IPL were compared to control. Statistical analysis of p53 expression data found a significant increase of p53 levels in both MCI (*P < 0.04) and AD (#P < 0.015), compared to controls. These results are consistent with the notion that neurons are degenerating in this particular region of the cerebral cortex by p53-dependent pathways from an early stage of AD. p53 levels are largely regulated in response to injury by changes in protein degradation, although this may not necessarily apply to post-mitotic cells such as neurons. In fact, most studies of p53 were performed on non-neuronal cells, but some laboratory investigated the role of p53 in neuronal cell death. They found in the primary culture of rat cerebellar granule cells an increase of p53-immunoreactivity and p53 DNA binding activity after exposure to excitatory conditions [39]. These data in total suggest that neuronal cells are very sensitive to apoptosis, and different triggers, such involved also in neurodegenerative conditions as excitotoxicity, increasing of intracellular calcium concentration, oxygen free radical production, could activate the apoptotic pathway by p53.
1.
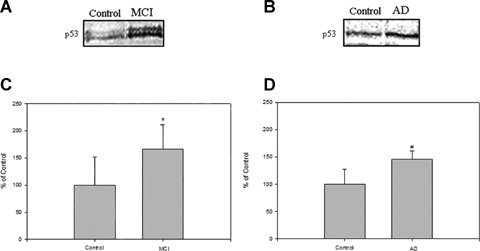
(A) and (B) represent blots of the levels of p53 in the IPL from MCI, AD and control, respectively.(C) and (D) represent densitometric analysis of (A) and (B), respectively. Equal amounts of protein (75 mg/lane) were electrophoresed using SDS-PAGE. Proteins were transferred to nitrocellulose membranes and probed with the primary anti-p53 antibody. (A) is a representative blot of data obtained from seven control and MCI samples, and (B) is a representative blot of data obtained from five control and AD samples, respectively. The control value was set to 100%, to which experimental values were compared.*P < 0.04;AD, #P < 0.015.
HNE has been reported to be one to the most cytotoxic products of lipid peroxidation [40]. The reactive sites on HNE (aldehyde functionality, double bond and hydroxy functionality) allow for reactions with cysteines, histidines and lysines by Michael addition to form covalent adducts [40–42]. HNE can cause further cross-linking through the side chain of lysine residues on other protein molecules via formation of a Schiff base [40, 42]. Modified and/or cross-linked proteins cause increased protein-protein interactions and/or lead to proteins with altered conformation. If generated at high concentrations, HNE can alter the physical state of lipids in the cell membranes, which can affect the function of transmembrane proteins [43]. HNE is a relative stable aldehyde that can diffuse to different sub-cellular compartments and interacts with many different cell proteins, like histones [44] and tau [45].
To determine if HNE also binds to p53 protein to potentially influence its pro-apoptotic activity, an immunoprecipitation experiment was performed using an antibody that binds to and precipitates p53. This complex was then probed with an antibody against HNE-bound proteins by Western blotting analysis. The results showed a significant difference in AD IPL compared to the controls (Fig. 2B; #P < 0.004), but no significant difference in HNE bound to p53 in MCI samples (Fig. 2A), though the mean level was elevated.
2.
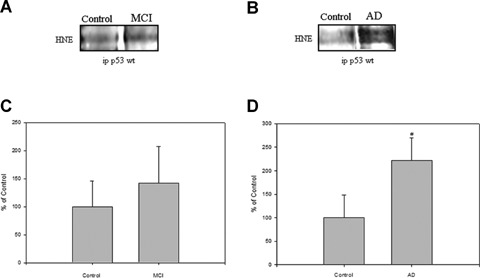
(A) and (B) represent blots of p53-HNE adduction studied from MCI, AD and control, respectively.(C) and (D) represent densitometric analysis of (A) and (B), respectively. Equal amount of protein (150 mg/lane) were immunoprecipitated by anti-p53 antibody, and immunoprecipitates were analyzed for HNE immunoreactivity by Western blotting.(A) is a representative blot of data obtained from seven control and MCI samples, and (B) is representative blot of data obtained from five control and AD samples, respectively. The control value was set to 100%, to which experimental values were compared. AD, #P < 0.004.
HNE is correlated to neuronal death since, when HNE is generated in response to apoptotic oxidative insults, it can induce neuronal apoptosis [30]. In addition, increased production of HNE is an index of increased level of oxidative damage [25, 32, 33, 46]. Recent work has shown that p53 was present in synapses and was increased in response to DNA damage, and oxidative and excitotoxic insults [6]. In many types of post-mitotic neurons, p53 may mediate apoptosis induced by a range of insults including DNA damage and oxidative stress [47, 48].
Lipid peroxidation is present in the brain of MCI patients [32] and suggests that oxidative stress may play a role in the pathogenesis of AD. HNE and acrolein are significantly increased in specific regions of MCI brains in comparison to control brains [32, 33]. Moreover, plasma, cerebrospinal fluid and urine of MCI patients showed significantly higher levels of isoprostanes, another product of lipid peroxidation, compared with cognitively normal elderly subjects [49, 50]. These results suggest that individuals with MCI have increased of oxidative damage before the onset of dementia.
The results in this report provide additional evidence confirming the link between oxidative stress and neuronal death of MCI and AD. For the first time, elevated p53 levels in MCI IPL have been demonstrated. Based on these studies, p53 is hypothesized to be involved in apoptotic pathways in brain from subjects with both MCI and AD, as shown by a significant increase of the levels of this transcription factor. HNE significantly binds p53 in AD IPL, but not in MCI IPL. This difference may suggest a direct involvement of lipid peroxidation product in p53 pathway in an advanced stage of neurodegeneration, but not in an earlier phase. It is not possible to speculate at this time if HNE causes activation of p53 and, as a consequence, apoptosis by changes in p53 conformation. Consequently, further studies of HNE in apoptotic neuronal death are ongoing to clarify its role in this important pathway for neurodegeneration and AD.
Acknowledgments
This work was supported in part by NIH grants AG-05119, AG-10836.
References
- 1.Katzman R, Saitoh T. Advances in Alzheimer's disease. FASEB J. 1991;5:278–86. [PubMed] [Google Scholar]
- 2.Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 3.Barcikowska M, Wisniewski HM, Bancher C, Grundke-Iqbal I. About the presence of paired helical filaments in dystrophic neurites participating in the plaque formation. Acta Neuropathol. 1989;78:225–31. doi: 10.1007/BF00687751. [DOI] [PubMed] [Google Scholar]
- 4.Terry RD, Hansen LA, DeTeresa R, Davies P, Tobias H, Katzman R. Senile dementia of the Alzheimer type without neocortical neurofibrillary tangles. J Neuropathol Exp Neurol. 1987;46:262–8. doi: 10.1097/00005072-198705000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–9. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 6.Gilman CP, Chan SL, Guo Z, Zhu X, Greig N, Mattson MP. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Med. 2003;3:159–72. doi: 10.1385/NMM:3:3:159. [DOI] [PubMed] [Google Scholar]
- 7.De La Monte SM, Sohn YK, Wands JR. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci. 1997;152:73–83. doi: 10.1016/s0022-510x(97)00131-7. [DOI] [PubMed] [Google Scholar]
- 8.Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T. Changes of p53 in the brains of patients with Alzheimer's disease. Biochem Biophys Res Commun. 1997;232:418–21. doi: 10.1006/bbrc.1997.6301. [DOI] [PubMed] [Google Scholar]
- 9.Alves da Costa C, Paitel E, Mattson MP, Amson R, Telerman A, Ancolio K, Checler F. Wild-Type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and murine neurons. Proc Natl Acad Sci USA. 2002;99:4043–8. doi: 10.1073/pnas.062059899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–72. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 11.Almog N, Rotter V. Involvement of p53 in cell differentiation and development. Biochim Biophys Acta. 1997;1333:F1–27. doi: 10.1016/s0304-419x(97)00012-7. [DOI] [PubMed] [Google Scholar]
- 12.Gottlieb TM, Oren M. p53 and apoptosis. Semin Cancer Biol. 1998;8:359–68. doi: 10.1006/scbi.1998.0098. [DOI] [PubMed] [Google Scholar]
- 13.Sansome C, Zaika A, Marchenko ND, Moll UM. Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett. 2001;488:110–5. doi: 10.1016/s0014-5793(00)02368-1. [DOI] [PubMed] [Google Scholar]
- 14.Marchenko ND, Zaika A, Moll UM. Death signalinduced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–12. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 15.Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem. 2000;275:7337–42. doi: 10.1074/jbc.275.10.7337. [DOI] [PubMed] [Google Scholar]
- 16.Meplan C, Richard MJ, Hainaut P. Redox signalling and transition metals in the control of the p53 pathway. Biochem Pharmacol. 2000;59:25–33. doi: 10.1016/s0006-2952(99)00297-x. [DOI] [PubMed] [Google Scholar]
- 17.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 18.McLure KG, Lee PW. p53 DNA binding can be modulated by factors that alter the conformational equilibrium. EMBO J. 1999;18:763–70. doi: 10.1093/emboj/18.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–77. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–60. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 21.Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM, Harman D. Oxygen radicals and human disease. Ann Intern Med. 1987;107:526–45. doi: 10.7326/0003-4819-107-4-526. [DOI] [PubMed] [Google Scholar]
- 22.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–47. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 23.Hensley K, MaidtML,Yu Z, Sangh H, Markesbery WR, Floyd RA. Electrochemical analysis of nitrotyrosine and dityrosine in Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–32. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–6. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 25.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1–42. J Neurochem. 2001;78:413–6. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 26.Hu W, Feng Z, Eveleigh J, Iyer G, Pan J, Amin S, Chung FL, Tang MS. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis. 2002;23:1781–9. doi: 10.1093/carcin/23.11.1781. [DOI] [PubMed] [Google Scholar]
- 27.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–43. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 28.Ji C, Amarnath V, Pietenpol JA, Marnett LJ. 4-hydroxynonenal induces apoptosis via caspase-3 activation and cytochrome c release. Chem Res Toxicol. 2001;14:1090–6. doi: 10.1021/tx000186f. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Sharma R, Patrick B, Sharma A, Jeyabal PV, Reddy PM, Saini MK, Dwivedi S, Dhanani S, Ansari NH, Zimniak P, Awasthi S, Awasthi YC. Regulation of CD95 (Fas) expression and Fas-mediated apoptotic signaling in HLE B-3 cells by 4-hydroxynonenal. Biochemistry. 2006;45:12253–64. doi: 10.1021/bi060780+. [DOI] [PubMed] [Google Scholar]
- 30.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petersen RC. Mild cognitive impairment clinical trials. Nat Rev Drug Discov. 2003;2:646–53. doi: 10.1038/nrd1155. [DOI] [PubMed] [Google Scholar]
- 32.Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006;397:170–3. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 33.Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging. 2006;27:1094–9. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 34.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 35.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–76. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 36.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22:223–32. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, Lovell M, Markesbery WR, Butterfield DA. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–56. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 38.Palmer AM, Burns MA. Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer's disease. Brain Res. 1994;645:338–42. doi: 10.1016/0006-8993(94)91670-5. [DOI] [PubMed] [Google Scholar]
- 39.Uberti D, Belloni M, Grilli M, Spano P, Memo M. Induction of tumour-suppressor phosphoprotein p53 in the apoptosis of cultured rat cerebellar neurones triggered by excitatory amino acids. Eur J Neurosci. 1998;10:246–54. doi: 10.1046/j.1460-9568.1998.00042.x. [DOI] [PubMed] [Google Scholar]
- 40.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 41.Uchida K, Osawa T, Hiai H, Toyokuni S. 4-Hydroxy-2-nonenal-trapping ELISA: direct evidence for the release of a cytotoxic aldehyde from oxidized low density lipoproteins. Biochem Biophys Res Commun. 1995;212:1068–73. doi: 10.1006/bbrc.1995.2078. [DOI] [PubMed] [Google Scholar]
- 42.Butterfield DA, Stadtman ER. Protein oxidation processes in aging brain. Adv Cell Aging Gerontol. 1997;2:161–91. [Google Scholar]
- 43.Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997;69:1161–9. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- 44.Drake J, Petroze R, Castegna A, Ding Q, Keller JN, Markesbery WR, Lovell MA, Butterfield DA. 4-Hydroxynonenal oxidatively modifies histones: implications for Alzheimer's disease. Neurosci Lett. 2004;356:155–8. doi: 10.1016/j.neulet.2003.11.047. [DOI] [PubMed] [Google Scholar]
- 45.Mattson MP, Fu W, Waeg G, Uchida K. 4-Hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. NeuroReport. 1997;8:2275–81. doi: 10.1097/00001756-199707070-00036. [DOI] [PubMed] [Google Scholar]
- 46.McGrath LT, McGleenon BM, Brennan S, McColl D, Mc IS, Passmore AP. Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM. 2001;94:485–90. doi: 10.1093/qjmed/94.9.485. [DOI] [PubMed] [Google Scholar]
- 47.Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA. p53-dependent cell death signaling in neurons. Neurochem Res. 2003;28:15–27. doi: 10.1023/a:1021687810103. [DOI] [PubMed] [Google Scholar]
- 48.Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. J Neurosci. 2003;23:8586–95. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pratico D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ. Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of Alzheimer disease. Arch Neurol. 2002;59:972–6. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]
- 50.Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Lipid peroxidation in aging brain and Alzheimer's disease. Free Radic Biol Med. 2002;33:620–6. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]